

Abstract
Angiogenesis is indispensable during fracture repair, and vascular endothelial growth factor (VEGF) is critical in this process. CCN1 (CYR61) is an extracellular matrix signaling molecule that has been implicated in neovascularization through its interactions with several endothelial integrin receptors. CCN1 has been shown to be up-regulated during the reparative phase of fracture healing; however, the role of CCN1 therein remains unclear. Here, the regulation of CCN1 expression in osteoblasts and the functional consequences thereof were studied. Stimulation of osteoblasts with VEGF resulted in a dose- and time-dependent up-regulation of CCN1 mRNA and protein. An up-regulation of both cell surface-associated CCN1 as well as extracellular matrix-associated CCN1 in osteoblasts was found. The supernatant of VEGF-prestimulated osteoblasts was chemotactic for endothelial cells, increasing their migration and stimulated capillary-like sprout formation. These effects could be attributed to the presence of CCN1 in the osteoblast supernatant as they were prevented by an antibody against CCN1 or by small interfering RNA-mediated knockdown of osteoblast CCN1. Moreover, the supernatant of VEGF-prestimulated osteoblasts induced angiogenesis in Matrigel plugs in vivo in a CCN1-dependent manner. In addition, blockade of CCN1 prevented bone fracture healing in mice. Taken together, the present work demonstrates a potential paracrine loop consisting of the VEGF-mediated up-regulation of CCN1 in osteoblasts that attracts endothelial cells and promotes angiogenesis. Such a loop could be operative during fracture healing.
Angiogenesis, the development of a microvascular network, is a strictly regulated process that is essential during embryonic development and healing mechanisms (1, 2). During bone growth, vascular structures invade the growth plate regions of the bone, providing a conduit for the entry of chondroclasts and osteoclasts that are involved in resorptive processes, whereas following bone injury, angiogenesis is transiently stimulated to promote repair mechanisms (3–5). Fracture repair is a multistep process involving migration, proliferation, and differentiation of several cell types such as endothelial cells, fibroblasts, chondroblasts, osteoblasts, and osteoclasts. Neovascularization in fracture repair is thought to be dependent on the functional cross-talk between bone-forming osteoblasts and endothelial cells within the bone microenvironment (6). However, although it was recognized very early that osteoblasts and osteoprogenitor cells are often located adjacent to endothelial cells in blood vessels at sites of new bone formation (7–9), it is not yet defined how osteoblasts may regulate angiogenesis-related endothelial cell functions.
Among a variety of proangiogenic growth factors, the vascular endothelial growth factor (VEGF)3 (2) plays a pivotal role in bone remodeling and healing. VEGF is produced by many cells in the bone environment and stimulates the invasion of new blood vessels into the cartilage, thereby playing a crucial role in regulating resorption of cartilage and bone formation (10). In the early stages of bone repair, large amounts of active VEGF are found in the fracture hematoma (11). VEGF is involved in conversion of the soft, cartilaginous callus to a hard, bony callus during fracture repair (12). Furthermore, VEGF has chemotactic effects on osteoblasts and may directly enhance their activity in vitro (13) or may indirectly induce their proliferation and differentiation by stimulating endothelial cells to produce osteoanabolic growth factors (14).
Emerging evidence points to a role of the CCN (CYR61, CTGF, Nov) family, comprising six members in humans, in both angiogenesis and osteogenesis. These proteins are secreted, extracellular matrix (ECM)-associated proteins that regulate cell adhesion, migration, differentiation, and survival (15). CCN1 (CYR61) is a cysteine-rich, heparin-binding protein, associated with both the ECM and the cell surface (16), which acts as an atypical non-Arg-Gly-Asp-containing ligand of integrin receptors of the β1- and αv-family. In particular, CCN1 promotes endothelial cell migration, survival, and tubule formation in an αvβ3-integrin-dependent manner (17). In addition to stimulating angiogenesis (18), CCN1 is implicated in chondrogenesis (19, 20). Interestingly, CCN1 expression is increased in the callus and the newly formed osteoid during the reparative phase of fracture healing (21); however, the role of CCN1 in this newly formed osteoid remains unclear. In particular, whether osteoblast-derived CCN1 may directly affect angiogenic functions of the endothelium has not been addressed yet.
These observations prompted us to investigate how VEGF and CCN1 could act in concert to link osteoblast and endothelial cell functions in the context of bone angiogenesis. The present study demonstrates a potential paracrine loop consisting of the VEGF-mediated up-regulation of CCN1 in osteoblasts that attracts vascular endothelial cells and promotes angiogenesis with potential implications in fracture healing.
EXPERIMENTAL PROCEDURES
Reagents
The following reagents were provided from the following sources. Platelet-derived growth factor (PDGF) AB, VEGF 165, transforming growth factor-β1 (TGF-β) were from R&D Systems (Wiesbaden, Germany); recombinant CCN1 protein was previously described (22) as well as purchased from Abnova (Taipei, Taiwan). Rabbit polyclonal antibody against human CCN1 and control rabbit IgG were from Santa Cruz Biotechnology (Heidelberg, Germany), whereas rabbit anti-CCN2 (connective tissue growth factor, CTGF) was from Abcam (Cambridge, MA), respectively. The polyclonal anti human CCN1 from rabbit was previously described (22). Fibrinogen and fibronectin and antibody to human actin were from Sigma-Aldrich (Munich, Germany); blocking monoclonal antibody LM609 against αv-integrin and blocking monoclonal antibody against β1-integrin (6S6) were from Chemicon (Hofheim, Germany).
Cell Culture
Human umbilical vein endothelial cells (HUVEC) were from Promocell (Heidelberg, Germany) and cultured as described by the supplier. HUVEC were used in low passages (up to the fourth passage). Primary human osteoblastic cells were obtained from femoral bone biopsies of healthy adults (23) and cultivated as described previously (24).
Generation of Osteoblast-conditioned Supernatant
Primary human osteoblastic cells were grown to confluency in 6-well plates, washed one time with PBS, and starved for 12 h in serum free Dulbecco’s modified Eagle’s medium (DMEM) low glucose (Invitrogen, Karlsruhe, Germany). Cells were then washed and incubated for an additional 1 h in DMEM 0.2% (v/v) fetal calf serum, in the presence or absence of 20 ng/ml VEGF, after which the medium was again changed to DMEM 0.2% fetal calf serum. After 6 h, the supernatant was collected and used in cell migration and the in vitro angiogenesis assay. For in vivo angiogenesis, supernatant was concentrated using a 30-kDa cut-off Amicon ultracentrifugal filter device (Millipore, Schwalbach, Germany).
siRNA-mediated Knockdown of CCN1 in Osteoblasts
For transfections, a chemically synthesized duplex siRNA directed against human CCN1 or a non-targeting control siRNA (Dharmacon, Chicago, IL) was engaged. siRNAs were transfected at a concentration of 100 nM using HiPerfect transfection reagent (Qiagen, Valencia, CA), as described (25). 48 h after transfection, cells were stimulated with VEGF, and the osteoblast-conditioned supernatants were collected as described above. CCN1 in the supernatants from non-transfected osteoblasts, from osteoblasts transfected with control siRNA, or from osteoblasts transfected with specific CCN1 siRNA was assessed by Western blot analysis.
Extracellular Matrix Preparation of Osteoblasts
ECM preparation was performed according to our previously described protocol (26). Briefly, primary human osteoblasts were grown to confluency in 48-well plates and washed three times with PBS containing 2% (w/v) bovine serum albumin and 1 mmol/liter CaCl2. Cells were removed with PBS containing 0.5% (w/v) Triton X-100 for 15 min followed by incubation with 0.5% (w/v) Triton-X100 and 0.1 mol/liter NH3 for another 15 min at 22 °C. Finally, wells were washed with PBS and blocked with PBS containing 3% (w/v) bovine serum albumin.
Cell Migration Assay
Migration of endothelial cells was tested using Transwell membranes (8-μm pore size and 6.5-mm diameter) in 24-well plates (Corning Costar), as described previously (27). The membranes were coated with fibrinogen or fibronectin (each at 10 μg/ml) in PBS for 16 h at 4 °C, washed, and air-dried before use. After gentle trypsinization, the cells were resuspended in DMEM containing 0.2% (v/v) fetal calf serum, and 100 μl of cell suspension at a density of 106/ml was added to the top chamber. Stimuli were added to the lower wells. Each treatment was performed in quadruplicates. After incubation of plates at 37 °C for 6 h, the number of migrated cells in the lower compartment was estimated with a cell counter (CASY counter, Schärfe System, Reutlingen, Germany).
In Vitro Angiogenesis Assay
In vitro angiogenesis in fibrin gels was quantitated using endothelial cell spheroids as described previously (28, 29). Briefly, spheroids containing ~750 cells were generated overnight, after which they were added to fibrin gels. Fibrin gels were prepared by mixing 750 μl of a solution containing 1.8 mg/ml fibrinogen in PBS with thrombin (0.65 units/ml). The spheroid-containing gel was allowed to polymerize for 30 min. Then, another 750 μl of serum-free medium with the corresponding test substance or supernatant from osteoblasts was added without or together with competitors, the plates were laid on top of the gel, and the spheroids were incubated for an additional 48 h at 37 °C, 5% CO2, and 100% humidity. Spheroids were evaluated microscopically, and sprout formation was expressed as sprouting spheroids/total spheroids.
Matrigel Plug Assay
After thawing on ice, Matrigel (BD Biosciences) containing 30 IU/ml heparin (Liquemin, Roche Applied Science, Mannheim, Germany), in the absence or presence of VEGF or recombinant CCN1 (100 ng/ml each), and supernatant from prestimulated or non-stimulated osteoblasts, without or with competitors, in a total volume of 400 μl was injected subcutaneously into the laterodorsal abdominal region of 8-week-old male C57Bl/6 mice. The Matrigel samples were recovered 5 days after implantation for photo documentation. To quantitate neovascularization the hemoglobin concentration in Matrigel, homogentisates were measured as described (29, 30).
Cell ELISA and Matrix ELISA
Cell and matrix ELISA were performed for the detection of CCN1 in primary human osteoblasts and in their ECM. Briefly, osteoblasts were grown to confluency on 96-well plates. Thereafter, cells were washed and incubated in the absence or presence of stimuli in 0.2% fetal calf serum-containing medium for different time periods at 37 °C, as indicated in the figure legends. Following incubation, cells were washed twice with PBS, fixed with methanol/acetone (1:1), and blocked with PBS containing 3% bovine serum albumin for 1 h at 22 °C and then incubated with the first antibody (against CCN1, dilution 1:1000) in Tris-buffered saline containing 0.3% bovine serum albumin, 0.05% Tween 20 followed by the addition of appropriate secondary peroxidase-conjugated antibody and the substrate 2,2′-azino-bis(3-ethylbenz-thiazoline-6-sulphonic acid) (ABTS). Total binding was quantitated at 405 nm, and nonspecific binding (binding of the secondary antibody to wells, in which the first antibody was omitted) was used as blank and was subtracted to calculate specific binding.
Reverse Transcription-PCR
Total RNA was extracted from cultured primary human osteoblasts using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and digested with RNase-Free DNase (Qiagen, Hilden, Germany) to remove any contaminating genomic DNA. cDNA synthesis from total RNA was performed by the first strand cDNA synthesis kit for reverse transcription-PCR (Roche Diagnostics GmbH) according to the manufacturer’s protocol. cDNA synthesis from total RNA was performed with avian myeloblastosis virus reverse transcriptase (Roche Diagnostics GmbH). To detect the expression of CCN1 and glyceraldehyde-3-phosphate dehydrogenase in primary human osteoblasts, the cDNA was used as a template for a normal PCR-reaction using primers specific for either CCN1 or glyceraldehyde-3-phosphate dehydrogenase, and it was incubated with Taq DNA polymerase (MBI-Fermentas, St. Leon-Rot, Germany) and the following primers: for CCN1, 5′-gcg ctc tcc acc tgc ccc-3′ and 5′-aac tcc acc tcg gag gca-3′; and for glyceraldehyde-3-phosphate dehydrogenase, 5′-cca ccc atg gca aat tcc atg gca-3′ and 5′-tct aga cgc cag gtc agg tcc acc-3′. All primers were synthesized by MWG-Biotech (Munich, Germany) and were of high purity salt-free quality. The polymerase was activated (3 min at 95 °C), and then up to 30 cycles (1 min at 95 °C, 1 min at 55 °C, 2 min at 72 °C) were performed on an MWG Primus 25 thermocycler. After the last cycle, all samples were incubated for an additional 10 min at 72 °C.
Bone Fracture Healing
Bone fractures were generated as described previously (31). 10-week-old female mice were used in each group. Briefly, on day 0, an anterior knee incision was made under general anesthesia with xylazine (0.2 mg/10 g of body weight, Bayer Leverkusen, Germany) and ketamine (0.5 mg/10 g of body weight, Sankyo, Osaka, Japan). A transverse osteotomy was then performed at the middle of the tibia with a bone saw (Volvere GX, NSK Nakanishi, Japan). Fractured bones were repositioned and stabilized by inserting the inner pin of a 23-gauge spinal needle intramedullary. Bone radiographs were taken by using a volumetric-computer tomography system (Siemens). Mice were treated with intraperitoneal injections of anti-CCN1 or control antibody (50 μg/mouse) at days 1, 2, 3, 5, 7, and 10. The fracture gap was visualized and measured at days 4 and 14 by the use of radiograph postprocessing software (ChiliPacs®, Chili, Heidelberg, Germany).
Statistical Analysis
Data were compared using the Student’s t test and analysis of variance as appropriate; p values of < 0.05 were regarded as significant.
RESULTS
VEGF Increases Expression of CCN1 mRNA and Protein in Osteoblasts
CCN1 protein is present at low or undetectable levels in quiescent cells but is rapidly synthesized upon stimulation (32). Upon synthesis, CCN1 may be detected on the cell surface as well as it may be secreted extracellularly and may associate with ECM (16, 33). We therefore studied the regulation of expression of CCN1 in primary osteoblasts. Stimulation of osteoblasts with VEGF resulted in a significant up-regulation of both CCN1 mRNA and protein (Fig. 1, A and B), whereas PDGF or TGF-β hardly affected CCN1 expression in osteoblasts. Up-regulated CCN1 was both detected in the cell surface (Fig. 1B) as well as associated with ECM preparations of the osteoblasts (Fig. 1C). In addition, the VEGF-mediated up-regulation of CCN1 was dose-dependent and time-dependent (Fig. 2, A and B). Interestingly, during the time course of osteoblast stimulation by VEGF, the pattern of up-regulation of ECM-associated CCN1 paralleled that of surface-associated CCN1.
FIGURE 1. VEGF induces CCN1 up-regulation in osteoblasts.
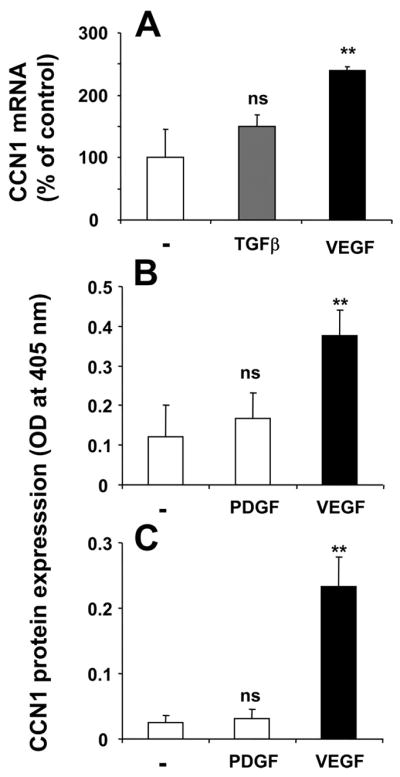
A, influence on CCN1 mRNA expression by TGF-β (20 ng/ml; 2 h; gray bar) and VEGF (20 ng/ml; 2 h; black bar) as indicated. CCN1 mRNA expression is presented as the percentage of control (in the absence of stimuli). B and C, the expression of CCN1 protein on the cell surface (B) and on the extracellular matrix (C) of osteoblasts was studied in the absence or presence of PDGF or VEGF (for 12 h), as indicated. Data are mean ± S.D. ns, not significant; **, p < 0.01.
FIGURE 2. VEGF induces CCN1 expression in a dose- and time-dependent manner.

A, CCN1 protein expression on the cell surface and extracellular matrix of osteoblasts was studied in the absence or presence of increasing concentrations of VEGF (for 12 h). B, time course of the effect of VEGF (20 ng/ml) on CCN1 protein expression on the cell surface and extracellular matrix of osteoblasts. CCN1 protein expression in osteoblasts is presented as the percentage of control (CCN1 expression in the absence of competitor represents the 100% control). Data are mean ± S.D. of a typical experiment; similar results were obtained in three separate experiments.
Osteoblast-derived CCN1 Promotes Endothelial Cell Migration
CCN1 has been reported to stimulate adhesive and migratory endothelial cell functions (17, 34). Consistently, recombinant CCN1 stimulated the migration of HUVEC toward fibronectin or fibrinogen in a β1-integrin- and αv-integrin-dependent manner, respectively, as observed by inhibition studies with specific blocking antibodies against these integrins (data not shown). The promigratory effect of CCN1 was comparable with the effect of VEGF (Fig. 3A). The up-regulation of CCN1 in osteoblasts by VEGF, as observed above, prompted us to investigate whether osteoblasts might exert proangiogenic activities. In particular, we studied whether the conditioned medium of VEGF-stimulated osteoblasts could also induce endothelial cell migration. The supernatant of osteoblasts prestimulated in the presence of VEGF promoted the migration of endothelial cells toward both fibronectin and fibrinogen by 2–3-fold (Fig. 3, B and C). The supernatant of non-stimulated osteoblasts only marginally promoted HUVEC migration (Fig. 3, B and C). Moreover, HUVEC migration induced by the conditioned medium of VEGF-prestimulated osteoblasts was abolished in the presence of an antibody against CCN1 (Fig. 3, B and C), whereas a control antibody or an antibody to another CCN family member, CTGF, were not effective at all.
FIGURE 3. Osteoblast-derived CCN1 promotes endothelial migration.

A, migration of HUVEC toward fibronectin (filled bars) or fibrinogen (open bars) (each 10 μg/ml) is shown in the absence (–) or presence of VEGF (100 ng/ml) or CCN1 (100 ng/ml) in the lower well. Data are mean ± S.E. of three experiments. *, p < 0.05 as compared with control (migration toward fibrinogen in the absence of stimulus); #, p < 0.02 as compared with control (migration toward fibronectin in the absence of stimulus). B and C, migration of HUVEC toward fibrinogen (B) and fibronectin (C) is shown in the absence of any stimulus (open bar) or in the presence of VEGF (100 ng/ml, filled bar), the supernatant from not prestimulated osteoblasts (OSN, gray bars), or the supernatant from VEGF-stimulated osteoblasts (VEGF-OSN, hatched bars) or, without or with anti-CCN1, anti-CTGF, or control antibody (con-IgG) (each 20 μg/ml). Data are mean ± S.E. of three experiments. *, p < 0.05; **, p < 0.01.
To provide further evidence for the specific role of CCN1 as the pro-migratory moiety of the osteoblast supernatant, we employed an alternative way to inhibit CCN1 by engaging siRNA-mediated knockdown. Significant down-regulation of CCN1 by 40–50% was achieved in cells transfected with siRNA-targeting CCN1 but not with control non-targeting siRNA (Fig. 4A). As demonstrated in Fig. 4, B and C, conditioned medium from VEGF-stimulated osteoblasts transfected with siRNA against CCN1 had reduced capacity of promoting endothelial cell migration toward fibrinogen or fibronectin, as compared with conditioned medium from VEGF-stimulated non-transfected osteoblasts or osteoblasts transfected with control non-targeting siRNA. Together, these data indicate that the pro-migratory effect of the osteoblast supernatant could be specifically attributed to the presence of CCN1 therein.
FIGURE 4.
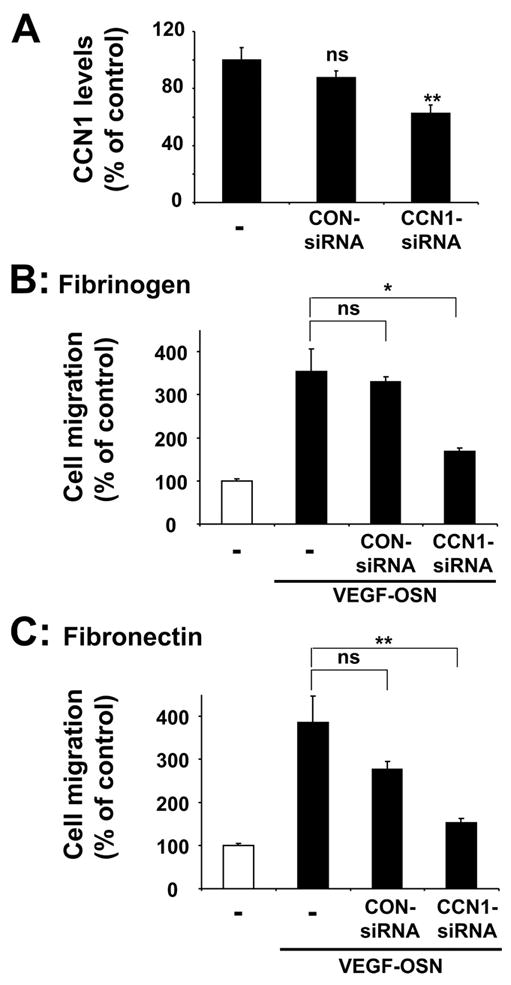
A, the levels of CCN1 in supernatants from VEGF-stimulated osteoblasts are shown. Supernatants of non-transfected (–) osteoblasts, osteoblasts transfected with control non-targeting siRNA (CON-siRNA), or osteoblasts transfected with specific CCN1 siRNA were assessed for their CCN1 levels by performing Western blot analysis. Data are expressed as the percentage of control (the CCN1 levels in supernatants from VEGF-stimulated non-transfected osteoblasts represent the 100% control). Significant down-regulation of CCN1 was observed only in osteoblasts transfected with specific CCN1 siRNA. ns, not significant, **, p < 0.01 as compared with control. B and C, migration of HUVEC toward fibrinogen (B) or fibronectin (C) (each 10 μg/ml) is shown in the absence (open bar) or presence of supernatant from VEGF-stimulated osteoblasts (VEGF-OSN, filled bars) in the lower well. Supernatants of non-transfected (–) osteoblasts, osteoblasts transfected with control non-targeting siRNA, or osteoblasts transfected with specific CCN1 siRNA were used. Data are mean ± S.E. of three experiments. ns, not significant; *, p < 0.05; **, p < 0.01.
Induction of Capillary Sprout Formation in Vitro by Osteoblast-derived CCN1
Above, we demonstrated that supernatant from VEGF-stimulated osteoblasts and especially CCN1 therein may promote migration of endothelial cells in vitro, a process essential for neovascularization. Therefore, we next examined the role of osteoblast-derived supernatant in a more complex in vitro assay that simulates angiogenesis. Capillary-like tube formation in three-dimensional fibrin gels depends on the invasive and migratory potential of endothelial cells. In this system, the supernatant of VEGF-prestimulated osteoblasts increased the number of capillary-like endothelial sprouts by 2-fold, whereas the supernatant of non-stimulated osteoblasts had no effect at all (Fig. 5). Interestingly, an antibody against CCN1 prevented the capillary-like sprout formation induced by the supernatant of VEGF-stimulated osteoblasts, whereas control antibody or antibody to CTGF failed to inhibit sprout formation (Fig. 5). These findings indicate that the VEGF-mediated up-regulation of CCN1 in osteoblasts accounts, at least partially, for the pro-angiogenic activity in the supernatant of osteoblasts.
FIGURE 5. Effect of osteoblast-derived CCN1 on in vitro capillary sprout formation.

HUVEC were incubated for 24 h in the absence (open bar) or presence of supernatant from not stimulated osteoblasts (OSN; gray bars) or from VEGF-prestimulated osteoblasts (VEGF-OSN; hatched bars) without or together with anti-CCN1, anti-CTGF, or control antibody (con-IgG) (each 20 μg/ml). Capillary-like tube formation is expressed relative to control, which is represented as sprout formation in the absence of any stimulus or competitor. Data are mean ± S.D. of three experiments. *, p < 0.05; **, p < 0.01.
Osteoblast-derived CCN1 Stimulates Angiogenesis in Vivo
The pro-angiogenic activity of CCN1 and the supernatant from VEGF-stimulated osteoblasts was then investigated in vivo using the Matrigel plug neovascularization assay. Matrigel was injected subcutaneously into the laterodorsal abdominal region of mice in the absence or presence of recombinant VEGF or recombinant CCN1 or the supernatant from osteoblasts that were prestimulated in the absence or presence of VEGF. The Matrigel matrixes were recovered 5 days later for analysis. The degree of neovascularization was quantified by determining the hemoglobin concentration in the Matrigel matrixes. Recombinant CCN1 induced a strong angiogenic response comparable with VEGF. Furthermore, the supernatant derived from VEGF-prestimulated osteoblasts induced neovascularization in the Matrigel plugs, whereas neovascularization was only weakly induced by the supernatant of non-stimulated osteoblasts (Fig. 6). Interestingly, the supernatant derived from VEGF-stimulated osteoblasts induced an almost maximal angiogenic response in the Matrigel plug assay that was comparable with the effect of recombinant VEGF (Fig. 6). Moreover, the use of an antibody against CCN1 prevented angiogenesis in response to the supernatant from VEGF-stimulated osteoblasts, whereas control antibody or antibody to CTGF were not effective at all (Fig. 6; data with anti-CTGF not shown). The specificity of the inhibitory effect of the antibody to CCN1 was also demonstrated by the fact that anti-CCN1 did not affect angiogenesis directly induced by recombinant VEGF (Fig. 6). These data indicate that the angiogenic effect of the osteoblast supernatant could be attributed to the presence of CCN1 therein. Taken together, these data indicate that VEGF up-regulates CCN1 in osteoblasts, which then promotes angiogenesis in vitro and in vivo.
FIGURE 6. Effect of osteoblast-derived CCN1 on in vivo angiogenesis.

Neo-vascularization in the Matrigel plug assay was studied in the absence (buffer; open bar) or presence of VEGF (100 ng/ml; filled bars), recombinant CCN1 (100 ng/ml; dotted bars), the supernatant from not stimulated osteoblasts (OSN; gray bars), or VEGF-prestimulated osteoblasts (VEGF-OSN; hatched bars) without (–) or together with anti-CCN1 or control antibody (con-IgG) (each 20 μg/ml). The quantitation of neovascularization in the Matrigel matrixes was performed by measuring their hemoglobin concentration. Hemoglobin concentration was expressed as mg of hemoglobin/g of wet tissue. Data are expressed as the percentage of the maximum (VEGF treatment in the absence of competitors). Data are mean ± S.D. of three experiments. ns, not significant; **, p < 0.01.
CCN1 Promotes Bone Fracture Healing in Vivo
The data obtained so far indicated that CCN1 may participate in angiogenesis in the context of bone fracture healing. To provide direct evidence for that, we studied the effect of CCN1 blockade in bone fracture healing model in mice. A transverse osteotomy was performed at the middle of the tibia, and fracture healing was followed by bone radiographs taken with a volumetric-CT system at days 4 and 14 after the fracture (31). By analyzing the size of the fracture gap, we found that fracture healing at day 14 was significantly inhibited by anti-CCN1, whereas no effect of the control antibody was observed (Fig. 7). Together, these data suggest that CCN1 is involved in fracture healing.
FIGURE 7. CCN1 blockade impairs fracture healing.

A, representative vCT images of tibial fractures 14 days after fracture. The arrows indicate the fracture gap. In the upper panel (control antibody (con-IgG) treatment), the fracture is almost completely healed, as indicated by the fact that the fracture gap is almost not measurable. In the lower panel (anti-CCN1 treatment), the fracture gap is still present. B, comparison of the fracture gap (in mm) at day 14 from control antibody treated (open bars) and anti-CCN1 treated (filled bars) mice. Data are mean ± S.D. (n = 3 mice/group). **, p < 0.01.
DISCUSSION
Neovascularization is indispensable for bone fracture repair, during which a coordinated cross-talk between osteoblasts and endothelial cells needs to take place in the local microenvironment (3–6). Here, we have demonstrated that VEGF and CCN1 may synergize in coupling osteoblasts and endothelial cells functionally. In particular, we found that osteoblast CCN1 expression is stimulated by VEGF, and this osteoblast-derived CCN1 may in turn stimulate proangiogenic functions in the endothelial cells, such as migration or capillary-like sprout formation. Additionally, we observed that osteoblast-derived CCN1 promotes angiogenesis in vivo, as well as fracture healing. Thus, our present findings point to a potential paracrine loop that may be important in the communication between osteoblasts and endothelial cells during bone angiogenesis.
Among several growth factors that are up-regulated during skeletal development and induced upon injury, a major role has been attributed to VEGF for bone development and fracture repair (4, 12, 35, 36). Unequivocally, VEGF is crucial in promoting neovascularization; however, VEGF may also be a key regulator in growth plate morphogenesis and cartilage remodeling. In vivo, VEGF blockade resulted not only in the vascular ablation in the growth plate but also in impaired trabecular bone formation (10), whereas a cartilage-specific VEGF deletion was accompanied by a similar phenotype (38). Moreover, VEGF inhibition in mice disrupted repair of femoral fractures and cortical bone defects (12). VEGF also directly regulates the proliferation and migration of osteoblasts that express both VEGF receptors, and systemic inhibition in mice led to a reduction in the osteoblast numbers at the growth plates (4, 39, 40). Over and above these direct effects of VEGF on osteoblasts and endothelial cells, VEGF may also have “indirect” paracrine effects that orchestrate osteogenesis and angiogenesis. The here described paracrine loop mediated by the proangiogenic activities of osteoblast-derived CCN1 that is induced by VEGF may represent such a pathway for the amplification of the proangiogenic and osteoanabolic effects of VEGF.
High CCN1 protein levels have been detected throughout the reparative phase of the callus, particularly in proliferating chondrocytes, osteoblasts, as well as within the newly formed osteoid (21). In fracture callus, VEGF expression is very early and tightly linked to the appearance of osteoblasts (41), and both VEGF and CCN1 have been found to co-localize in the edematous area of osteonecrosis (42). Thus, our present findings suggest that VEGF-stimulated osteoblasts may represent a major source for the increased CCN1, and the functional interplay between VEGF and CCN1 shown here is consistent with these previous histological data. Additionally, although we cannot exclude that another proangiogenic moiety besides CCN1 is induced in osteoblasts by VEGF, CCN1 inhibition almost abolished the proangiogenic activities of the supernatant from VEGF-stimulated osteoblasts. As demonstrated here, CCN1 unequivocally mediates the proangiogenic activities of osteoblasts as well as fracture healing. Although these two functions of CCN1 are likely to be intimately linked with each other, it is still conceivable that the role of CCN1 in fracture healing may be the result of further not yet identified actions of CCN1 separable from its proangiogenic activities.
Interestingly, CCN1-deficient mice displayed embryonic lethality due to placental vascular insufficiency and reduced vessel integrity. This effect was attributed to the reduced expression of VEGF-C in the allantoic mesoderm of CCN1-deficient mice (43). In addition, CCN1 may stimulate the expression of proangiogenic growth factors including VEGF or the basic fibroblast growth factor, as well as metalloproteases in different cells (44). Thus, the regulation of CCN1 by VEGF in osteoblasts, as presented here, may be more complex. The intriguing observation that VEGF up-regulates CCN1, and vice versa, strengthens the hypothesis that a paracrine loop exists between CCN1 and VEGF that may regulate angiogenesis in several (patho-)physiological processes including fracture healing.
The up-regulated osteoblast-derived CCN1 was associated with both the cell surface and the extracellular matrix of the osteoblasts. This extracellular matrix-associated CCN1 in the bone microenvironement may be released by proteases such as plasmin, thereby allowing it to act onto endothelial cells at more distant sites. In fact, the plasmin-mediated cleavage of CCN1 from the matrix of tumor cells was recently demonstrated (22).
The signaling intermediates responsible for the VEGF-induced up-regulation of CCN1 in osteoblasts were not addressed in this study. Recently, CCN1 expression in osteoblasts was demonstrated to be regulated by the Wnt/β-catenin signaling pathway (45). Moreover, in bladder smooth muscle cells, CCN1 overexpression increased the expression of VEGF (37). Whether CCN1 may also induce VEGF expression in osteoblasts, which would implicate the existence of a VEGF-CCN1 autocrine loop in these cells, is worth addressing in a future study. Nevertheless, our present findings provide clear evidence that VEGF and CCN1 cooperate to mediate a crosstalk between osteoblasts and endothelial cells, which may be operative in bone angiogenesis and fracture healing.
Acknowledgments
We thank Y. Mueller and Dr. V. Haxsen for technical assistance, as well as Dr. V. Orlova and Dr. A. Bierhaus for help with siRNA experiments.
Footnotes
The abbreviations used are: VEGF, vascular endothelial growth factor; CTGF, connective tissue growth factor; PDGF, platelet-derived growth factor; TGF, transforming growth factor; ECM, extracellular matrix; siRNA, small interfering RNA; HUVEC, human umbilical vein endothelial cells; PBS, phosphate-buffered saline; ELISA, enzyme-linked immunosorbent assay.
References
- 1.Carmeliet P. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 2.Risau W. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 3.Winet H. Bone. 1996;19:39S–57S. doi: 10.1016/s8756-3282(96)00133-0. [DOI] [PubMed] [Google Scholar]
- 4.Gerber HP, Ferrara N. Trends Cardiovasc Med. 2000;10:223–228. doi: 10.1016/s1050-1738(00)00074-8. [DOI] [PubMed] [Google Scholar]
- 5.Olsen BR, Reginato AM, Wang W. Annu Rev Cell Dev Biol. 2000;16:191–220. doi: 10.1146/annurev.cellbio.16.1.191. [DOI] [PubMed] [Google Scholar]
- 6.Mandracchia VJ, Nelson SC, Barp EA. Clin Podiatr Med Surg. 2001;18:55–77. [PubMed] [Google Scholar]
- 7.Decker B, Bartels H, Decker S. Anat Rec. 1995;242:310–320. doi: 10.1002/ar.1092420304. [DOI] [PubMed] [Google Scholar]
- 8.Trueta J, Little K. J Bone Jt Surg Br. 1960;42:367–376. doi: 10.1302/0301-620X.42B2.367. [DOI] [PubMed] [Google Scholar]
- 9.Trueta J. J Bone Jt Surg. 1963;45B:402–418. [Google Scholar]
- 10.Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. Nat Med. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- 11.Street J, Winter D, Wang JH, Wakai A, McGuiness A, Redmont HP. Clin Orthop Relat Res. 2000;378:224–237. doi: 10.1097/00003086-200009000-00033. [DOI] [PubMed] [Google Scholar]
- 12.Street J, Bao M, deGuzman L, Bunting S, Peale FV, Ferrara N, Steinmetz H, Hoeffel J, Cleland JL, Daugherty A, van Bruggen N, Redmont HP, Carano RAD, Filvaroff EH. Proc Natl Acad Sci U S A. 2002;99:9656–9661. doi: 10.1073/pnas.152324099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayr-Wohlfart U, Waltenberger J, Hausser H, Kessler S, Gunther KP, Dehio C, Puhl W, Brenner RE. Bone. 2002;30:472–477. doi: 10.1016/s8756-3282(01)00690-1. [DOI] [PubMed] [Google Scholar]
- 14.Wang DS, Miura M, Demura H, Sato K. Endocrinology. 1997;138:2953–2962. doi: 10.1210/endo.138.7.5275. [DOI] [PubMed] [Google Scholar]
- 15.Lau LF, Lam SC. Exp Cell Res. 1999;248:44–57. doi: 10.1006/excr.1999.4456. [DOI] [PubMed] [Google Scholar]
- 16.Yang PD, Lau FL. Cell Growth & Differ. 1991;2:351–357. [PubMed] [Google Scholar]
- 17.Leu SJ, Lam SC, Lau LF. J Biol Chem. 2002;277:46248–46255. doi: 10.1074/jbc.M209288200. [DOI] [PubMed] [Google Scholar]
- 18.Babic AM, Kireeva ML, Kolesnikova TV, Lau LF. Proc Natl Acad Sci U S A. 1998;95:6355–6360. doi: 10.1073/pnas.95.11.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Brien TP, Lau LF. Cell Growth & Differ. 1992;3:645–654. [PubMed] [Google Scholar]
- 20.Wong M, Kireeva ML, Kolesnikova TV, Lau LF. Dev Biol. 1997;192:492–508. doi: 10.1006/dbio.1997.8766. [DOI] [PubMed] [Google Scholar]
- 21.Hadjiargyrou M, Ahrens W, Rubin CT. J Bone Miner Res. 2000;15:1014–1023. doi: 10.1359/jbmr.2000.15.6.1014. [DOI] [PubMed] [Google Scholar]
- 22.Pendurthi UR, Tran TT, Post M, Rao LV. Cancer Res. 2005;65:9705–9711. doi: 10.1158/0008-5472.CAN-05-0982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kasperk C, Helmboldt A, Börcsök I, Heuthe S, Cloos O, Niethard F, Ziegler R. Calcif Tissue Int. 1997;61:464–473. doi: 10.1007/s002239900369. [DOI] [PubMed] [Google Scholar]
- 24.Liegibel UM, Sommer U, Tomakidi P, Hilscher U, van den Heuvel L, Pirzer R, Hillmeier J, Nawroth P, Kasperk C. J Ex Med. 2002;196:1387–1392. doi: 10.1084/jem.20021017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orlova VV, Economopoulou M, Lupu F, Santoso S, Chavakis T. J Exp Med. 2006;203:2703–2714. doi: 10.1084/jem.20051730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chavakis T, Kanse SM, Yutzy B, Lijnen HR, Preissner KT. Blood. 1998;91:2305–2312. [PubMed] [Google Scholar]
- 27.Chavakis T, Cines DB, Rhee JS, Liang OD, Schubert U, Hammes HP, Higazi AA, Nawroth PP, Preissner KT, Bdeir K. FASEB J. 2004;18:1306–1308. doi: 10.1096/fj.03-1009fje. [DOI] [PubMed] [Google Scholar]
- 28.Korff T, Augustin HG. J Cell Sci. 1999;112:3249–3258. doi: 10.1242/jcs.112.19.3249. [DOI] [PubMed] [Google Scholar]
- 29.Athanasopoulos AN, Economopoulou M, Orlova VV, Sobke A, Schneider D, Weber H, Augustin HG, Eming SA, Schubert U, Linn T, Nawroth PP, Hussain M, Hammes HP, Herrmann M, Preissner KT, Chavakis T. Blood. 2006;107:2720–2727. doi: 10.1182/blood-2005-08-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barcelos LS, Talvani A, Teixeira AS, Vieira LQ, Cassali GD, Andrade SP, Teixeira MM. J Leukocyte Biol. 2005;78:352–358. doi: 10.1189/jlb.1104682. [DOI] [PubMed] [Google Scholar]
- 31.Shimoaka T, Kamekura S, Chikuda H, Hoshi K, Chung UI, Akune T, Maruyama Z, Komori T, Matsumoto M, Ogawa W, Terauchi Y, Kadowaki T, Nakamura K, Kawaguchi H. J Biol Chem. 2004;279:15314–15322. doi: 10.1074/jbc.M312525200. [DOI] [PubMed] [Google Scholar]
- 32.O’Brien TP, Yang GP, Sanders L, Lau LF. Mol Cell Biol. 1990;10:3569–3577. doi: 10.1128/mcb.10.7.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kireeva ML, Latinkic BV, Kolesnikova TV, Chen CC, Yang GP, Abler AS, Lau LF. Exp Cell Res. 1997;233:63–77. doi: 10.1006/excr.1997.3548. [DOI] [PubMed] [Google Scholar]
- 34.Kireeva ML, Mo FE, Yang GP, Lau LF. Mol Cell Biol. 1996;16:1326–1334. doi: 10.1128/mcb.16.4.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carano RAD, Filvaroff EH. Drug Discov Today. 2003;8:980–989. doi: 10.1016/s1359-6446(03)02866-6. [DOI] [PubMed] [Google Scholar]
- 36.Peng H, Wright V, Usas A, Gearhart B, Shen HC, Cummins J, Huard J. J Clin Investig. 2002;110:751–759. doi: 10.1172/JCI15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou D, Herrick DJ, Rosenbloom J, Chaqour B. J Appl Physiol. 2005;98:2344–2354. doi: 10.1152/japplphysiol.01093.2004. [DOI] [PubMed] [Google Scholar]
- 38.Haigh JJ, Gerber HP, Ferrara N, Wagner EF. Development (Camb) 2000;127:1445–1453. doi: 10.1242/dev.127.7.1445. [DOI] [PubMed] [Google Scholar]
- 39.Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M. Proc Natl Acad Sci U S A. 1998;95:9349–9354. doi: 10.1073/pnas.95.16.9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Midy V, Plouet J. Biochem Biophys Res Commu. 1994;199:380–386. doi: 10.1006/bbrc.1994.1240. [DOI] [PubMed] [Google Scholar]
- 41.Zelzer E, McLean W, Ng YS, Fukai N, Reginato AM, Lovejoy S, D’Amore PA, Olsen BR. Development (Camb) 2002;129:1893–1904. doi: 10.1242/dev.129.8.1893. [DOI] [PubMed] [Google Scholar]
- 42.Radke S, Battmann A, Jatzke S, Eulert J, Jakob F, Schutze N. J Orthop Res. 2006;24:945–952. doi: 10.1002/jor.20097. [DOI] [PubMed] [Google Scholar]
- 43.Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF. Mol Cell Biol. 2002;22:8709–8720. doi: 10.1128/MCB.22.24.8709-8720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brigstock DR. Angiogenesis. 2002;5:153–165. doi: 10.1023/a:1023823803510. [DOI] [PubMed] [Google Scholar]
- 45.Si W, Kang Q, Luu HH, Park JK, Luo Q, Song WX, Jiang W, Luo X, Li X, Yin H, Montag AG, Haydon RC, He TC. Mol Cell Biol. 2006;26:2955–2964. doi: 10.1128/MCB.26.8.2955-2964.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]