

Abstract
The impact of infection by the low-virulent ASFV/NH/P68 (NHV) and the highly virulent ASFV/L60 (L60) isolates on porcine macrophages was assessed through the quantification of IFNα, TNFα, IL12p40, TGFβ and ASFV genes by real-time PCR at 2, 4 and 6 h post-infection. Increased IFNα, TNFα and IL12p40 expression was found in infection with NHV, in which expression of TGFβ was lower than in infection with L60. Principal component analysis showed a positive interaction of cytokines involved in cellular immune mechanisms, namely IFNα and IL12p40 in the NHV infection. Quantification by ELISA confirmed higher production of IFNα, TNFα and IL12p40 in the NHV-infected macrophages. Overall, our studies reinforce and clarify the effect of the NHV infection by targeting cellular and cellular-based immune responses relevant for pig survival against ASFV infection.
Introduction
African swine fever, first described by Montgomery [24, 25], is one of the most threatening diseases for pig husbandry. African swine fever virus (ASFV) is an icosahedral cytoplasmic double-stranded DNA virus which is the only member of the family Asfarviridae [9]. ASFV infects all the members of the Suidae family and is non-pathogenic in its natural hosts, bushpigs (Potamochoerus porcus) and warthogs (Phacochoerus aetiopicus), which are kept infected through a natural cycle involving soft ticks (Ornithodorusmoubata) [30, 39]. In domestic pigs, African swine fever (ASF) may vary from acute to chronic and unapparent clinical forms in parallel with development of distinct pathological lesions [31].
ASFV preferentially infects pig monocytes and macrophages, which are the main targets for in vivo viral replication [5, 6, 14, 15, 20]. In vitro studies demonstrate that ASFV of different virulence infect and induce lysis of blood-derived macrophages [4, 11, 20] and bone marrow and alveolar macrophages [3, 20]. Macrophage infection by ASFV is of foremost relevance for pathogenesis, taking into consideration that macrophages play important roles in both innate and acquired immune responses. Relevant in the role of macrophages as orchestrators of immune responses, they synthesize cytokines that have an impact on the development of inflammatory responses (pro-inflammatory cytokines such as IFN type I, IL1, IL6 and TNFα) and cytokines that participate in the development of specific immune mechanisms (immunoregulatory cytokines such as IL12, IL15, IL18) through the activation of the Th1 and Th2 responses [26].
Previous studies have shown that ASFV inhibits phagocytosis, antibody-mediated phagocytosis and chemotaxis in porcine macrophages cultivated in vitro, although no changes were observed in the expression of Fc receptors, nor in the capacity to induce antibody-dependent cytotoxicity (ADCC) [21]. In identical circumstances, the expression of SLA antigens was not changed on those cells [13].
Our previous studies using porcine blood-derived macrophages infected in vitro with two ASFV isolates of different virulence, the highly virulent L60 and the low-virulent NHV, demonstrated a particular effect of infection by the latter isolate in that significantly increased levels of transcripts for TNFα, IL6, IL12 and IL15 were identified at 6 hours post-infection, in contrast to the effect of infection with ASFV/L60 [12]. The differential expression of those cytokines on macrophages infected with either ASFV isolate suggests a role of ASFV/NHV in inducing inflammatory and cellular-based responses in the natural host, such as the previously described ASFV-specific CTL activity [22] and NK activity [19]. In the work reported here, we have extended our previous studies to evaluate the impact of the infection of porcine macrophages by the two abovementioned ASFV isolates in the expression of IFNα, TNFα, IL12p40 (mRNA and protein) and TGFβ (mRNA) at different times post-infection (2, 4, 6 and 18 h). In parallel, we have assessed the course of infection through the quantification of the expression of the ASFV genes VP32, VP72 and A238L and through the quantification of viral yields at 18 h post-infection.
Materials and methods
Experimental design
Porcine blood-derived macrophage cultures from seven pig donors were prepared and infected in vitro with either the low-virulent ASFV/NH/P68 (NHV), or the highly virulent ASFV/L60 (L60) at a multiplicity of 3 (MOI = 3), or with pseudorabies virus (PrV) (used as control of the infection responses to ASFV). Duplicates of 54 cDNA samples from individual macrophage cultures collected at 2, 4 and 6 h post-infection were used to quantify mRNA expression of the cytokines IFNα, TNFα, IL12p40 and TGFβ and the expression of the ASFV genes A238L, VP32 and VP72. The characterization of mRNA expression of different cytokines and viral genes was conducted by quantitative real-time RT-PCR. Supernatants of macrophage cultures, collected at 18 h post-infection (overnight incubation) with the abovementioned virus were used to quantify final product for IFNα, TNFα and IL12p40 by quantitative ELISA.
Enriched porcine blood-derived macrophage cultures and viral infection
Suspensions of peripheral blood mononuclear cells (PBMC) from seven crossbred Large White × Landrace pigs (6 months old) were prepared from heparinised peripheral blood samples collected under aseptic conditions during bleeding at the abattoir, using slight modifications of protocols described previously [12]. Briefly, heparinised blood samples were incubated (37°C, 15 min), with 10% (v/v) of a 5% (v/v) Dextran T 500 solution in Hank’s balanced saline solution (HBSS), and supernatants were collected and centrifuged (400 g, 30 min, room temperature) over Ficoll-Hypaque (d = 1.077, Seromed). The interface PBMCs were collected, washed twice and resuspended in complete culture medium (RPMI 1640 with 100 IU/ml penicillin, 100 µg/ml streptomycin and 20 mM HEPES) supplemented with 10% foetal bovine serum (FBS), (Gibco BRL, 102770-106) and seeded in T175 tissue culture flasks (Nunc, 156502) at 5 × 106 cells/ml. Cultures were incubated (37°C, 5% CO2 and > 90% relative humidity) for 72 h. After incubation, non-adherent cells were removed by serial washing with pre-warmed phosphate-buffered saline (PBS) (8 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4, 0.24 g KH2PO4 per litre, pH 7.4), and adherent macrophages were harvested by treatment with 0.8 mM EDTA in this buffer. Cell viability counting by trypan blue dye exclusion was always > 90%. Macrophages were re-suspended at 5 × 106/ml in complete culture medium in sterile polypropylene centrifuge tubes (Nunc, 366060) and inoculated with working suspensions of NHV, L60 or PrV at a multiplicity of infection of three (MOI = 3). Tubes were kept horizontal and incubated (37°C, 5% CO2 and > 90% relative humidity) with slight mechanical shaking for 90 min to facilitate virus adsorption. Cells were washed, resuspended in complete culture medium and incubation was continued for 2, 4, 6, and 18 h. At 2, 4 and 6 h, cells were washed, and pellets were used for mRNA quantification. Cells incubated for 18 h were treated similarly and supernatants and pellets were used to quantify virus yields; supernatants were used to quantify final products of different cytokines. For all experiments, non-inoculated macrophages from the same donors, cultured in complete culture medium, were used as controls.
Virus
Two naturally occurring ASFV isolates were used: the low-virulent non-hemadsorbing ASFV/NH/P68 (NHV) and the highly virulent ASFV/L60 (L60) [19]. Virus stocks were grown in blood-derived macrophage cultures as described previously [21] for no more than six passages. Supernatants from macrophage cultures infected with stock virus at an approximate multiplicity of infection of 5 (MOI = 5) were collected when CPE was observed in more than 80% of cells (around 72 h post-infection), clarified by centrifugation and further centrifuged overnight in a GSA rotor at 8,000 g, 4°C. Pellets were suspended (Tris 20 mM, EDTA 1 mM, NaCl 1 M) and re-sedimented by centrifugation (10,000 g, SW 28 rotor, Sorvall, 90 min, 4°C), through a cushion of 25% sucrose. The resulting pellet was finally re-suspended in culture medium. Viral suspensions were titrated by observation of cytopathic effect (CPE) at end-point dilutions [21]. Suspensions of pseudorabies virus (PrV) were used as clarified supernatants of Vero cell cultures infected and lysed by the virus, obtained after an undetermined number of passages in these cells (kindly supplied by Benedita Cruz, LNIV, Lisbon, Portugal). Virus titration was performed in Vero cells as above for ASFV.
RNA extraction
RNA from macrophage cultures was obtained using the commercial kit “High Pure RNA extraction kit” (ROCHE) in accordance with the manufacturer’s instructions. The obtained RNA was dissolved in diethylpyrocarbonate (DEPC)-treated water and stored at −70°C until use. Samples of RNA were quantified in a spectrophotometer (DU40 Beckman), at 260 and 280 nm (A). Total RNA was calculated as described in the literature [33].
Reverse transcription of mRNA into cDNA
Ten μg of total RNA was mixed with oligo (dT)12–18 primers (Gibco BRL, 18064-014) and incubated (70°C, 10 min). The mixture was chilled on ice and incubated (45°C, 50 min) with fivefold concentrated reverse transcriptase buffer (50 mM Tris–HCl, pH 8.4, 75 mM KCl (Gibco BRL, 18064-014), 3 mM MgCl2 (Gibco BRL, 18064-014), 500 µM of each deoxynucleotide, 10 mM dithiothreitol (Gibco BRL, 18064-014) and 200 U of Superscript II, RT Moloney Murine Leukemia virus reverse-transcriptase (Gibco BRL, 18064-014). The reactions were incubated at 90°C for 5 min, and the samples of the cDNA obtained were stored at −20°C until further use.
Primers
Primers for the mRNA amplification of cyclophilin, IFNα, TNFα, IL12p40, TGF-beta, VP32, VP72 and A238L were designed using the sequences obtained from the Genebank Data as the follows:
Cyclophilin (forward—ACGGGTCCTGGCATCTTG and reverse—AAATGAAAAACTGGGAACCGTTT,
IFNα (forward—CCAGGTCCAGAAGGCTCAAG and reverse—GCAGCCGAGCCCTCTGT), TNFα (forward—TGGCCCCTTGAGCATCA and reverse—ACGGGCTTATCTGAGGTTTGAG),
IL12p40 (forward—CCAAATCTCAGCCAAGGTTACAT and reverse—TAGAACCTAATTGCAGGACACAGATG),
TGFβ (forward—AGGACCTGGGCTGGAAGTG and reverse—GGGCCCCAGGCAGAAAT),
VP32 (forward—TGCACATCCTCCTTTGAAACA T and reverse—TCTTTTGTGCAAGCATATACAGCTT),
VP72 (forward—ACGGCGCCCTCTAAAGGT and reverse—CATGGTCAGCTTCAAACGTTTC) and
A238L (forward—GGCCGTACTTCAAATGTGCT and reverse—CCAGTCCCAAGCAGTAAAGC).
To ensure that amplified cDNA could be distinguished from any amplified genomic DNA contaminants, each pair of primers was selected in order to include an intron in the sequence to be amplified, as determined from sequences available in Genbank. When porcine sequences were not available, consensus DNA sequences from other species were used. Cyclophilin was used as the housekeeping gene in order to normalize cytokine quantification results. The choice of cyclophilin was based on previous studies (unpublished data) in which no differences in the level of expression of this gene were found on PBMCs from pigs experimentally inoculated with ASFV of different virulence.
Plasmids for quantification in RT-PCR
Plasmids for cyclophilin, IFNα, TNFα, TGFβ, VP32, VP72 and A238L were obtained as described previously [12]. Colonies of recombinant E. coli were collected and incubated at 37°C overnight in suspension with vigorous shaking in LB broth medium (10 g of Bacto-trypton, 5 g of yeast-extract and 10 g of NaCl per litre) supplemented with ampicillin (0.1 mg/ml). Extracted DNA was used for amplification with the appropriate primers (as described above) using a conventional PCR machine (Perkin–Elmer) and sequenced to confirm the identity of the amplicon. Dilutions of PCR amplicons were quantified by spectrophotometry [33] and used as standards in real-time PCR for the quantification of mRNA expression of different cytokines. Quantification of mRNA expression of IL12p40 was assessed using purified amplicons, and the concentration was calculated as described above.
Optimisation of standard curves for quantification of cytokine mRNA
Quantification of cyclophilin, IFNα, TNFα, TGFβ, IL12p40, VP32, VP72 and A238L mRNA expression was assessed by quantitative real-time RT-PCR against the corresponding standard curves obtained with 101–107 DNA dilutions of recombinant plasmids (or purified amplicons for IL12p40). Results of cytokine quantification from macrophage cultures were expressed as microgram equivalents of the corresponding cytokine standard. The quantification was obtained with the fluorescence data in the log-phase portion of the curve, and a crossing line was set above the level of the noise band to give a value for the crossing point. The crossing point values of the standards (plasmids/purified amplicons) at known concentrations were expressed by a standard curve obtained as a fractional cycle number determined at the log-phase of the product amplification, plotted against log concentration, which was used to determine the concentration in μg of the PCR products. The melting curves obtained after amplification were used to verify the specificity of the PCR products/amplicons by comparison with the melting curves of the corresponding recombinant plasmids/amplicons.
Viral titration in macrophage supernatants
Supernatants and pellets of macrophage cultures infected with NHV or L60 obtained at 18 h post-infection were titrated by observation of cytopathic effect (CPE) at end-point dilutions as described above [21].
Real-time RT-PCR for mRNA quantification of cyclophilin, IFNα, TNFα, IL12p40, and TGFβ, in macrophages infected with NHV, L60 or PrV and VP32, VP72 and A238L in macrophages infected with NHV or L60
Samples of cDNA were used for mRNA quantification of cyclophilin, IFNα, TNFα, IL12p40, TGFβ, VP32, VP72 and A238L using the Applied Biosystems 7300 Real Time PCR (Perkin Elmer) following the manufacturer’s instructions: [12.5 µl of master mix (Power SYBR Green PCR master mix), Applied Biosystems cat no 4367659, 2.5 µl of forward and reverse primer (50 nM each), 5 µl of MILLI-Q water and 2.5 µl of cDNA). The cycles for each run were optimized as follows for cyclophilin, IFNα, TNFα, TGFβ, IL12p40, VP32, VP72 and A238L: 1 cycle of denaturation (95°C for 600 s), 50 cycles of denaturation (95°C for 15 s) and hybridization and extension (60°C for 60 s), conditions of final denaturation: 65°C for 5 s with 20°C/s ramp rate and subsequent heating of the samples to 95°C with 0.1°C/s ramp rate.
Quantitative enzyme-linked immunosorbent assay (ELISA) for IFNα, TNFα and IL12p40
Quantitative immunoassay kits were used to measure protein levels of IFNα (PBL Interferon Source, cat no. 41100), TNFα and IL12p40 (both from R&D Systems, cat no. PTA00 and P1240, respectively), following the manufacturer’s instructions. Results are shown as the ratio between values in μg/ml obtained in infected cells and non-infected control cells.
Expression of results and statistical analysis
The quantification of mRNA expression of the different genes was obtained as the ratio between the amount of each cytokine or VP and the amount of the housekeeping gene (cyclophilin), both in µg. For each group of macrophages infected with NHV, L60 and PrV results are shown as the ratio between the values obtained in infected and non-infected control cells (C). The validation of the use of the housekeeping gene was confirmed using the ANOVA test. Since there are many variables under analysis, a principal component analysis (PCA) [16] based on the correlation matrix was performed to assess the overall mRNA cytokine profile in both viral infections. A principal component is a linear combination of the original variables. There are as many principal components as variables under analysis. For sake of simplicity, we only analysed the first principal component, which explains most variability of the data. By interpreting the linear coefficients of the principal component, one can recognize which cytokines are more important during infection. The Wilcoxon signed ranks test for paired samples was used to compare ELISA results in different viral infections and for the study of VP (VP32, VP72 and A238L) dynamics. The significance level was set at 5%. Differences in virus yields between NHV- and L60-infected macrophages were analysed using a T test.
Results
Viral titration in infected macrophages
Results of virus titration from supernatants and pellets of NHV- and L60-infected macrophages are summarized in Fig. 1. Briefly, yields from supernatants and pellets from NHV-infected macrophage cultures were higher than in L60-infected macrophage cultures (AVG for NHV supernatants was 5.6 and 4.6 for L60, with P = 0.026, and AVG for NHV pellets was 5 and 3.7 for L60, with P = 0.025).
Fig. 1.
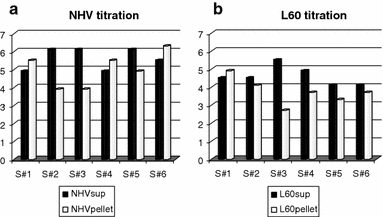
Virus titration from supernatants and pellets of NHV- (a) and L60- (b) infected macrophages cultures from each pig donor (S#). Titres are expressed as log TCID50
Quantification of the expression of IFNα, TNFα, IL12p40 (mRNA and protein) and TGFβ (mRNA) in macrophages infected with ASFV of different virulence at different times of infection (2, 4 and 6 h) using real-time PCR and quantitative ELISA
Porcine blood-derived macrophage cultures from seven pig donors were prepared and infected in vitro with either the low-virulent ASFV/NH/P68 (NHV), the highly virulent ASFV/L60 (L60) at a multiplicity of 3 (MOI = 3) or with pseudorabies virus (PrV) (used as control for the responses to ASFV infection). Duplicates of 54 cDNA samples from individual macrophage cultures collected at 2, 4 and 6 h post-infection were used to quantify mRNA expression of the cytokines IFNα, TNFα, IL12p40 and TGFβ and the expression of the ASFV genes A238L, VP32 and VP72. Their expression was compared to that in non-infected control cells (C) using a ratio calculation (NHV/C, L60/C and PrV/C). Results of the mRNA expression of IFNα, showed an increase of 81× (at 2 h post-infection) and 85× (at 4 h post-infection) in macrophages infected with the low-virulent NHV in comparison with the control cells. In macrophages infected with the highly virulent L60 or PrV, the increase of expression of this cytokine was only evident at 2 h post-infection (33× and 32×, respectively) and lower than in the NHV-infected macrophages (Table 1A). mRNA expression of TNFα showed an increase of 1.93× (at 4 h post-infection) and 5× (at 6 h post-infection) in the macrophages infected with the low-virulent NHV in comparison with the control cells. In macrophages infected with the highly virulent L60 or PrV, the increase of expression of this cytokine was never higher than 2×, even at 6 h post-infection (Table 1B).
Table 1.
Quantification of mRNA expression of IFNα (A), TNFα (B), IL12p40 (C) and TGFβ (D)
| NHV/C | L60/C | PrV/C | |
|---|---|---|---|
| A | |||
| 2 h | 81.18 | 33.18 | 32.66 |
| 4 h | 85.40 | 1.57 | 2.64 |
| 6 h | 1.52 | 1.62 | 7.89 |
| B | |||
| 2 h | 1.45 | 1.94 | 0.86 |
| 4 h | 1.93 | 2.08 | 1.85 |
| 6 h | 5.03 | 1.98 | 1.81 |
| C | |||
| 2 h | 10.3 | 6.6 | 5.6 |
| 4 h | 9.8 | 1.6 | 1.6 |
| 6 h | 1.1 | 1.1 | 8.4 |
| D | |||
| 2 h | 0.66 | 0.93 | 0.83 |
| 4 h | 0.75 | 1.00 | 1.16 |
| 6 h | 1.35 | 1.68 | 2.29 |
Relative mRNA expression of the different cytokines is shown as the ratio between the amount of each cytokine and the amount of the housekeeping gene (cyclophilin), both in μg. For each group of infected macrophages with NHV, L60 and PrV at 2, 4 and 6 hours post-infection, results are shown as the ratio between the values obtained in infected and non-infected control cells (C) (NHV/C, L60/C and PrV/C)
Quantification of the mRNA expression of IL12p40 showed an increase of around 10× (at 2 and 4 h post-infection) of the mRNA expression of this cytokine in the macrophages infected with the low-virulent NHV in comparison with the control cells. In macrophages infected with the highly virulent L60, the increase of expression of this cytokine was never higher than 6.6× (observed at 2 h post-infection). In PrV-infected macrophages, the mRNA expression of this cytokine was enhanced 8.4× later in infection (6 h post-infection) (Table 1C).
mRNA expression of TGFβ showed an increase of 2× (at 6 h post-infection) in the macrophages infected with PrV in comparison with control cells. In macrophages infected with the highly virulent L60, an increase of expression (1.6×) was observed at 6 h post-infection. The lower levels of mRNA expression of this cytokine in comparison with the control values were found in the NHV-infected macrophages (Table 1D).
Cytokine profiles in both NHV and L60 infections were assessed by principal component analysis (Table 3). At 2 h post-infection with NHV, the coefficient of TGFβ is close to zero (0.003), which suggests that this cytokine is not important in the first principal component. The remaining cytokines have coefficients with the same sign and similar magnitude. Thus, this principal component can be interpreted as a sort of mean effect of IFNα, TNFα, IL12p40 and A238L. At 2 h post-infection with L60, TGFβ has the smallest coefficient, but not as close to zero as for NHV at the same time point. Since all variables have coefficients with the same sign and similar values, this principal component is related to a mean effect of all variables. At 4 h post-infection with NHV, TGFβ, TNFα and A238L have the highest coefficients in absolute terms. Since TGFβ has a negative coefficient as opposed to TNFα and A238L, this principal component shows that TGFβ seems to have an opposite effect to TNFα and A238L. In L60, all variables are important at 4 h post-infection, because the respective coefficients, are similar in absolute terms. TGFβ, TNFα and IL12p40 show negative coefficients while IFNα and A238L exhibit positive coefficient. Therefore, TGFβ, TNFα and IL12p40 seem to have opposite effects to IFNα and A238L. With respect to 6 h post-infection with NHV, similar results were obtained to the ones at 4 h. At 6 h post-infection with L60, the highest coefficients in absolute terms are associated with IFNα, TGFβ and A238L. Since TGFβ has a negative coefficient as opposed to the IFNα and A238L, this principal component compares the effects of TGFβ to those of IFNα and A238L.
Table 3.
Assessement of the overall mRNA cytokine expression in NHV and L60 infection by principal component analysis
| Variables | 2 h | 4 h | 6 h | |||
|---|---|---|---|---|---|---|
| NHV | L60 | NHV | L60 | NHV | L60 | |
| IFNα | −0.584 | 0.538 | 0.011 | 0.460 | 0.137 | 0.553 |
| TGFβ | 0.003 | 0.202 | −0.545 | −0.411 | −0.561 | −0.543 |
| TNFα | −0.367 | 0.489 | 0.580 | −0.506 | 0.514 | 0.129 |
| IL12p40 | −0.525 | 0.561 | −0.157 | −0.361 | 0.390 | −0.139 |
| A238L | −0.499 | 0.346 | 0.585 | 0.478 | 0.501 | 0.603 |
| Var. Exp. % | 51.4 | 55.7 | 55.5 | 65.4 | 51.6 | 49.7 |
ELISA results showed a tendency for higher levels in the protein expression for IFNα in the NHV-infected macrophage cultures in comparison with the L60-infected macrophage cultures. By comparison with the control cells an increase of 0.7× was observed in the NHV infection (P = 0.02). The higher results obtained for the quantification of the IFNα protein at 18 h post-infection were observed with PrV-infected macrophages (Fig. 2).
Fig. 2.
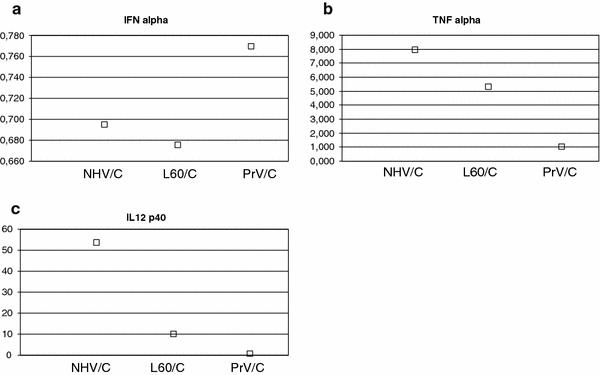
Quantification of the final product (protein) for IFNα (a), TNFα (b) and IL12p40 (c) in macrophage cultures infected with NH/P68, L60 and PrV. For each group of macrophages infected with NHV, L60 and PrV results are shown as the ratio between the values obtained in infected and non-infected control cells (C) (NHV/C, L60/C and PrV/C)
The ELISA quantification of TNFα showed an increase by a factor of 8 and 5 in the supernatants of the macrophages infected with NHV and L60, respectively (Table 2) in comparison with control cells. These increases are statistically significant (P = 0.02 and 0.03 for NHV and L60, respectively).
Table 2.
Statistical analysis of ELISA results of cytokine production between the different groups tested (control, NHV, L60 and PrV) using the Wilcoxon signed ranks test
| Cytokine | P Values (one-tailed) | ||
|---|---|---|---|
| Friedman test | Wilcoxon signed ranks test | ||
| IFNα | 0.22 | NHV-Control | 0.02 |
| L60-Control | 0.06 | ||
| PrV-Control | 0.22 | ||
| L60-NHV | 0.31 | ||
| PrV-NHV | 0.22 | ||
| PrV-L60 | 0.50 | ||
| IL12 | 0.06 | NHV-Control | 0.22 |
| L60-Control | 0.31 | ||
| PrV-Control | 0.42 | ||
| L60-NHV | 0.03 | ||
| PrV-NHV | 0.02 | ||
| PrV-L60 | 0.41 | ||
| TNFα | 0.0005 | NHV-Control | 0.02 |
| L60-Control | 0.03 | ||
| PrV-Control | 0.34 | ||
| L60-NHV | 0.06 | ||
| PrV-NHV | 0.02 | ||
| PrV-L60 | 0.03 | ||
The IL12p40 protein was increased 53.3 and 10 times in the supernatants of the macrophages infected with NHV and L60, respectively, in comparison with control cells (Table 2). Expression of IL12p40 in the NHV supernatants was significantly higher than in the L60 supernatants (P = 0.03). Lower values of expression of this cytokine were observed in PrV infection, which were significantly different from the NHV infection (P = 0.02).
Quantification of the expression of VP32, VP72 and A238L in macrophages infected with ASFV of different virulence at different times of infection (2, 4 and 6 h)
The quantification of mRNA expression of VP32 was clearly observed at early stages of infection (from 2 h post-infection) in macrophages infected with both the low-virulent NHV and the highly virulent L60. Results showed identical levels of mRNA expression of VP32 at 2 (P = 0.078), 4 (P = 0.578) and 6 (P = 0.625) hours post-infection in macrophage cultures infected with both isolates (Fig. 3a). mRNA expression of VP72 and A238L in the seven macrophage cultures was increased at 4 h post-infection in macrophages infected with the low-virulent NHV in comparison with results from macrophages infected with L60 (P = 0.016 and P = 0.031, respectively) (Fig. 3b, c). mRNA expression of VP72 and A238L at 2 and 6 h post-infection was identical in the macrophages infected with both isolates.
Fig. 3.

Quantification of mRNA expression of VP32 (a), VP72 (b) and A238L (c) in NHV- and L60-infected macrophage cultures. Results are show as average ratio ± standard error (AVG ± SE) between the values in μg of each VP and the values in μg of the housekeeping gene cyclophilin
Discussion
Depending on the viral isolate, ASFV may induce acute to chronic, or even unapparent, clinical forms of disease in domestic pigs, in parallel with the development of distinct pathological lesions. Different aspects of virus biology may contribute to this, and during the past decades, several studies have shown that viral–host interactions are orchestrated by a complex viral genome, which encodes more than one hundred proteins, some of which are known to interfere with or evade host immune responses [8].
Cellular-based immune mechanisms have been demonstrated to be relevant in protection, as shown in pigs experimentally inoculated with the low-virulent ASFV/NH/P68 (NHV), which developed ASFV-specific cytotoxic T lymphocytes (CTL) activity [17, 18, 22] enhanced natural killer (NK) cell activity, and survival of challenge with the highly virulent ASFV/L60 (L60) [19]. More recently, the role of specific ASFV CTL in protection was further studied in pigs inoculated the low-virulent OUR/T88/3 and challenged with the virulent OUR/T88/1 [29]. In this study, it was demonstrated that pigs exposed to OUR/T88/3 and then depleted of CD8+ lymphocytes were no longer fully protected against OUR/T88/1 challenge. These results reinforce the role of CD8+ lymphocytes in protective immunity to ASFV infection.
Macrophage-derived cytokines determine the development of inflammatory responses through the expression of pro-inflammatory cytokines, which are the first defence against infection, and the induction of specific immune responses through the expression of immunoregulatory cytokines. Our previous studies using the L60 and NHV model of infection of pig macrophages [12] demonstrated significantly increased levels of transcripts for TNFα, IL6, IL12 and IL15 in macrophages infected with the low-virulent virus NHV in comparison to the effect of infection by L60 at 6 h post-infection.
The studies reported here were conducted to characterize the impact of infection in porcine macrophages by NHV and by L60 on the expression and production of different cytokines at 2, 4 and 6 h post-infection. At these different time points, viral infection was assessed through the quantification of different mRNA expression of viral proteins VP32, VP72 and A238L, which, except for the values observed at 4 h post-infection for VP72 and A238L, were similar in both infections. To better assess the efficiency of infection by each isolate, virus yields obtained at 18 h post-infection, when at least one viral replication cycle is considered to be completed, have shown higher progeny levels of NHV in comparison to L60 infection.
To date, little is known about the impact of interferon type I induction and regulation of ASFV infection. The interferon family (IFNs) is one important family of inducible cytokines that stimulate the early or innate antiviral response to limit viral replication [2, 23]. The multiple subtypes of α-interferons and the single β-interferon belong to the type I family of interferons (IFNs), while type II interferons have a single member: IFN-γ [37]. Type I IFNs have important biological functions, ranging from immune cell development and activation to tumour cell killing and, most importantly, inhibition of replication of many RNA and DNA virus [35]. IFNα and β regulate the homeostatic differentiation of hematopoietic cells such as B cells, T cells, osteoclasts, myeloid dendritic cells (DCs) and natural killer (NK) cells. In the case of immature DCs, their activation and maturation can be induced by IFNα/β, leading to the up-regulation of major histocompatibility complex (MHC) molecules (especially class I MHC), chemokines, chemokine receptors and costimulatory molecules (CD40, CD80, CD86), which in turn leads to efficient homing in secondary lymphoid organs and CD8+ and CD4+ T cell responses. In NK cells, IFNα and β increase levels of perforin and lead to the induction of cytotoxic activity. T lymphocyte responses are also modulated through IFNα/β promotion of Th1 differentiation [37].
In our studies, quantification of IFNα mRNA expression was conducted in seven cultures of porcine blood-derived macrophages from different pig donors. The increase of IFNα mRNA and protein expression compared to control cells was higher with NHV than with L60. The reasons for these differences between the two ASFV isolates are not known. However, recent studies using ASFV with MGF360/530 deleted [1] demonstrated the implication of these multigenic families in the transcription inhibition of the interferon gene and in the interferon activation pathways. Other authors using virulent ASFV without these multigenic families deleted also reported an inhibition in the IFN transcripts [32, 38, 40, 41]. In contrast to the presence of those genes in the L60 isolate, NHV virus has a naturally occurring deletion in the MGF360/530 genes [10], which could be related to the large enhancement of IFNα mRNA expression in macrophages infected with the NHV isolate. Moreover, type I IFNs are potent activators of anti-viral pathways in infected cells and are strong inducers of NK cell cytotoxicity, which is well characterized in NHV infection [19].
mRNA and protein expression of TNFα was increased in macrophages infected with the low-virulent NHV in comparison with the control cells. In macrophages infected with the highly virulent L60 or PrV, the increase in expression of this cytokine was never higher than with NHV. The increase in the expression of TNFα has already been demonstrated in our previous studies using the low-virulent NHV virus [12]. Induction of inflammatory responses in NHV infection may be regulated through the enhancement of the expression of this cytokine, which can activate the cellular immune mechanisms described previously [19, 22] and may contribute to the protection of the host against viral infection.
Quantification of the mRNA and protein expression of IL12p40 showed a higher increase in this cytokine with NHV compared to L60 and PrV. IL12p40 is a very important cytokine in the activation of the Th1 lymphocytes and in the differentiation of the CD4+/CD8+ subpopulations [36]. The enhancement of this cytokine is clearly evident in the NHV infection and in accordance with our previous studies [12]; it may be involved in the enhancement of T cell proliferation in the development of a specific CTL and NK response [19, 22] and in the induction of LAK cells, as reported for NHV infection [34].
An increase of 2× (at 6 h post-infection) in the mRNA expression of TGFβ was observed in macrophages infected with PrV in comparison with control cells. Lower levels of the mRNA expression of this cytokine in comparison with the control values were found in NHV-infected macrophages. In macrophages infected with the highly virulent L60, an increase in expression (1.6 times) was observed at 6 h post-infection. An increase in the expression of the anti-inflammatory cytokine TGFβ was previously reported in the infection with the virulent ASFV/Malawi/Lil [40].
A principal component analysis was used to clarify the interpretation of the abovementioned results (Table 3). The first principal components can be interpreted differently when analyzing either NHV and L60, or different time points. This shows that the two viral infections induce different cytokine profiles. According to this analysis, the viral protein A238L is very important during both infections, since it appears in every principal component with a large (absolute) value. At 2 h post-infection, the role of the cytokines studied is not well defined for either virus, since the principal components are a sort of average effect of the variables. After this time point, some relevant changes are observed. In the NHV infection, there is somehow a counteracting effect of TGFβ against the expression of TNFα and A238L at 4 and 6 h post-infection. With respect to L60 infection at 4 h, TGFβ, TNFα and IL12p40 have an opposite effect to IFNα and A238L. At 6 h post-infection, TNFα and IL12p40 seem to loose their relevance in the course of the infection. More importantly, TGFβ appears to counteract IFNα and A238L expression. From 4 to 6 h, the difference between cytokine profiles in the L60 infection is probably related to innate cytokine responses that include IFN type I, TNFα and IL12p40, which can induce anti-viral mechanisms in virus-infected cells. Other authors using lymphocytic choriomeningitis virus [27] have suggested that it may be important to limit IL12p40 during viral infections through IFN type I. In fact, even when IL12p40 is observed in early stages of infection, it is only expressed during a short window of time [28], and interestingly, as may happen in ASFV infection, its production can be blocked by IFN type I [7].
Although IFN type I can negatively regulate IL12p40 expression, IL12p40 is also induced in certain viral infections and is required for the activation of NK cells (2), which may explain the enhancement of this cellular activity in the NHV in vivo infections [19].
In conclusion, the NHV isolate induces in porcine macrophages higher mRNA expression and production of IFNα, TNFα and IL12p40 and a positive interaction of cytokines involved in cellular immune mechanisms, namely IFNα and IL12p40.
Overall, our studies reinforce and clarify the effect of the NHV infection by targeting cellular and cellular-based immune responses relevant for pig survival against ASFV infection.
Acknowledgments
This work was supported by the project POCI/CVT/59122/2004, by the postdoctoral fellowship FCT/BPD/14676/2003 and by the Welcome Trust Project AHDW/03/04, no. 075813: “African swine fever virus: Development of vaccines and epidemiological investigations”. The authors are grateful to Ana Rita Chaves, Jú Silva and Clara Cartaxeiro for technical assistance.
References
- 1.Afonso CL, Piccone ME, Zaffuto KM, Neilan J, Kutish GF, Lu Z, Balinsky CA, Gigg TR, Bean TJ, Zsak L, Rock DL. African swine fever virus multigene family 360 and 530 genes affect host interferon response. J Virol. 2004;78:1858–1864. doi: 10.1128/JVI.78.4.1858-1864.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biron CA. Role of early cytokines, including alpha and beta interferons (IFN-α/β), in innate and adaptive immune responses to viral infections. Sem Immunol. 1998;10:383–390. doi: 10.1006/smim.1998.0138. [DOI] [PubMed] [Google Scholar]
- 3.Carrascosa AL, Santarén JF, Viñuela E. Production and titration of African swine fever virus in porcine alveolar macrophages. J Virol Methods. 1982;3:303–310. doi: 10.1016/0166-0934(82)90034-9. [DOI] [PubMed] [Google Scholar]
- 4.Casal I, Enjuanes L, Viñuela E. Porcine leucocyte cellular subsets sensitive to African swine fever virus in vitro. J Virol. 1984;52:37–46. doi: 10.1128/jvi.52.1.37-46.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colgrove GS. Immunofluorescence and inclusion bodies in circulating leukocytes of pigs infected with African swine fever virus. Bull Epizoot Dis Africa. 1968;16:341–343. [PubMed] [Google Scholar]
- 6.Colgrove GS, Haelterman O, Coggins L. Pathogenesis of African swine fever virus in young pigs. Am J Vet Res. 1969;30:1343–1359. [PubMed] [Google Scholar]
- 7.Cousens LP, Orange JS, Su HC, Biron CA. Interferon-α/β inhibition of interleukin 12 and interferon-γ production in vitro and endogenously during viral infection. Proc Natl Acad Sci USA. 1997;94:634–639. doi: 10.1073/pnas.94.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixon LK, Abrams CC, Bowick G, Goatley LC, Kay-Jackson PC, Chapman D, Liverani E, Nix R, Silk R, Zhang F. African swine fever virus proteins involved in evading host defence systems. Vet Immunol Immunopathol. 2004;100(3–4):117–134. doi: 10.1016/j.vetimm.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 9.Dixon LK, Escribano JM, Martins C, Rock DL, Salas ML, Wilkinson PJ (2005) In: Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA (eds) Vírus Taxonomy, VIIIth Report of the ICTV135–143. Elsevier/Academic Press, London
- 10.Duarte M (2000) Bases moleculares da virulência e hemadsorção nos isolados nacionais Lisboa 60 e NH/68 do vírus da peste suína Africana. PhD Thesis, Universidade Nova de Lisboa, Instituto de Tecnologia Química e Biológica, Oeiras, Portugal
- 11.Enjuanes L, Cubero I, Viñuela E. Sensitivity of macrophages from different species to African swine fever (ASF) virus. J Gen Virol. 1977;34:455–463. doi: 10.1099/0022-1317-34-3-455. [DOI] [PubMed] [Google Scholar]
- 12.Gil S, Spagnuolo-Weaver M, Canals A, Sepúlveda N, Oliveira J, Aleixo A, Allan G, Leitão AC, Martins CLV. Expression at mRNA level of cytokines and A238L gene in porcine blood-derived macrophages infected in vitro with African swine fever virus (ASFV) isolates of different virulence. Arch Virol. 2003;184(11):2077–2097. doi: 10.1007/s00705-003-0182-x. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez-Juarrero M, Mebus CA, Pan R, Revilla Y, Alonso JM, Lunney JK. Swine leukocyte antigen (SLA) and macrophage marker expression on both African swine fever virus (ASFV) infected and non infected primary porcine macrophage cultures. Vet Immunol Immunopathol. 1992;32:243–259. doi: 10.1016/0165-2427(92)90049-V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heuschele WP, Coggins L, Stone SS. Fluorescent antibody studies on African swine fever. Am J Vet Res. 1966;27:477–484. [PubMed] [Google Scholar]
- 15.Heuschele WP. Studies on the pathogenesis of African swine fever. I quantitative studies on the sequential development of virus in pig tissues. Arch Ges Virusforsch. 1967;21:349–356. doi: 10.1007/BF01241735. [DOI] [PubMed] [Google Scholar]
- 16.Johnson RA, Wichern DW. Applied multivariate statistical analysis. New Jersey: Prentice-Hall Inc.; 1998. [Google Scholar]
- 17.Leitão A, Malur A, Cornelis P, Martins CL. Identification of a 25-Aminoacid sequence from the major African swine fever virus (ASFV) structural protein VP72 recognised by porcine cytotoxic T lymphocytes using a lipoprotein based expression system. J Virol Methods. 1998;75:113–119. doi: 10.1016/S0166-0934(98)00105-0. [DOI] [PubMed] [Google Scholar]
- 18.Leitão A, Malur A, Cartaxeiro C, Vasco G, Cruz B, Cornelis P, Martins CL. Bacterial lipoprotein based expression vectors as tools for the characterisation of African swine fever virus (ASFV) antigens. Arch Virol. 2000;145:1639–1657. doi: 10.1007/s007050070081. [DOI] [PubMed] [Google Scholar]
- 19.Leitão A, Cartaxeiro C, Coelho R, Cruz B, Parkhouse RME, Portugal FC, Vigário JD, Martins CLV. The non-hemadsorbing African swine fever virus isolate ASFV/NH/P68 provides a model for defining the protective anti-virus immune response. J Gen Virol. 2001;82:513–523. doi: 10.1099/0022-1317-82-3-513. [DOI] [PubMed] [Google Scholar]
- 20.Malmquist WA, Hay D. Hemadsorption and cytophatic effect produced by African swine fever virus in swine bone marrow and buffy coat cultures. Am J Vet Res. 1960;21:104–108. [PubMed] [Google Scholar]
- 21.Martins C, Scholl T, Mebus CA, Fish H, Lawman MJP. Modulation of porcine peripheral blood derived macrophages functions by in vitro infection with ASFV isolates of different virulence. Viral Immunol. 1988;1:177–190. doi: 10.1089/vim.1987.1.177. [DOI] [PubMed] [Google Scholar]
- 22.Martins C, Lawman M, Scholl T, Mebus C, Lunney J. African swine fever virus specific porcine cytotoxic T cell activity. Arch Virol. 1993;129:211–225. doi: 10.1007/BF01316896. [DOI] [PubMed] [Google Scholar]
- 23.Mbow ML, Sarisky RT. What is disrupting IFN-alpha’s antiviral activity? Trends Biotechnol. 2004;22(8):395–399. doi: 10.1016/j.tibtech.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 24.Montgomery RE. On a form of swine fever occurring in British East (Kenia Colony) J Comp Pathol Ther. 1921;34:159–191. [Google Scholar]
- 25.Montgomery RE. On a form of swine fever occurring in British East (Kenia Colony) J Comp Pathol Ther. 1921;34:243–262. [Google Scholar]
- 26.Murtaugh MP, Baarsch MJ, Zhou Y, Scamurra RW, Lin G. Inflammatory cytokines in animal health and disease. Vet Immunol Immunopathol. 1996;54(1–4):45–55. doi: 10.1016/S0165-2427(96)05698-X. [DOI] [PubMed] [Google Scholar]
- 27.Orange JS, Salazar-Mather TP, Opal SM, Spencer RL, Miller AH, McEwen BS, Biron CA. Mechanism of interleukin 12-mediated toxicities during experimental viral infections: Role of tumor necrosis factor and glucocorticoids. J Exp Med. 1995;181:901–914. doi: 10.1084/jem.181.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orange JS, Biron CA. An absolute and restricted requirement for IL12 in natural killer cell IFNγ production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J Immunol. 1996;156:1138–1142. [PubMed] [Google Scholar]
- 29.Oura CAL, Denyer MS, Takamatsu H, Parkhouse RME. In vivo depletion of CD8+ T lymphocytes abrogates protective immunity to African swine fever virus. J Gen Virol. 2005;86:2445–2450. doi: 10.1099/vir.0.81038-0. [DOI] [PubMed] [Google Scholar]
- 30.Plowright W, Parker J, Peirce MA. The epizootology of African swine fever in Africa. Vet Rec. 1969;85:668–674. [PubMed] [Google Scholar]
- 31.Plowright W, Thomson GR, Neser JA (1994) African swine fever. In: Coetzer JAW, Thomson GR, Tustin RC (eds) Infectious diseases of livestock, 1st edn, vol 1, Chap 51. Oxford University Press, Southern Africa, pp 568–599
- 32.Powell PP, Dixon LK, Parkhouse RM. An IkappaB homologue encoded by African swine fever virus provides a novel mechanism for downregulation of proinflamatory cytokine responses in host macrophages. J Virol. 1996;70:8527–8533. doi: 10.1128/jvi.70.12.8527-8533.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. A laboratory manual. 2. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 34.Scholl T, Lunney JK, Mebus CA, Duffy E, Martins CLV. Virus-specific cellular blastogenesis and interleukin-2 production in swine after recovery from African swine fever. Am J Vet Res. 1989;50:1781–1786. [PubMed] [Google Scholar]
- 35.Solis M, Goubau D, Romieu-Mourez R, Genin P, Civas A, Hiscott J. Distinct functions of IRF-3 and IRF-7 in IFN-alpha gene regulation and control of anti-tumor activity in primary macrophages. Biochem Pharm. 2006;72:1469–1476. doi: 10.1016/j.bcp.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 36.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptative immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 37.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–335. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 38.Vallée I, Tait SWG, Powell P. African swine fever virus infection of porcine aortic endothelial cells leads to inhibition of inflammatory responses, activation of the thrombitc state, and apoptosis. J Virol. 2001;75:10372–10382. doi: 10.1128/JVI.75.21.10372-10382.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wardley RC, Andrade CM, Black DN, Castro Portugal FL, Enjuanes L, Hess WR, Mebus C, Ordas A, Rutili D, Sanchez Vizcaino J, Vigario JD, Wilkinson PJ, Moura Nunes JF, Thomson G. African Swine Fever virus. Brief review. Arch Virol. 1983;76:73–90. doi: 10.1007/BF01311692. [DOI] [PubMed] [Google Scholar]
- 40.Whittall JTD, Parkhouse RME. Changes in swine macrophage phenotype after infection with African swine fever virus: cytokine production and responsiveness to interferon-γ and lipoplysaccharide. Immunology. 1997;91:444–449. doi: 10.1046/j.1365-2567.1997.00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zsak L, Lu Z, Burrage TG, Neilan JG, Kutish GF, Moore DM, Rock DL. African swine fever virus multigene family 360 and 530 genes are novel macrophage host range determinants. J Virol. 2001;75:3066–3076. doi: 10.1128/JVI.75.7.3066-3076.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]