

Abstract
BACKGROUND & AIMS
The NF-κB/IKKβ pathway has been shown to represent a key link between inflammation and cancer, inducing pro-inflammatory cytokines in myeloid cells and antiapoptotic pathways in epithelial cells. However, the role of NF-κB pathway in gastric carcinogenesis and injury has not been well defined. We derived mice with a conditional knockout of Ikkβ in gastric epithelial cells (GECs) and myeloid cells, and examined responses to ionizing radiation (IR) and Helicobacter felis (Hf) infection.
METHODS
IkkβΔstom mice were generated by crossing Foxa3-Cre mice to IkkβF/F mice. Cellular stress was induced with IR and Hf in IkkβΔstom, IkkβF/F, and cis-NF-κB-EGFP reporter mice. Gastric histopathology, apoptosis, proliferation, necrosis, reactive oxygen species (ROS) and the expression of cytokines, chemokines and antiapoptotic genes were assessed for up to 18 months post-infection. The role of myeloid IKKβ in these models was studied by crosses with LysM-Cre mice.
RESULTS
NF-κB activity was upregulated in myeloid cells with acute Hf infection, but in epithelial cells by IR or long-term Hf infection during progression to dysplasia. Deletion of IKKβ in GECs led to increased apoptosis, ROS and cellular necrosis, and resulted in upregulation of IL-1α and downregulation of antiapoptotic genes. Loss of IKKβ in GECs resulted in worse inflammation and more rapid progression to gastric preneoplasia, while loss of IKKβ in myeloid cells inhibited the development of gastric atrophy.
CONCLUSIONS
The loss of IKKβ/NF-κB signaling in GECs results in increased apoptosis and necrosis in response to cellular stress, and accelerated development of dysplasia by Helicobacter infection.
Keywords: Helicobcater, NF-κB, interleukin-1α, apoptosis, gastric cancer
Introduction
The gastrointestinal tract is under continuous exposure to injurious agents that can lead to cellular stress and trigger epithelial damage and cell death. Epithelial cells can die in different ways that can be distinguished on the basis of cell morphology as well as the intracellular signaling pathways.1 Apoptosis is a naturally occurring form of cell death that can be initiated through the extrinsic and/or the intrinsic pathway, and deregulation of this process contributes to the pathogenesis of a wide spectrum of disorders represented by cancer.2 Ionizing radiation (IR) is known to be a specific trigger of apoptosis, particularly in the gastrointestinal tract3 and as such IR is often used to treat advanced cancers which are often more resistant to apoptosis.4 Apoptotic cells send out signals to phagocytic cells that locate and engulf the dying cells in a generally non-inflammatory manner. In contrast to apoptosis, cellular necrosis lacks the features of apoptosis or autophagy and does not occur normally.5 Cellular necrosis is induced by various external factors, and typically leads to oxidative stress and a strong inflammatory response.6 Although the relationship of necrosis to inflammation has for a while been poorly understood, recent studies suggest that IL-1α acts as a mediator that translates signals from necrotic cells to induce the recruitment of immune cells to the site of injury.7, 8
The IKKβ/NF-κB signaling complex represents a key pathway that regulates apoptosis in epithelial cells and modulates the response of the gastrointestinal mucosa to external stimuli.9 NF-κB is a transcriptional complex that consists of a heterodimer of p50 and RelA/p65 that in leukocytes can activate numerous downstream target genes, many of which play a role in immune responses.10 Under resting conditions, NF-κB forms a complex with the inhibitor of κBα (IκBα) which serves as a negative regulator of the complex, confining it to the cytoplasm and preventing nuclear localization. Many stimulatory factors, including microbial infection (LPS) and proinflammatory cytokines such as TNF–α and IL-1β, can activate NF-κB, primarily through IKKβ-dependent phosphorylation and degradation of IκBα proteins.11 Degradation of IκBα allows translocation of NF-κB into the nucleus where it can activated transcription of downstream targets. Indeed, in the murine intestinal epithelium, the NF-κB pathway was shown to be an important mechanism that allows for protection from apoptosis induced by IR and dextran sulfate sodium (DSS) treatment.12, 13
Recently, the role of NF-κB in the development of cancer has become more clearly defined through conditional deletion of IKKβ in specific cell types. In the colonic epithelium, deletion of IKKβ results in markedly reduced progression of colon cancer in the AOM+DSS model.14 In contrast, deletion of IKKβ in hepatocytes results in acceleration of liver cancer in the diethylnitrosamine(DEN) model.15 A similar increase in cancer has been observed in models of skin cancer.16 In the liver model, the increase in cancer was due to enhanced reactive oxygen species (ROS) production, increased hepatocyte death, and augmented compensatory proliferation of surviving hepatocytes.15 Thus, the early increase in cell death in some of these models has been in a large part attributed to the loss of the antiapoptotic function of IKKβ/NF-κB in these epithelial cells types. In addition, IKKβ has been deleted from myeloid cells in a number of these cancer models that resulted in marked reductions in tumor development, indicating a key role for proinflammatory signals, particularly IL-6, in driving cancer progression.17 Thus, the IKK/NF-κB pathway does appear to be a key bridge between inflammation and cancer.
Little is known about the role of IKKβ/NF-κB in the gastric epithelium and in the predisposition to gastric cancer which is strongly associated with chronic infection with Helicobacter pylori.18 Gastric cancer is thought to be induced to a large extent by the H. pylori-dependent immune response and the upregulation of inflammatory cytokines such as IL-1β in myeloid cells,19, 20 although direct interactions between GECs and H. pylori may also play role. In addition, inhibition of apoptosis in Fas null mice blocks gastric cancer progression,21 suggesting that early induction of apoptosis could drive the development of gastric cancer. Chronic infection by H. pylori has been shown to activate NF-κB signaling in vitro,22 but the precise localization of NF-κB activation in vivo has not been well defined. Activation of NF-κB by Helicobacter infection is believed to occur at lower levels in GECs,23, 24 although the precise significance of this interaction is unclear.
To determine the role of NF-κB in response to cellular stress associated with induction of apoptosis, we employed two different models (IR and H. felis infection) in combination with NF-κB reporter mice25 and IKKβ conditional knockout mice.26–28 These studies confirm a key role for IKKβ/NF-κB in epithelial cells in inhibition of both apoptotic and necrotic cell death, and thus in prevention of progression to gastric dysplasia.
Materials and methods
Mice
All animal studies were approved by the Institutional Animal Care and Use Committee at Columbia University. Foxa3-Cre mice on a C57BL6/J × CD1 mixed background were used to direct expression of Cre recombinase to the gastric mucosa.27 cis-NF-κBEGFP 25 LysM-Cre,26 IkkβF/F,28 and Rosa26r reporter mice,29 on a pure C57BL6/J background were described previously. All protocols for bacterial culture, acute injury model induced by IR, chronic H. felis infection model, histological evaluation, immunohistochemical studies, ELISA, real-time qRT-PCR assay of H. felis infection in mouse stomachs, proinflammatory CC-chemokines, and cytokines are detailed in the Supplementary Methods.
Results
NF-κB is Upregulated in Response to Gastric Epithelial Injury by Ionizing Radiation or Chronic Helicobacter Infection
In order to investigate the role of IKKβ/NF-κB in GECs in vivo, we first studied the activation of NF-κB in the stomach under two conditions of cellular stress, IR and H. felis infection, using the cis-NF-κBEGFP mice. While there were few detectable EGFP expressions under basal non-stressed conditions, we observed a marked induction of EGFP expression 4 hrs after 12 Gy IR, primarily in GECs (Figure 1A). We confirmed the marked activation of NF-κB after IR using immunohistochemistry for phospho-IκBα, and nuclear p65-NF-κB, which was higher in IkkβF/F mice, compared to mice lacking IKKβ in GECs (IkkβΔstom) (Figure 1B, Supplementary Figure 6D). IR-induced NF-κB activation was also paralleled by a 2.2-fold increase in mRNA levels of TNF-α after 4h IR (Figure 1C).
Figure 1.
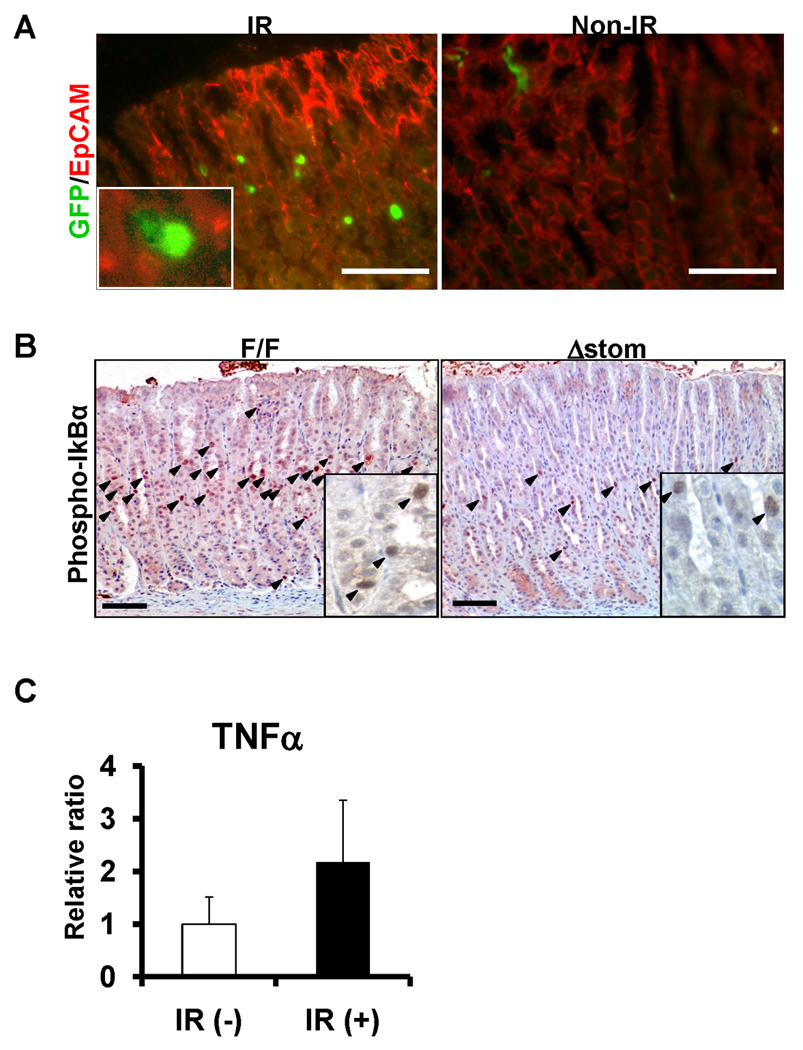
Activation of NF-κB in murine gastric epithelium after IR. (A) cis-NF-κBEGFP mice were exposed to whole-body IR, and sacrificed at 4h. EGFP expression was detected by immunofluorescent microscopy. (red) EpCAM- immunohistochemistry as a marker for epithelial cells, original magnification 400×. Inset, 600×. Scale bars, 50µm. (B) Activation of NF-κB in IkkβF/F and IkkβΔstom mice after IR. Arrows indicated the positive cells for phosphor-IκBα, the marker of NF-κB activation. Original magnification 200×. Inset, 600×. Scale bars, 50µm. (C) TNFα mRNA expression in stomach 4 hrs after 12Gy IR. Values represent the mean ± SE (n = 3 per group).
As a second model of gastric epithelial injury, we studied long-term H. felis infected mice for evidence of NF-κB activation. Whereas few EGFP expressing cells were detectable in early H. felis-infected mice, greater EGFP expression was seen in later stages of infection (Figure 2). As suggested in previous studies,20 NF-κB activation at early time points was found mainly in stromal cells in mice showing mild-to-moderate inflammation. However, at 24-mo post-infection, NF-κB activation was detected in both stromal cells and GECs, particularly in animals with gastric dysplasia, as determined by EGFP expression and nuclear NF-κB-p65 (Figure 2). Thus, cellular stress due to both IR and chronic infection results in NF-κB activation in GECs, although the latter case occurs with a delayed time course.
Figure 2.
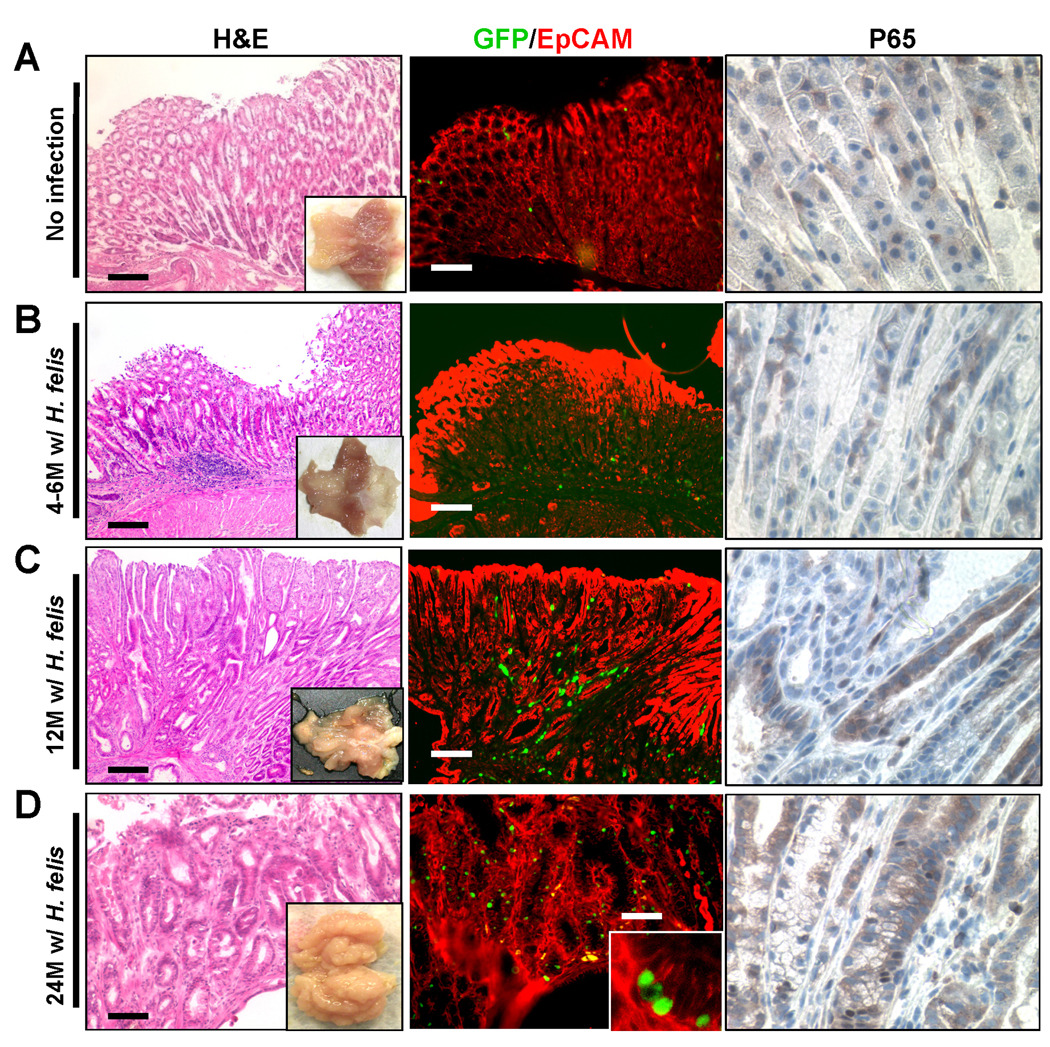
NF-κB activation in murine GECs in cis-NF-κBEGFP mice infected with H. felis. (A) non-infected mice, H. felis infected mice for 4–6 months (B), 12 months (C) and 24 months (D). HandE (lt.), and macrograph of the stomach (lt. inset). EGFP expression was detected by immunofluorescent microscopy (middle), and immunohistochemistry for NF-κB-p65 (rt., 600×). (Red) EpCAM immunohistochemistry, original magnification 100×. Scale bars, 100µm (n = 5 per period).
Deletion of Ikkβ in GECs Using the Foxa3-Cre to Generate IkkβΔstom Mice
In order to investigate the functional significance of NF-κB in murine GECs, we performed GEC-specific gene ablation of Ikkβ using crosses to Foxa3-Cre mice.27 Foxa3-Cre mice were initially crossed with Rosa26R mice to see whether Cre specifically recombined the loxP sites in GECs (Supplementary Figure 1A). Next, we crossed Foxa3-Cre mice with Ikkβ floxed (IkkβF/F) mice to delete Ikkβ gene in GECs (IkkβΔstom) (Supplementary Figure 1B).28 In Foxa3-Cre mice homozygous for the IkkβF allele, complete recombination of this locus (producing the IkkβΔ allele) occurred in GECs (Supplementary Figure 1C and 1D). Quantification of residual IkkβF using qRT-PCR of genomic DNA obtained from whole gastric tissue revealed that <50% retained the non-recombined locus. Since GECs represent only a fraction of the cells in the stomach, the residual Ikkβ expression can be attributed largely to non-epithelial cell types. To confirm this, we isolated GECs and performed qRT-PCR analysis which revealed that Ikkβ mRNA expression in IkkβΔstom mice was less than 20% of that in control mice (Supplementary Figure 1E). In subsequent studies, we compared IkkβΔstom mice to control, Cre-negative-IkkβF/F littermates. IkkβΔstom mice appeared normal, fertile, gained weight at the same rate as IkkβF/F mice, and showed no significant phenotypes in the organs analyzed (Supplementary Figure 2).
Mice with Gastric Deficiency in IKKβ Show More Rapid Progression to Preneoplasia and Neoplasia
In order to explore the long-term consequences of altered gastric epithelial homeostasis in the setting of Ikkβ deficiency, we carried out long-term observational studies in two groups of mice: (1) H. felis-infected IkkβΔstom; and (2) H. felis-infected IkkβF/F mice. The mice were all on the same genetic background (C57BL/6 × CD1), and examined at 3, 6, 12, and 18 months after H. felis infection.
Significant gastric lesions were not observed in uninfected mice at any time point up to 18 months of observation (data not shown). In contrast, in all infected groups, gastric mucosal inflammation, epithelial cell defects, pseudopyloric metaplasia, and foveolar hyperplasia were detected, however, oxyntic atrophy was detected only in H. felis-infected IkkβΔstom mice at 3 months post-infection. At 6 months post-infection, IkkβΔstom mice had marked distortions of the gastric units, decreased number of parietal cells, and increased inflammatory cell infiltration (Figure 3A). Every histopathological parameter was more severe in IkkβΔstom than in IkkβF/F mice. Neither metaplasia nor dysplasia was prominent at this early time point. At 12 months post-infection, there were moderate to severe inflammation, mucous neck cell hyperplasia with dramatic loss of parietal cell confirmed by immunohistochemistry for HK-ATPaseβ and TFF2 in IkkβΔstom compared with IkkβF/F mice (Figure 3B, Supplementary Figure 3). Although metaplastic glands were also seen extending throughout the mucosa of in IkkβΔstom, dysplasia was barely detected in IkkβΔstom up to 12 months post-infection. The slow progression to dysplasia in this study was due to the mixed CD1/C57BL/6 background which is moderately resistant to Helicobacter-dependent carcinogenesis.
Figure 3.
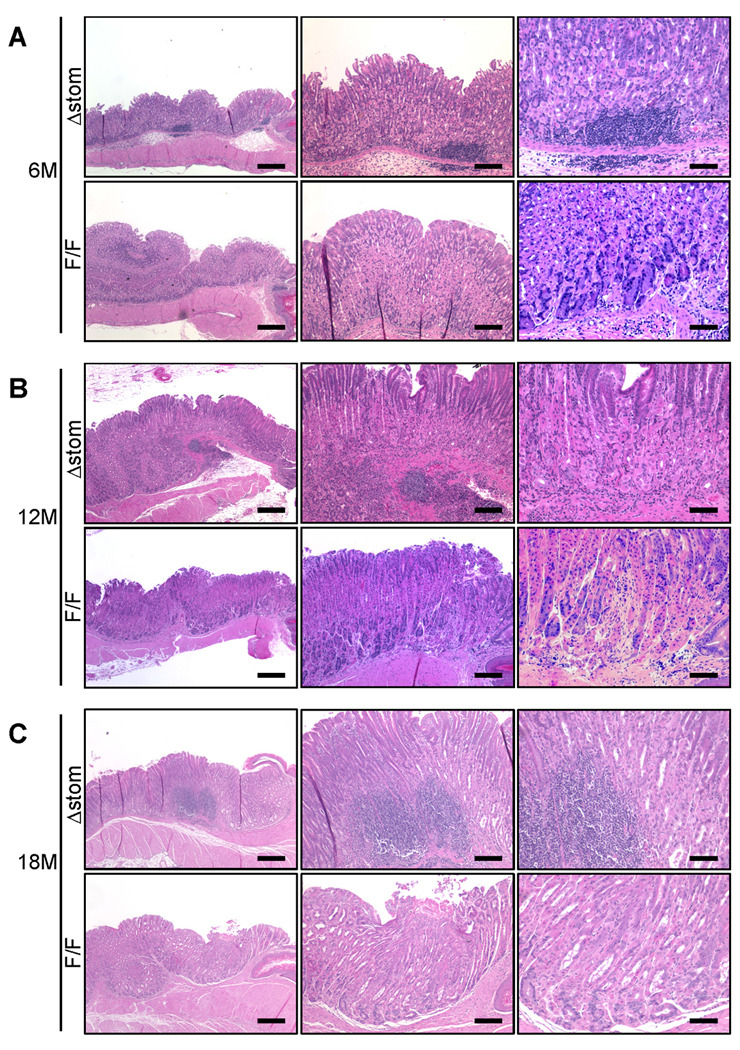
Natural progression of H. felis infection in the IkkβF/F and IkkβΔstom mice infected with H. felis for 6 months (A) (n=6), 12 months (B) (n=8), and 18 months (C) (n=14 in IkkβF/F, and n=12 in IkkβΔstom group). Representative HandE staining were shown. IkkβΔstom and IkkβF/F mice HandE staining. Original magnifications and Scale bars; (lt.) 40×, 250µm, (middle) 100×, 100µm, and (rt.) 200×, 50µm.
Then, we further observed the mice until 18 months post-infection. There were still significant differences, with greater degrees of pseudopyloric metaplasia, oxyntic atrophy and foveolar hyperplasia in IkkβΔstom mice (Figure 3C). Importantly, only in IkkβΔstom mice showed gastric dysplasia at 18 months post-infection (33%; 4 out of 12), while no mice developed dysplasia in IkkβF/F. Consistent with the presence of dysplasia, IkkβΔstom mice showed a significant increase in CD44+ cells, a marker for gastric cancer stem cells,4 particularly in areas with severe gastric inflammation (Figure 4A), and we observed increases in DCAMKL-1+ cells, a putative marker for gastric progenitor cells (Figure 4B).30 Finally, we performed immunostaining for a number of stromal markers, and confirmed that alpha-SMA-, collagen-I-and S100A-positive cells were significantly increased in IkkβΔstom mice than IkkβF/F mice (Supplementary Figure 4), suggesting that gastric inflammation found in IkkβΔstom mice was accompanied by severe stromal reaction. The acceleration in progression to atrophy in the IkkβΔstom mice did not appear to be due to alterations in colonization or immune responses against H. felis (Supplementary Figure 5). There were no gender differences in phenotypes. Histological changes were summarized in Figure 4C to 4F.
Figure 4.

Immunohistochemistry for CD44 (A), and DCAMKL-1 (B) in IkkβΔstom mice (top) and IkkβF/F mice (bottom) 18 months post-infection. Magnification: 100×. Scale bars, 100µm. Summary of histological scores in mice infected with H. felis for 3 months (C), 6 months (D), 12 months (E), and 18 months (F). Each parameter was scored as described in the supplementary methods. *Indicates a significant differences (p<0.05) compared with IkkβF/F and IkkβΔstom mice.
IKKβ Deletion in GECs Results in Increased Apoptosis and Cellular Proliferation Following Either IR or H. felis Infection
Previous studies have suggested that a key role for the IKKβ/NF-κB pathway in epithelial cells is the inhibition of apoptosis.12, 13 To test the hypothesis that NF-κB functions primarily in anti-apoptotic responses to stressful stimuli in GECs, we examined the apoptotic susceptibility of IkkβΔstom mice in comparison to IkkβF/F mice following IR or chronic H. felis infection (Figure 5). At baseline, apoptotic GECs in non-challenged IkkβF/F and IkkβΔstom mice were infrequent. IkkβΔstom mice treated with 12-Gy IR showed significantly increased apoptosis at 48 h compared to IkkβF/F mice with a 3–4-fold increase (Figure 5A and 5B). Cell proliferation as assessed by Brd-U incorporation was significantly increased by two-fold in IkkβΔstom mice compared to IkkβF/F mice (Figure 5C and 5D). In the chronic model of long-term (6–18 mo) H. felis infection, IkkβΔstom mice showed increased apoptosis that extended to regions not observed in the IkkβF/F mice, such as in the lower third of the gastric glands (Figure 5E and Supplementary Figure 6A). Since increased apoptosis is the initial response to gastric injury that appears to be amplified after Ikkβ loss, we assessed the expression of anti-apoptotic genes that were known to be NF-κB downstream targets. The prominent apoptotic responses in IkkβΔstom mice after IR or chronic H. felis infection were associated with decreased mRNA expression of c-Flip and c-IAP2, well-known anti-apoptotic genes (Supplementary Figure 6B and 6C), suggesting that loss of antiapoptotic gene expression after Ikkβ loss might be a key factor leading to increased apoptosis.
Figure 5.
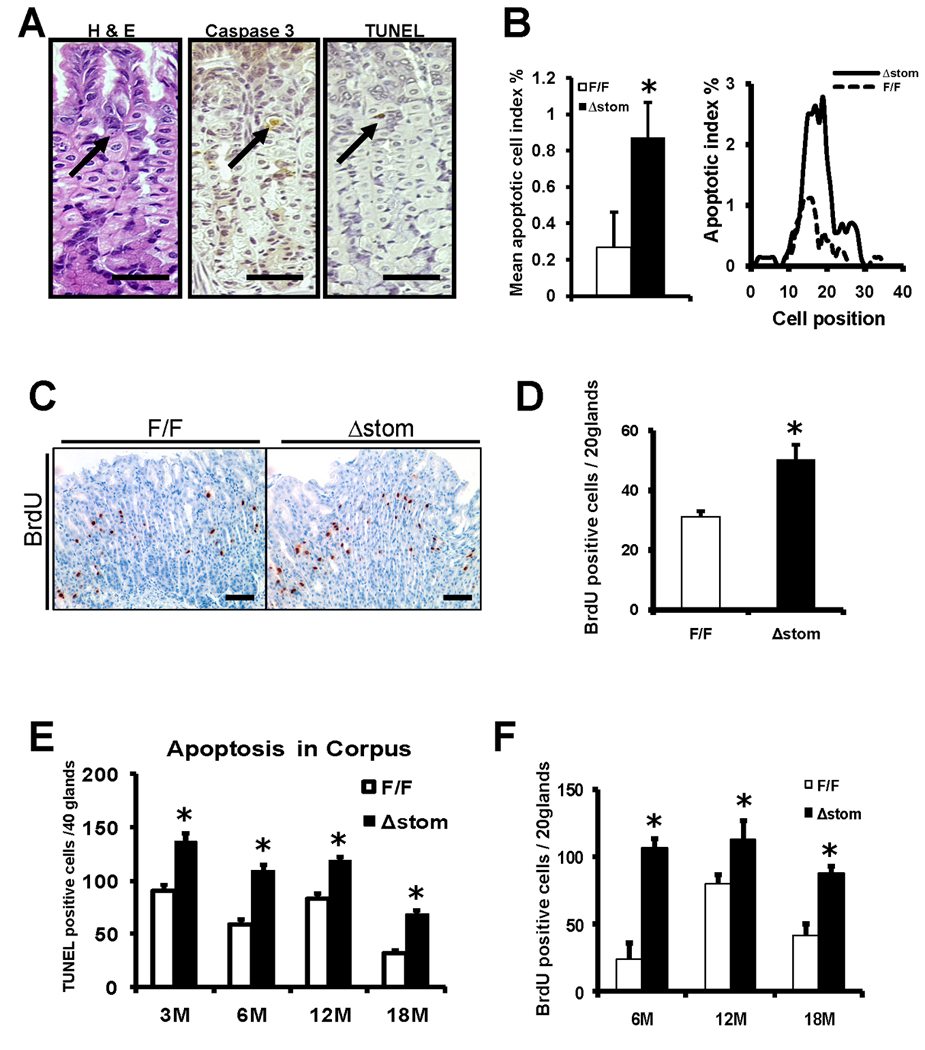
(A) Representative photomicrographs of apoptotic cells after IR. HandE, caspase-3, and TUNEL in serial section of gastric corpus 48 h after 12Gy IR. (arrow) apoptotic cells positive for caspase-3 and TUNEL. Original magnification 400×. Scale bars, 50µm. (B) Mean apoptotic cell index % and cell positions assessed by HandE. (C, D) Increased epithelial cell proliferation in IkkβΔstom mice 120 hr after 12Gy IR. Scale bars, 50µm. Apoptosis scores (E) and Brd-U labeling index (F) for IkkβF/F and IkkβΔstom mice infected with H. felis. *Indicates a significant differences (p<0.05) compared with IkkβF/F and IkkβΔstom mice (n = 6 per group).
As in the case with IR, H. felis-infected IkkβΔstom mice showed greater increases in cell proliferation compared to H. felis-infected IkkβF/F mice (Figure 5F and Supplementary Figure 6A). An increase in gastric proliferation has been shown to be largely secondary, representing compensatory responses that occur in reason to increased rates of apoptosis.31 Taken together, these data suggest that loss of Ikkβ in GECs results in deregulation of apoptosis and proliferation following cellular stress, and thus impaired epithelial homeostasis. This notion was further supported by the finding that 120 h post IR, IkkβΔstom mice showed persistent epithelial cell damage, whereas IkkβF/F mice showed full epithelial restitution and recovery (Figure 6A).
Figure 6.

(A) Representative photographs of acute injury after IR. IkkβF/F (lt.) and IkkβΔstom (rt.) 120 h after 12 Gy IR. HandE (top, 2nd), immunohistochemistry for DHE (3rd), and 8-OHdG (4th). Original magnifications; 200×(top, bottom), and 400× (2nd, 3rd). Scale bars, 50µm. (n = 6) (B) Immunohistochemistry for 8-OHdG in the stomach 18 months post-infection. (Black arrows) Positive staining for 8-OHdG. 8-OHdG positive cells were scored per 40 corpus glands. Scale bars, 100µm. (C) mRNA expression for pro-inflammatory cytokines/chemokines 120 h after 12 Gy IR, and (D) time course of IL-1alpha mRNA expression after H. felis infection determined by qRT-PCR.
IKKβ-deficient GECs Show Increased Oxidative Stress, Cellular Necrosis and IL-1α Release After IR and Chronic Infection
Previous reports have indicated that hepatocyte Ikkβ inhibits hepatocarcinogenesis in part by suppressing accumulation of reactive oxygen species (ROS) that can lead to severe liver damage and hepatocellular necrosis.8, 15 In order to further explore the consequences of Ikkβ loss in GECs after IR, we analyzed ROS associated DNA damage and the degree of cellular necrosis in response to IR. To assess the accumulation of superoxide anions, we stained freshly frozen tissue sections with dihydroethidine (DHE), showing greater staining of superoxide anions in GECs of IkkβΔstom than IkkβF/F mice after IR (Figure 6A). ROS production can lead to oxidative DNA damage, which can be assessed by immunostaining for 8-hydroxydeoxyguanosine (8-OHdG). The gastric mucosa of IkkβΔstom mice exhibited a higher number of anti-8-OHdG-positive cells than IkkβF/F mice 120hrs after IR (Figure 6A). In addition, IkkβΔstom mice displayed increased necrotic cell damage manifested by abnormal mitotic nuclei and marked increases in nuclear size (Figure 6A). Thus, the loss of Ikkβ resulted in increased oxidative stress, DNA damage and cellular necrosis. Oxidative stress induced by ROS has an important role in the formation of gastric injury by Helicobacter infection.32 18 months post H. felis infection, the gastric mucosa of IkkβΔstom mice also exhibited a higher number of anti-8-OHdG-positive cells than IkkβF/F mice (Figure 6B), suggesting that loss of Ikkβ is likely associated with the ROS accumulation and subsequent DNA damage in this model.
Recently, it has been demonstrated that liver inflammation due to acetaminophen administration is mediated by IL-1α release from necrotic cells.7 We therefore examined whether IR-induced or H. felis infected GECs also released IL-1α. We found that 120 hrs after IR, the level of IL-1α along with several other pro-inflammatory cytokines (IL-1β, IL-6, CCL2), was much higher in IkkβΔstom mice compared to IkkβF/F mice (Figure 6C). In addition, we also found that IL-1α was higher in IkkβΔstom than IkkβF/F mice 3 to 18 month post-infection (Figure 6D), suggesting that loss of IKKβ in GECs could induce the release of IL-1α which might contribute to more severe inflammatory phenotypes observed in IkkβΔstom mice following epithelial stress.
We have proposed that pathologic elevation of a single proinflammatory cytokine, IL-1β, can induce gastric inflammation and cancer in mice.20 However, while NF-κB signaling is a critical transcription factor for activation of a number of proinflammatory cytokines, we noted only moderate increases in the expression of proinflammatory cytokines in the gastric mucosa of IkkβΔstom mice (data not shown). Given the importance of CC chemokine responses in the pathogenesis of H. felis-induced gastric carcinogenesis, we analyzed CC chemokine expression profiles by qRT-PCR in the 2 groups of mice infected with H. felis. MCP-1 (CCL-2), MIP-1α (CCL3), and RANTES (CCL5) were significantly higher in the IkkβΔstom mice as compared with IkkβF/F mice at early time points (1.5 to 3 months post-infection) (Supplementary Figure 7). In addition, the circulating levels of interleukin-6 (IL-6) were significantly increased in the IkkβΔstom as compared with those of IkkβF/F mice at 6 and 12 months post-infection (Supplementary Figure 7D). Since ROS accumulation is one of the key mechanisms mediating gastric injury after Ikkβ loss, we assessed the expression levels of free radical deactivating enzymes which were typically seen in the setting of ROS accumulation. As expected, they were generally increased in IkkβΔstom than IkkβF/F mice (except for MnSOD which was regulated by NF-κB) suggesting that IR stress induced ROS accumulation (Supplementary Figure 7E).
Loss of IKKβ in GECs Results in Accumulation of Myeloid Cells that Promote Progression of Gastric Neoplasia
Recent studies have linked cancer progression to pro-inflammatory cytokines and thus to myeloid cells. To determine whether the loss of Ikkβ in GECs led to any alteration in resident macrophages, we purified thioglycollate-elicited peritoneal macrophages from IkkβΔstom and IkkβF/F mice, and examined IL-6 production in response to LPS or H. felis. However, there were no significant differences between the two groups (Supplementary Figure 8). Next, to examine whether the loss of Ikkβ in GECs directly affects macrophage recruitment to the gastric mucosa, we performed immunohistochemistry for F4/80+ macrophages (Figure 7A and 7B). The number of F4/80+ cells was significantly greater in IkkβΔstom mice compared to IkkβF/F mice. Thus, these data suggest that a difference in the rate of recruitment of macrophages into the gastric mucosa might account for differences in the severity of the gastric phenotype in vivo.
Figure 7.
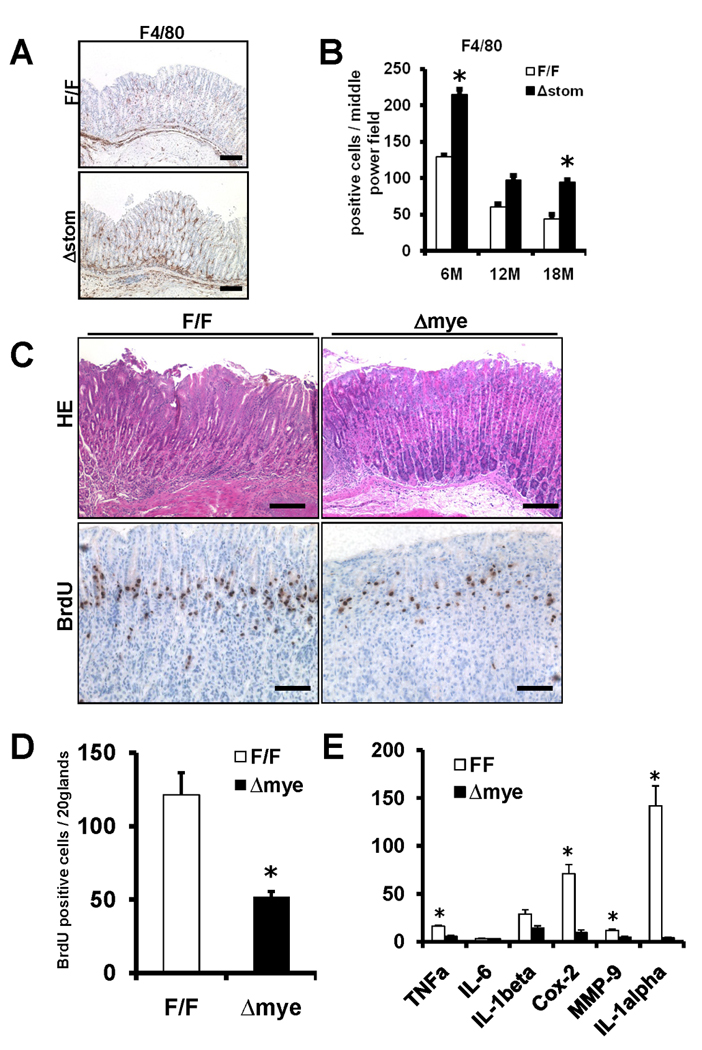
(A) Representative F4/80 staining for IkkβF/F and IkkβΔmye mice 6 months post-infection. Original magnification 100×, Scale bars, 100µm. (B) F4/80 positive cells were counted per high power fields (20 corpus glands). (C) Representative H&E staining (top) and Brd-U staining (bottom) for IkkβF/F and IkkβΔmye mice 6 months post-infection. Scale bars, 100µm (top), 50µm (bottom). (D) Brd-U labeling index. (E) Expression of indicated genes in the stomach IkkβF/F and IkkβΔmye was determined by qRT-PCR. Results were relative expression compared with 104 GAPDH expressions. *Indicates a significant differences (p<0.05) compared with IkkβF/F and IkkβΔstom mice (n=6).
To address the functional significance of the Ikkβ in myeloid cells, we used mice specifically lacking Ikkβ in the myeloid lineage (IkkβΔmye).26 We infected IkkβΔmye and IkkβF/F mice with H. felis, and analyzed gastric histology and proinflammatory cytokine expression. IkkβΔmye mice showed milder gastric inflammation and decreased proliferation rates after H. felis infection (Figure 7C and 7D), although there were no differences in terms of apoptosis (data not shown). IkkβΔmye also showed reduced mRNA expression of TNFα, IL-1beta, Cox-2, MMP-9, and IL-1alpha (Figure 7E). Thus, as opposed to GECs, Ikkβ deletion in the myeloid lineage showed the critical role of Ikkβ in myeloid cells in progression to gastric neoplasia.
Discussion
In this study, we derived mice with conditional deletion of IKKβ in GECs, and have shown that loss of IKKβ in the stomach results in more rapid H. felis-dependent progression to dysplasia in the setting of H. felis infection. In contrast, deletion of IKKβ in myeloid cells inhibited H. felis-dependent progression to atrophy and dysplasia. The rapid progression to dysplasia observed in the IkkβΔstom mice was associated with greater susceptibility to apoptosis and necrosis after cellular stress induced by H. felis infection. The increased necrosis was associated with upregulation of IL-1α, and CXCL2, increased infiltration with myeloid cells and more severe chronic inflammation. Taken together, the findings strengthen the association between not only chronic inflammation and cancer, and suggest that IKKβ/NF-κB signaling in GECs suppresses chronic inflammation by inhibiting cellular apoptosis and necrosis in response to severe cellular stress.
Although NF-κB has a well-established role in the activation of numerous cytokines and chemokines that can promote inflammation, this role has been best established in monocytes and macrophages.20, 33 In contrast, the role of NF-κB signaling in epithelial cells has been somewhat less clear. Although a number of studies have suggested that H. pylori can induce NF-κB activation in gastric cancer cell lines leading to increased cytokine and chemokine expression,22 there has been limited evidence that Helicobacter can activate NF-κB in primary, non-transformed GECs either in vitro34 or in vivo.23, 35 In the current study, analysis of NF-κB activation using NF-κB-EGFP reporter mice showed little or no NF-κB activation in GECs within 12 months H. felis infection, although increased NF-κB activity could be detected in dysplasia. Considering the evidence that shows NF-κB activation in human gastric epithelial cells infected with H. pylori,22 NF-κB activation might be underestimated somewhat by the NF-κB-EGFP reporter mice. In addition, deletion of IKKβ and thus inactivation of NF-κB signaling led to a worsening, rather than inhibiting, of chronic inflammation. Similar findings in the intestine were reported by Greten and colleagues using villin-Cre to delete IKKβ from intestinal epithelial cells.14 In the DSS colitis model, loss of IKKβ worsened, rather than inhibited, the inflammatory response to injury. Taken together, these data suggest that NF-κB signaling in the gastric epithelium plays very limited roles in the immune response to infection or injury.
In contrast to a lack of a role for NF-κB in pro-inflammatory responses in the gastric epithelial compartment, NF-κB is likely to be important in the reaction to apoptotic stimuli such as Helicobacter infection. NF-κB activation appears to be a key signaling event required for protection against apoptosis in the gastric epithelium in vivo. Indeed, our data suggest that the defective anti-apoptotic response of epithelial cells in the absence of NF-κB activation lead to increased gastric apoptosis with severe epithelial damage after IR or chronic Helicobacter infection. Previous studies have shown that NF-κB is a critical regulator of survival of intestinal epithelial cells after IR.12 In addition, the absence of IKKβ in intestinal epithelial cells or hepatocytes resulted in increased apoptosis after exposure to AOM+DSS or DEN, respectively.14, 15 In our study, deletion of the Ikkβ gene in GECs resulted in marked decreases in the mRNA expression of c-FLIP and c-IAP2, established anti-apoptotic genes and known NF-κB targets, although significant changes in c-IAP2 could not be correlated precisely with the time course of the apoptosis. NF-κB likely regulates other genes or pathways that account for the remainder of the apoptotic resistance normally observed in WT mice. Thus, IKKβ/NF-κB pathways appear to be critical for inhibition of apoptosis and maintenance or homeostasis in the gastric epithelium.
While NF-κB represents a key link between inflammation and cancer, NF-κB deficiency in prior studies has resulted in both decreased14 and increased15, 16 susceptibility to cancer, depending upon the tissue and the model employed. In the gastric mucosa, increased apoptotic rates in the setting of Helicobacter infection have been linked to compensatory increases in proliferation and more rapid progression to gastric neoplasia. Indeed IkkβΔstom mice showed higher rates of progression to dysplasia. High grade dysplasia, associated with increased CD44+ expression, which has been described as a marker of gastric cancer stem cells,4 and increased DCAMKL1+ cells,30 which is a possible gastric progenitor, was observed in IkkβΔstom mice but not in IkkβF/F. Collectively, our data suggests that loss of Ikkβ might induce proliferation in the number of gastric progenitor cells following loss of parietal cells. Thus, the findings in the Helicobacter-associated cancer model mimics earlier findings of increased cancer with IKKβ deficiency in the DEN hepatic cancer model and the skin cancer models,15, 16 but differs from the finding of decreased cancer in the AOM+DSS colon cancer model.14 In these earlier reports, the disparate results in the different models were accounted for by the inflammatory nature of the colorectal cancer model, and the lack of inflammation in the DEN model. However, H. felis, similar to AOM+DSS, induces cancer in a large part through induction of a chronic inflammatory state.18 The endpoint of our current study was dysplasia, not more advanced cancer, and given our finding of increased NF-κB activity at the dysplastic stage, we cannot rule out that further progression to cancer may be inhibited by the absence of IKKβ.36
In conclusion, the current study emphasizes the somewhat greater importance of IKKβ/NF-κB signaling in the myeloid compartment as opposed to the epithelial compartment. Numerous studies have demonstrated correlations between the recruitment of myeloid cells, including macrophages, monocytes and myeloid progenitors, and the development of cancer.20, 33 Deletion of IKKβ using LysM-Cre resulted in downregulation of proliferative responses, and marked inhibition of the expression of numerous NF-κB-dependent genes, such as TNFα, Cox-2 and IL-1alpha. In particular, IL-1alpha, a key inflammatory cytokine and mainly produced by myeloid cells, has been reported to be associated with enhanced angiogenesis and liver metastasis in humans gastric cancers, suggesting that IL-1alpha could be the possible target to treat gastric cancer.37
In contrast, loss of IKKβ from the epithelial compartment resulted in more severe inflammatory responses to H. felis infection, and increased influx of macrophages to the gastric mucosa, and we attribute this finding to increased epithelial necrosis in the IkkβΔstom mice. While much attention has been given to the modulation of apoptosis, a generally non-inflammatory form of cell death, the loss of IKKβ also appears to predispose to increased necrosis. Elevated rates of 8-OHdG staining, a sign of oxidative DNA damage, were observed in both IR and H. felis models of cellular stress, and this increased oxidative stress was associated with increased cellular necrosis. Previous studies have shown that hepatocellular necrosis results in increased IL-1α release, which can mediate the recruitment of inflammatory cells to sites of injury.7, 8 In the current study, we demonstrated increases in both IL-1α and CXCL2 in IKKβ deficient animals associated with increased chronic inflammation confirmed by histological analysis. Consequently, loss of IKKβ from GEC resulted in increased necrosis, IL-1α release, and recruitment of inflammatory cells that accelerated progression through stages of gastric atrophy, metaplasia and dysplasia. Overall, these findings highlight the important role that IKKβ/NF-κB play in gastric epithelial cell homeostasis and the response to cellular injury. IKKβ/NF-κB regulates a variety of signaling pathways, including proinflammatory cytokines and apoptosis. Proper regulation of IKKβ/NF-κB on the specific cell types would be required in targeting this transcription factor to treat cancer.
Supplementary Material
Acknowledgments
The authors thank D.A. Brenner for providing NF-κBEGFP mice, Shanisha AK Gordon, and Anthony Mitchell for their help with animal procedure, and thank Rong-zhen Chen and Hiroyuki Tomita for their excellent histological assistance.
Funding: This research was supported by grants from the National Institute of Health grants 5R01CA093405-08 (T.C.W). W.S. was supported by Kanae Foundation for the Promotion of the Medical Science 2008, and Japan Society for the Promotion of Science 2009.
Abbreviations used in this paper
- GECs
gastric epithelial cells
- IR
ionizing radiation
- ELISA
enzyme-linked immunosorbent assay
- qRT-PCR
quantitative reverse transcriptional polymerase chain reaction
- DEN
diethylnitrosamine
- IKKβ
I-kappa-B-kinase-β
- TNF
tumor necrosis factor
- IL
interleukin
- TFF2
trefoil factor 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors contributions: Study concept and design; T.C.W, acquisition of data; W.S, S.T, A.B.R, J.G.F, M.T.W, S.M, M.D.P, analysis and interpretation of data; T.C.W, and W.S, drafting of the manuscript; W.S, K.B, and T.C.W, obtained funding; T.C.W, technical, and material support; K.H.K and K.M, study supervision; T.C.W
Conflict of interest: The authors disclose no conflicts.
References
- 1.Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–1243. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 2.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–921. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 3.Przemeck SM, Duckworth CA, Pritchard DM. Radiation-induced gastric epithelial apoptosis occurs in the proliferative zone and is regulated by p53, bak, bax, and bcl-2. Am J Physiol Gastrointest Liver Physiol. 2007;292:G620–G627. doi: 10.1152/ajpgi.00391.2006. [DOI] [PubMed] [Google Scholar]
- 4.Takaishi S, Okumura T, Tu S, Wang SS, Shibata W, Vigneshwaran R, Gordon SA, Shimada Y, Wang TC. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells. 2009;27:1006–1020. doi: 10.1002/stem.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 7.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 8.Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, Karin M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spehlmann ME, Eckmann L. Nuclear factor-kappa B in intestinal protection and destruction. Curr Opin Gastroenterol. 2009;25:92–99. doi: 10.1097/MOG.0b013e328324f857. [DOI] [PubMed] [Google Scholar]
- 10.Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Egan LJ, Eckmann L, Greten FR, Chae S, Li ZW, Myhre GM, Robine S, Karin M, Kagnoff MF. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci U S A. 2004;101:2452–2457. doi: 10.1073/pnas.0306734101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eckmann L, Nebelsiek T, Fingerle AA, Dann SM, Mages J, Lang R, Robine S, Kagnoff MF, Schmid RM, Karin M, Arkan MC, Greten FR. Opposing functions of IKKbeta during acute and chronic intestinal inflammation. Proc Natl Acad Sci U S A. 2008;105:15058–15063. doi: 10.1073/pnas.0808216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 15.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 16.Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, Marinkovich MP, Tao S, Lin Q, Kubo Y, Khavari PA. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–643. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- 17.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007;117:60–69. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni JF, Jr, Rabkin CS. The role of interleukin-1 polymorphisms in the pathogenesis of gastric cancer. Nature. 2001;412:99. doi: 10.1038/35083631. [DOI] [PubMed] [Google Scholar]
- 20.Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl O, Fox JG, Wang TC. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–419. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai X, Stoicov C, Li H, Carlson J, Whary M, Fox JG, Houghton J. Overcoming Fas-mediated apoptosis accelerates Helicobacter-induced gastric cancer in mice. Cancer Res. 2005;65:10912–10920. doi: 10.1158/0008-5472.CAN-05-1802. [DOI] [PubMed] [Google Scholar]
- 22.Keates S, Hitti YS, Upton M, Kelly CP. Helicobacter pylori infection activates NF-kappa B in gastric epithelial cells. Gastroenterology. 1997;113:1099–1109. doi: 10.1053/gast.1997.v113.pm9322504. [DOI] [PubMed] [Google Scholar]
- 23.van Den Brink GR, ten Kate FJ, Ponsioen CY, Rive MM, Tytgat GN, van Deventer SJ, Peppelenbosch MP. Expression and activation of NF-kappa B in the antrum of the human stomach. J Immunol. 2000;164:3353–3359. doi: 10.4049/jimmunol.164.6.3353. [DOI] [PubMed] [Google Scholar]
- 24.Shibata W, Hirata Y, Maeda S, Ogura K, Ohmae T, Yanai A, Mitsuno Y, Yamaji Y, Okamoto M, Yoshida H, Kawabe T, Omata M. CagA protein secreted by the intact type IV secretion system leads to gastric epithelial inflammation in the Mongolian gerbil model. J Pathol. 2006;210:306–314. doi: 10.1002/path.2040. [DOI] [PubMed] [Google Scholar]
- 25.Magness ST, Jijon H, Van Houten Fisher N, Sharpless NE, Brenner DA, Jobin C. In vivo pattern of lipopolysaccharide and anti-CD3-induced NF-kappa B activation using a novel gene-targeted enhanced GFP reporter gene mouse. J Immunol. 2004;173:1561–1570. doi: 10.4049/jimmunol.173.3.1561. [DOI] [PubMed] [Google Scholar]
- 26.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 27.Katz JP, Perreault N, Goldstein BG, Actman L, McNally SR, Silberg DG, Furth EE, Kaestner KH. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology. 2005;128:935–945. doi: 10.1053/j.gastro.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 28.Li ZW, Omori SA, Labuda T, Karin M, Rickert RC. IKK beta is required for peripheral B cell survival and proliferation. J Immunol. 2003;170:4630–4637. doi: 10.4049/jimmunol.170.9.4630. [DOI] [PubMed] [Google Scholar]
- 29.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 30.Giannakis M, Stappenbeck TS, Mills JC, Leip DG, Lovett M, Clifton SW, Ippolito JE, Glasscock JI, Arumugam M, Brent MR, Gordon JI. Molecular properties of adult mouse gastric and intestinal epithelial progenitors in their niches. J Biol Chem. 2006;281:11292–11300. doi: 10.1074/jbc.M512118200. [DOI] [PubMed] [Google Scholar]
- 31.Wang TC, Goldenring JR, Dangler C, Ito S, Mueller A, Jeon WK, Koh TJ, Fox JG. Mice lacking secretory phospholipase A2 show altered apoptosis and differentiation with Helicobacter felis infection. Gastroenterology. 1998;114:675–689. doi: 10.1016/s0016-5085(98)70581-5. [DOI] [PubMed] [Google Scholar]
- 32.Kawasaki K, Nishio A, Nakamura H, Uchida K, Fukui T, Ohana M, Yoshizawa H, Ohashi S, Tamaki H, Matsuura M, Asada M, Nishi T, Nakase H, Toyokuni S, Liu W, Yodoi J, Okazaki K, Chiba T. Helicobacter felis-induced gastritis was suppressed in mice overexpressing thioredoxin-1. Lab Invest. 2005;85:1104–1117. doi: 10.1038/labinvest.3700305. [DOI] [PubMed] [Google Scholar]
- 33.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim JM, Kim JS, Jung HC, Oh YK, Chung HY, Lee CH, Song IS. Helicobacter pylori infection activates NF-kappaB signaling pathway to induce iNOS and protect human gastric epithelial cells from apoptosis. Am J Physiol Gastrointest Liver Physiol. 2003;285:G1171–G1180. doi: 10.1152/ajpgi.00502.2002. [DOI] [PubMed] [Google Scholar]
- 35.Ferrero RL, Ave P, Ndiaye D, Bambou JC, Huerre MR, Philpott DJ, Memet S. NF-kappaB activation during acute Helicobacter pylori infection in mice. Infect Immun. 2008;76:551–561. doi: 10.1128/IAI.01107-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sasaki N, Morisaki T, Hashizume K, Yao T, Tsuneyoshi M, Noshiro H, Nakamura K, Yamanaka T, Uchiyama A, Tanaka M, Katano M. Nuclear factor-kappaB p65 (RelA) transcription factor is constitutively activated in human gastric carcinoma tissue. Clin Cancer Res. 2001;7:4136–4142. [PubMed] [Google Scholar]
- 37.Ma J, Sawai H, Matsuo Y, Ochi N, Yasuda A, Takahashi H, Wakasugi T, Funahashi H, Sato M, Okada Y, Takeyama H, Manabe T. Interleukin-1alpha enhances angiogenesis and is associated with liver metastatic potential in human gastric cancer cell lines. J Surg Res. 2008;148:197–204. doi: 10.1016/j.jss.2007.08.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.