

Abstract
Reported ocular findings in the 22q11.2 deletion syndrome (which encompasses the phenotypes of DiGeorge, velocardiofacial, and Takao (conotruncal-anomaly-face) syndromes) have included posterior embryotoxon (prominent, anteriorly displaced Schwalbe’s line at the corneal limbus or edge), retinal vascular tortuosity, eyelid hooding, strabismus, and astigmatism. We present seven 22q11.2 patients from multiple centers with sclerocornea, an eye finding previously unreported in the literature. Four boys and three girls were identified with sclerocornea, systemic DGS/VCFS findings, and fluorescence in situ hybridization (FISH)-confirmed microdeletion at chromosome 22q11.2. FISH diagnosis was perinatal in six patients but at 2 years of age in one child. Sclerocornea was bilateral in five patients. Findings included descemetocele (five eyes), microophthalmos (one eye), iridocorneal adhesions (one bilateral case), and severe anterior segment dysgenesis (one eye). Two patients underwent bilateral corneal transplantation; another two were scheduled for possible unilateral transplant. Sclerocornea is a static congenital condition in which the cornea is opaque and vascularized and resembles the sclera. The novel finding of sclerocornea suggests that a genetic locus at 22q11.2 may be involved in anterior segment embryogenesis. In most of our patients, the diagnostic process was underway, but in one patient 22q11.2 deletion was not suspected until after the child had already been undergoing treatment for sclerocornea for 2 years. Sclerocornea should be added to the clinical manifestations of the 22q11.2 deletion syndrome. Ophthalmologists diagnosing sclerocornea in children with systemic findings suggestive of 22q11.2 deletion should ensure appropriate genetic referral.
Keywords: sclerocornea, corneal opacity, chromosome 22q11.2 deletion syndrome, DiGeorge syndrome, velocardiofacial syndrome, Opitz G/BBB syndrome, Cayler cardiofacial syndrome, Catch 22
INTRODUCTION
The chromosome 22q11.2 deletion syndrome encompasses phenotypes previously called DiGeorge syndrome, velocardiofacial (Shprintzen) syndrome, Conotruncal anomaly face (Takao) syndrome, and some cases of autosomal dominant Optiz G/BBB syndrome and Cayler cardiofacial syndrome. [Driscoll et al., 1993; Wilson et al., 1993; Matsuoka et al., 1994; McDonald-McGinn et al., 1995; LaCassie and Arriaza, 1996; Wulfsberg et al., 1996; McDonald-McGinn et al., 1997a,b, 1999]. The 22q11.2 deletion syndrome occurs in about 1 in 4,000 live births. [Devriendt et al., 1998; Oskarsdottir et al., 2004] and is a contiguous gene deletion syndrome detected by fluorescence in situ hybridization (FISH) in 95% of the cases.
The clinical manifestations are varied and include congenital heart malformations (74%), in particular conotruncal malformations (tetralogy of Fallot, interrupted aortic arch, ventricular septal defect, and truncus arteriosus), palatal abnormalities (69%) such as velopharyngeal incompetence, submucous cleft palate and overt cleft palate, characteristic facial features in the majority of Caucasians (e.g., overfolded/squared-off helices, bulbous nasal tip, small mouth and chin), learning difficulties (70–90%), and thymus and parathyroid hypoplasia with immune (T-cell) deficiency (77%) and hypocalcemia (50%). [McDonald-McGinn et al., 1996,1997a, 1999a, 2001; Sullivan, 2004; Bassett et al., 2005; McDonald-McGinn et al., 2005, 2006] Other findings include feeding problems, growth hormone deficiency, autoimmune disorders, hearing loss, seizures, and renal, musculoskeletal, and laryngotracheoesophageal abnormalities.
Ophthalmologic findings in the 22q11.2 deletion syndrome have included posterior embryotoxon (a prominent, anteriorly displaced Schwalbe’s line at the corneal limbus or edge), retinal vascular tortuosity, eyelid hooding, strabismus, and astigmatism. [Fitch, 1983; Shprintzen et al., 1985; Beemer et al., 1986; Mansour et al., 1987; Ryan et al., 1997; Forbes et al., 2006] We present a multicenter case series of seven patients with the 22q11.2 deletion syndrome and sclerocornea, a finding previously unreported in the literature.
MATERIALS AND METHODS
Clinical data were collected on seven patients with sclerocornea and the 22q11.2 microdeletion diagnosed by FISH presenting to the Children’s Hospital of Philadelphia, Philadelphia, PA; Children’s Healthcare of Atlanta at Egleston, GA; the University of Iowa, Iowa City, IA; the Lucile Salter Packard Children’s Hospital, Palo Alto, CA; and the Marshfield Clinic, Saint Joseph’s Hospital, Marshfield, WI. All patients were examined by an ophthalmologist. FISH diagnosis was carried out using the N25 probe or the TUPLE 1 probe in the chromosome 22q11.2 region to demonstrate a single 22 homolog signal, contrasted with a control probe, for example, ARSA in 22q12.3, present on both chromosome 22 homologs.
The study was approved by the Institutional Review Board of the University of Pennsylvania/Children’s Hospital of Philadelphia as part of studies on patients with the Chromosome 22q11.2 deletion syndrome, and the study data were handled in compliance with HIPAA regulations.
RESULTS
Four boys and three girls were identified with sclerocornea, systemic 22q11.2 deletion findings, and FISH-confirmed microdeletion at 22q11.2 (Table I). Diagnosis of the 22q11.2 deletion was perinatal in six patients and at 2 years of age in one patient (Patient 3), who had undergone corneal transplantation earlier in life and presented with a perforated corneal ulcer requiring surgical repair, after which a post-operative fever work-up led to the discovery of systemic abnormalities and the 22q11.2 microdeletion. Sclerocornea was bilateral in five patients. Additional ocular findings included descemetocele (five eyes), microophthalmos (one eye), iridocorneal adhesions (one bilateral case), and severe anterior segment dysgenesis (one eye). Three patients underwent corneal transplantation (two bilateral, one unilateral), and another patient was scheduled for a possible unilateral transplant. Figure 1
TABLE I.
Clinical Findings of Seven Patients With 22q11.2 Deletion and Sclerocornea
| Case | BW; GA; Sex | Family history | 22q Diagnosis | Systemic findings | Ocular findings | Management | |
|---|---|---|---|---|---|---|---|
| 1 | 2.9 kg; 35 weeks; Male | Mother – ASD | Prenatal | Dextrocardia, complete AV canal, secundum ASD, PDA, round broad face, small ears, cleft lip and palate, mild thymic hypoplasia | Bilateral sclerocornea with vascularization (corneas appeared ectatic and at risk for rupture), microphthalmia | Corneal transplantation delayed due to poor health. At 2 months of age, roving eye movements, left eye with central descemetocele, severe corneal protrusion but no perforation. At 1 year of age, did not survive cardiac surgery | |
| 2 | 2.9 kg; FT; Female | Mother – epilepsy (carbamazapine during pregnancy), myopia, esotropia, hearing loss, Father – cleft lip and heart defect, Maternal aunt – ASD or VSD, died 16 years of age | Postnatal | IAA, LV outflow tract obstruction, secundum ASD, VSD, craniosynostosis of metopic and bilateral coronal sutures, low set ears, stenotic ear canals, left preauricular skin tag, cleft palate, micrognathia, anal stenosis, hypocalcemia | Bilateral sclerocornea, iridocorneal adhesions, ectopic pupils; OD – corneal descemetocele | Corneal transplantation and lysis of adhesion OS (24 days old), OD (2 months old), pathology – corneal stromal fibrosis, mild inflammation and neovascularization, abnormally thin Descemet’s membrane. Uncomplicated post-op course | |
| 3 | N/A; FT; Male | 2.5 years of age | Right aortic arch, anomalous retropharyngeal subclavian artery contributing to vascular ring, slightly dysmorphic ears, bilateral hearing loss. Note: Post-surgical fever work-up led to chest radiograph abnormality and discovery of systemic findings and 22q11.2 diagnosis | Bilateral sclerocornea, failed corneal transplants (OD 5 months, OS 18 months). Presented with fever, cough, and right eye discharge. EUA revealed bilaterally opacified corneas, bulging cornea OD with perforated descemetocele, culture grew Haemophilus influenzae (Aegyptius), ocular ultrasound showed shallow choroidal detachment OD | Perforation sealed with cyanoacrylate glue and contact lens. Intravitreal Ancef and Ceftazidin. Topical cipro-floxacin and vancomycin | ||
| 4 | 2.7 kg; FT; Male | Preterm labor at 34 weeks, requiring nifedipine, magnesium sulfate, Mother – scoliosis, Paternal great uncle – unilateral congential glaucoma | Postnatal | Small PDA, enlarged aortic root, overfolded superior helices, bilateral epicanthal folds, broad high nasal bridge, laterally built up nose, small nares, mild micrognathia, left vocal cord paralysis, laryn-gomalacia, long fingers and toes, decreased T-cell count, recurrent otitis media | Right sclerocornea involving the visual axis and peripheral vascularization. Normal iris visible superiorly. Right fundus was not visible. Left eye was normal | Patching OS. Parents refused corneal transplantation. At 2.5 years of age, vision light perception OD | |
| 5 | 2.5 kg; FT; Female | Father and maternal uncle – “turned in feet” | Postnatal | Large outlet VSD, OAA, secundum ASD, normal karyotype | OD – severe anterior segment dysgenesis with extreme descemetocele protruding forward and preventing eyelid closure. OS – sclerocornea with vascularization | OD – aggressive topical lubricants until eyelid closure was possible. OS – planned corneal transplantation | |
| 6 | N/A; FT; Male | Sibling – VSD, 22q11.2 deletion | Postnatal | VSD | Bilateral posterior embryotoxon and sclerocornea. Faint corneal opacification inferiorly OD, denser white stromal opacity with vascularization OS from inferior limbus to near visual axis | Observe | |
| 7 | 3.5 kg; FT; Female | Father – 22q11.2 deletion | Prenatal | VSD, IAA type, rear pointed overfolded helix, anteriorly placed anus, low T-cell count | Sclerocornea right eye – inferior opacification extending up into visual axis (Fig. 1). | Corneal transplantation OD at 7 weeks of age | |
ASD, atrial septal defect; AV, atrioventricular; BW, birth weight; EUA, examination under anesthesia; FT, full term; GA, gestational age; IAA, interupted aortic arch; LV, left ventricular; N/A, not available; OD, right eye; OS, left eye; PDA, patent ductus arteriosis.
Fig. 1.
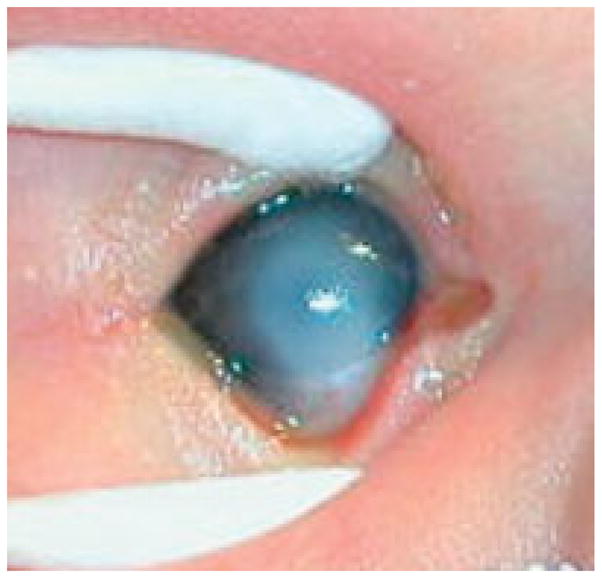
Sclerocornea of the right eye in a newborn with Chromosome 22q11.2 microdeletion (Patient 7). The limbus (corneal–scleral border) is poorly defined and there is opacification of the inferior cornea with extension up into the visual axis. This child subsequently underwent corneal transplantation at the age of 7 weeks. The left eye was unaffected.
DISCUSSION
Sclerocornea is a congenital, non-progressive, non-inflammatory condition in which one or both corneas demonstrate some degree of opacification coupled with flattening of the normal corneal curvature. In all patients, opacification is more pronounced in the corneal periphery with varying involvement of the visual axis, in contrast with the primarily central opacification found in Peter anomaly. Sclerocornea is most commonly bilateral and asymmetric, although unilateral cases have been reported. [Krachmer et al., 1997] Flattening of corneal curvature may be severe; indeed, corneal curvatures equal to or less than scleral curvature are pathognomonic for the condition. [Desvignes et al., 1967] The association between opacification and flattening is so strong that most authors consider sclerocornea and cornea plana to be phenotypic manifestations of the same condition.
Sclerocornea may be sporadic or familial, without gender predilection, and when inherited, the recessive form typically demonstrates a more severe phenotype than the dominant form. [Bloch, 1965; Howard and Abrahams, 1971; Eliott et al., 1985; Tahvanainen et al., 1995] Associated genetic loci that have been reported include Xp22.31 when sclerocornea is present with microphthalmia and dermal aplasia as part of the MIDAS syndrome [Happle et al., 1993]; 18q21.3 in a 12-year-old child with autism, anophthalmia, microphthalmia, and sclerocornea and a mutation in the RAX gene [Voronina et al., 2004]; and 6p22-24 in a dysmorphic infant with an interstitial deletion [Moriarty and Kerr-Muir, 1992].
The finding of sclerocornea in association with the chromosome 22q11.2 deletion syndrome suggests that a genetic locus at 22q11.2 influences anterior segment embryogenesis. The anterior segment of the eye includes the cornea, anterior chamber, iris, lens, ciliary body, and related structures. The embryologic basis for sclerocornea lies in the absence of normal migration of neural crest cells between weeks seven and ten of gestation. Lack of migration results in failure of formation of the limbal anlage, resulting in absence of the limbus, lack of differentiation between corneal and scleral architecture, and development of corneal opacification and flattening. [Friedman et al., 1975; Townsend, 1998] The link between the 22q11.2 deletion and anterior segment embryogenesis is also supported by a recent report of Peter anomaly in a patient with a 22q11.2 deletion. [Casteels and Devriendt, 2005] Further, posterior embryotoxon has been reported to be the most common ocular finding in patients with the chromosome 22q11.2 deletion syndrome, present in half of patients. [Forbes et al., 2006] Posterior embryotoxon is characterized by an anteriorly displaced and thickened Schwalbe’s ring, which is the anterior boundary of the trabecular meshwork and the posterior boundary of Descemet’s membrane, and is seen at greater frequency in patients with anterior segment dysgenesis anomalies (e.g., Axenfeld anomaly and Rieger syndrome). It is possible that 22q11.2 and the other genetic loci reported in association with sclerocornea play multifactorial and interrelated roles in neural crest migration and differentiation.
Based upon our report of isolated cases from multiple hospitals, it is not possible to accurately report the prevalence of sclerocornea in patients with the 22q11.2 deletion syndrome. However, sclerocornea is likely to be infrequent, as it was not an identified ocular finding in a recent series of 90 patients with the 22q11.2 deletion at the Children’s Hospital of Philadelphia. [Forbes et al., 2006]. Extrapolating upon the number of patients with 22q11.2 deletion syndrome examined by one of the ophthalmologist authors (B. J. F.), we estimate the prevalence of sclerocornea to be approximately 1 in 240 patients.
Treatment of sclerocornea is often complex, and outcomes may be poor. Correction of refractive errors, monitoring and treatment of amblyopia, clearing of central opacification via corneal transplantation, treatment of glaucoma, and management of other associated ocular abnormalities result in lifelong relationships with many different ophthalmic subspecialists. Allografts have a high rate of rejection in the cases of sclerocornea, especially when performed in the pediatric population, and the treatment of glaucoma in these cases is most often surgical due to abnormalities of the anterior chamber angle, which contains the aqueous outflow tracts of the eye. [Gregersen, 1981; Kampik et al., 1986] In six of our seven reported patients, the 22q11.2 genetic diagnostic process was already underway. However, in one patient (Patient 3) the 22q11.2 deletion was not suspected until after the child had already been undergoing treatment for sclerocornea for 2 years, including bilateral corneal transplantation. The delay in diagnosis in this case underscores the importance of recognizing this new syndromic association for sclerocornea and ensuring that pediatric ophthalmologists ask appropriate questions to determine if a systemic evaluation is indicated. Sclerocornea should be added to the clinical manifestations of the 22q11.2 deletion syndrome, and ophthalmologists diagnosing sclerocornea in children with systemic findings suggestive of 22q11.2 deletion should ensure appropriate genetic referral.
Acknowledgments
Heed Ophthalmic Foundation.
Footnotes
Data from this study were presented at the American Association of Pediatric Ophthalmology and Strabismus Annual Meeting in Keystone, Colorado, March 2006 and the Fifth International 22q11.2 Deletion Syndrome Conference, Marseilles, France, July 2006.
References
- Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, Gatzoulis MA. Clinical features of 78 adults with 22q11 deletion syndrome. Am J Med Genet Part A. 2005;138A:307–313. doi: 10.1002/ajmg.a.30984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beemer FA, de Nef JJ, Delleman JW, Bleeker-Wagemakers EM, Shprintzen RJ. Additional eye findings in a girl with the velo-cardio-facial syndrome. Am J Med Genet. 1986;24:541–542. doi: 10.1002/ajmg.1320240319. [DOI] [PubMed] [Google Scholar]
- Bloch N. Les differents types de sclerocornáee, leurs modes d’hereditáe et les malformations congáenitales concomitantes. J Genet Hum. 1965;14:133–172. [PubMed] [Google Scholar]
- Casteels I, Devriendt K. Unilateral Peter anomaly in a patient with DiGeorge syndrome. J Pediatr Ophthalmol Strabismus. 2005;42:311–313. doi: 10.3928/0191-3913-20050901-17. [DOI] [PubMed] [Google Scholar]
- Desvignes P, Pouliquen Y, Legras M, Guyot JD. Aspect iconographique d’une cornea plana dans une maladie de Lobstein. Arch Ophthalmol. 1967;27:585–592. [PubMed] [Google Scholar]
- Devriendt K, Fryns JP, Mortier G. The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet. 1998;35:789–790. doi: 10.1136/jmg.35.9.789-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll DA, Salvin J, Sellinger B, Budarf ML, McDonald-McGinn DM, Zackai EH, Emanuel BS. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: Implications for genetic counselling and prenatal diagnosis. J Med Genet. 1993;30:813–817. doi: 10.1136/jmg.30.10.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliott JH, Ferman SS, O’Day DM, Garber M. Hereditary sclerocornea. Arch Ophthalmol. 1985;103:676–679. doi: 10.1001/archopht.1985.01050050068020. [DOI] [PubMed] [Google Scholar]
- Fitch N. Velo-cardio-facial syndrome and eye abnormality [letter] Am J Med Genet. 1983;15:669. [Google Scholar]
- Forbes BJ, Binenbaum G, Edmund JC, Delarato N, McDonald-McGinn DM, Zackai EH. Ocular findings in the chromosome 22q11.2 deletion syndrome. J AAPOS. 2006 doi: 10.1016/j.jaapos.2006.08.006. (in press) [DOI] [PubMed] [Google Scholar]
- Friedman AH, Weingeist S, Brackup A, Marinoff G. Sclerocornea and defective mesodermal migration. Br J Ophthalmol. 1975;59:638. doi: 10.1136/bjo.59.11.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen E. Keratoplasty in babies with sclerocornea: To do or not to do? Report of an unsuccessful case. Dev Ophthalmol. 1981;5:52. [PubMed] [Google Scholar]
- Happle R, Daniels O, Koopman RJJ. MIDAS syndrome (microphthalmia, dermal aplasia, and sclerocornea): An X-linked phenotype distinct from Goltz syndrome. Am J Med Genet. 1993;47:710–713. doi: 10.1002/ajmg.1320470525. [DOI] [PubMed] [Google Scholar]
- Howard RO, Abrahams IW. Sclerocornea. Am J Ophthalmol. 1971;71:1254. doi: 10.1016/0002-9394(71)90972-x. [DOI] [PubMed] [Google Scholar]
- Kampik A, Lund OE, Halbig W. Penetrating keratoplasty in congenital corneal opacities. Klin Monatsbl Augenheilkd. 1986;188:188–192. doi: 10.1055/s-2008-1050611. [DOI] [PubMed] [Google Scholar]
- LaCassie Y, Arriaza MI. Opitz GBBB syndrome and the 22q11 deletion syndrome [letter] Am J Med Genet. 1996;62:318. doi: 10.1002/(SICI)1096-8628(19960329)62:3<318::AID-AJMG21>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Mansour AM, Goldberg RB, Wang FM, Shprintzen RJ. Ocular findings in the velocardial facial syndrome. J Pediatr Ophthalmol Strabismus. 1987;24:263–266. doi: 10.3928/0191-3913-19870901-16. [DOI] [PubMed] [Google Scholar]
- Matsuoka R, Takao A, Kimura M, Imamura S, Kondo C, Joh-o K, Ikeda K, Nishibatake M, Ando M, Momma K. Confirmation that the conotruncal anomaly face syndrome is associated with a deletion within 22q11.2. Am J Med Genet. 1994;53:285–289. doi: 10.1002/ajmg.1320530314. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Driscoll DA, Bason L, Christensen K, Lynch D, Sullivan K, Canning D, Zavod W, Quinn N, Rome J. Autosomal dominant “Opitz” GBBB syndrome due to a 22q11. 2 deletion. Am J Med Genet. 1995;59:103–113. doi: 10.1002/ajmg.1320590122. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Driscoll DA, Emanuel BS, Christensen K, Lynch D, Sullivan K, Canning D, Zavod W, Quinn N, Rome J. The 22q11 deletion in African-American patients: An underdiagnosed population. Am J Hum Genet. 1996;59:A90. [Google Scholar]
- McDonald-McGinn DM, LaRossa D, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, Moss E, Wang P, Solot C, Schultz P, Lynch D, Bingham P, Keenan G, Weinzimer S, Ming JE, Driscoll D, Clark BJ, 3rd, Markowitz R, Cohen A, Moshang T, Pasquariello P, Randall P, Emanuel BS, Zackai EH. The 22q11.2 Deletion: Screening, diagnostic work up, and outcome of results; report of 181 patients. Genetic Test. 1997;1:99–108. doi: 10.1089/gte.1997.1.99. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Zackai EH, Low D. What’s in a name? The 22q11.2 deletion [letter] Am J Med Genet. 1997;72:247–249. [PubMed] [Google Scholar]
- McDonald-McGinn DM, Kirschner R, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, Moss E, Solot C, Wang P, Jacobs I, Handler S, Knightly C, Heher K, Wilson M, Ming JE, Grace K, Driscoll D, Pasquariello P, Randall P, Larossa D, Emanuel BS, Zackai EH. The Philadelphia story: The 22q11.2 deletion: Report on 250 patients. Genet Couns. 1999;10:11–24. [PubMed] [Google Scholar]
- McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, Finucane B, Driscoll DA, Emanuel BS, Zackai EH. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: Cast a wide FISHing net. Genet Med. 2001;3:23–29. doi: 10.1097/00125817-200101000-00006. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Minugh-Purvis N, Kirschner RE, Jawad A, Tonnesen MK, Catanzaro JR, Goldmuntz E, Driscoll D, Larossa D, Emanuel BS, Zackai EH. The 22q11.2 deletion in African-American patients: An underdiagnosed population? Am J Med Genet Part A. 2005;134A:242–246. doi: 10.1002/ajmg.a.30069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11.2 Deletion Syndrome. [Accessed Feb 2006];2006 www.genetests.org. [PubMed]
- Moriarty AP, Kerr-Muir MG. Sclerocornea and interstitial deletion of the short arm of chromosome 6–(46XY del[6] [p22 p24]) J Pediatr Ophthalmol Strabismus. 1992;29:177–179. doi: 10.3928/0191-3913-19920501-12. [DOI] [PubMed] [Google Scholar]
- Oskarsdottir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: A population-based study in Western Sweden. Arch Dis Child. 2004;89:148–151. doi: 10.1136/adc.2003.026880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, Aurias A, Raymond FL, Clayton-Smith J, Hatchwell E, McKeown C, Beemer FA, Dallapiccola B, Novelli G, Hurst JA, Ignatius J, Green AJ, Winter RM, Brueton L, Brondum-Nielsen K, Scambler PJ. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: A European collaborative study. J Med Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shprintzen RJ, Wang F, Goldberg R, Marion R. The expanded velo-cardio-facial syndrome (VCF): Additional features of the most common clefting syndrome. Am J Hum Genet. 1985;37:A77. [Google Scholar]
- Sullivan KE. The clinical, immunological, and molecular spectrum of chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Curr Opin Allergy Clin Immunol. 2004;4:505–512. doi: 10.1097/00130832-200412000-00006. [DOI] [PubMed] [Google Scholar]
- Tahvanainen E, Forsius H, Karila E, Ranta S, Eerola M, Weissenbach J, Sistonen P, de la Chapelle A. Cornea plana congenita gene assigned to the long arm of chromosome 12 by linkage analysis. Genomics. 1995;26:290–293. doi: 10.1016/0888-7543(95)80213-6. [DOI] [PubMed] [Google Scholar]
- Townsend WM. Congential corneal opacification. In: Kaufman HE, Barron BA, McDonald MB, editors. The Cornea. 2. Boston: Butterworth-Heinmann; 1998. pp. 371–373. [Google Scholar]
- Voronina VA, Kozhemyakina EA, O’Kernick CM, Kahn ND, Wenger SL, Linberg JV, Schneider AS, Mathers PH. Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Hum Mol Genet. 2004;13:315–322. doi: 10.1093/hmg/ddh025. [DOI] [PubMed] [Google Scholar]
- Wilson DI, Burn J, Scambler P, Goodship J. DiGeorge syndrome: Part of CATCH 22. J Med Genet. 1993;30:852–856. doi: 10.1136/jmg.30.10.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulfsberg EA, Leana-Cox J, Neri G. What’s in a name? Chromosome 22q abnormalities and the DiGeorge, velocardiofacial, and conotruncal anomalies face syndromes. Am J Med Genet. 1996;65:317–319. doi: 10.1002/(SICI)1096-8628(19961111)65:4<317::AID-AJMG13>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]