

Abstract
After intracerebral hemorrhage (ICH), the brain parenchyma is exposed to blood containing red blood cells (RBCs) and consequently to its lysis products. Iron-rich hemoglobin (Hb) is the most abundant protein in RBCs. When released into the brain parenchyma during hemolysis, Hb becomes a central mediator of cytotoxicity. Our study indicates that haptoglobin (Hp), an acute-phase response protein primarily synthesized in the liver and known to bind and neutralize Hb in the bloodstream, is also expressed in brain in which it plays an important role in defending neurons from damage induced by hemolytic products after ICH. We demonstrate that the Hb-induced hypohaptoglobinemia aggravates ICH-induced brain damage while pharmacologic intervention with sulforaphane to induce brain Hp is linked to a reduction in brain damage. In agreement with these findings, Hp deficiency worsens whereas Hp overexpression alleviates ICH-mediated brain injury. We also identified that oligodendroglia are the primary source of brain-derived Hp among brain cells and that oligodendroglia-released Hp plays protective roles against Hb-mediated toxicity to neurons and oligodendrocytes. We conclude that Hp, particularly the brain-derived Hp, plays cytoprotective roles and represents a potential therapeutic target for ICH treatment.
Introduction
Intracerebral hemorrhage (ICH) is a common and often fatal stroke subtype that burdens ∼1 in 5000 people every year (Woo et al., 2002) and for which there is no treatment. Even if the patient survives the ictus, the resulting hematoma within the brain parenchyma triggers a series of events termed secondary injury, leading to brain edema, progression of neurological deficits, or death.
The red blood cell (RBC), a main cellular component of the hematoma, lyses within days and releases hemoglobin (Hb), which subsequently is degraded into heme and iron, cytotoxins representing a key contributor to secondary brain injury after ICH (Wagner et al., 2003; Wu et al., 2003; Xi et al., 2006). In agreement with this scheme, intracerebral injection of lysed erythrocytes or purified Hb produces massive brain edema within <24 h (Xi et al., 1998; Huang et al., 2002). The cell-free Hb is a potent neurotoxin capable of initiating reactions such as oxidative damage to lipids, DNA, and proteins (Nakamura et al., 2005, 2006; Zhao et al., 2007b), caspase activation (Wang et al., 2002), blood–brain barrier disruption (Xi et al., 1998; Bhasin et al., 2002; Keep et al., 2008), neuronal death (Regan and Guo, 1998; Wang et al., 2002), and irreversible brain damage (Aronowski and Hall, 2005; Xi et al., 2006). Thus, approaches aimed at neutralizing Hb could represent an important clinically relevant target for the treatment of ICH.
Haptoglobin (Hp) is an acute-phase α2-acid response glycoprotein known to tightly bind Hb and form stable Hb–Hp complexes (Wada et al., 1970). By forming Hp–Hb complexes, Hp stabilizes and shields heme iron within the hydrophobic pocket of Hb, thereby preventing its pro-oxidative properties and cytotoxicity (Yang et al., 2003; Zuwala-Jagiello, 2006). Hp is produced principally in liver by hepatocytes (Peters and Alper, 1966; Bowman and Kurosky, 1982; Hoj et al., 1984) and secreted into blood circulation. Although the tissue-specific expression of Hp in some peripheral organs such as the lung, skin, or kidney was demonstrated (Dobrotina and Ezhova, 1977; Dobryszycka, 1997; Lim et al., 1998; Yang et al., 2003), the role of Hp in brain tissue, including in context of ICH-associated blood/hemoglobin toxicity, has not been studied yet (D'Armiento et al., 1997). Limited studies suggest the presence of Hp in neural retina (Chen et al., 1998), astrocytes after ischemia (Lee et al., 2002), and subarachnoid blood clot (Borsody et al., 2006), thus raising the speculation that, under some conditions, in addition to its brain entry through disrupted blood–brain barrier, Hp might be synthesized locally in the brain. As such, locally produced brain Hp may protect the brain from Hb-mediated damage in the case of ICH.
The objective of this study was to establish the role of Hp in defending brain from blood toxicity after ICH. Our hypothesis was that Hp expression in the brain is upregulated in response to ICH to help combat blood toxicity and that manipulation aiming at upregulation of Hp expression will reduce ICH-mediated brain injury.
Materials and Methods
All animal studies followed the guidelines outlined in Guide for the Care and Use of Laboratory Animals from the National Institutes of Health and were approved by the University of Texas–Houston Health Science Center Animal Welfare Committee.
Modified ICH and ICH model in rodents.
We have characterized an ICH model in mouse/rat based on intrastriatal injection of autologous blood (Zhao et al., 2007b, 2009). Here, to elucidate the role of Hp in the detoxification of hemolytic products (e.g., hemoglobin), we used a modified ICH (M-ICH) model in mouse/rat that is based on injection of lysed RBCs into the left striatum. Briefly, whole blood was collected via heart (intraventricular) puncture and instantly lysed using a Sonicator (model W-10) equipped with a metal probe. Randomly selected male and female (equally distributed among groups) mice (weighing 25∼30 g) or male Sprague Dawley (SD) rats (weighing 200∼275 g) under isoflurane anesthesia (delivered by the face mask in a 1:1 mixture of oxygen and nitrous oxide) were immobilized onto a stereotaxic frame. A 1-mm-diameter burr hole was drilled in the skull and a 31 gauge stainless-steel cannula was inserted for the lysed blood (12 μl for mouse and 35 μl for rat) infusion at a speed of 3 μl/min. Whole blood without sonication was used to produce ICH. The core body temperature was maintained at 37.0 ± 0.5°C during surgery and the first 3 h after surgery.
Induction of hypohaptoglobinemia.
To induce hypohaptoglobinemia (HHp), rats were subjected to intraperitoneal injection of purified Hb (Sigma-Aldrich; 50 mg in 1 ml saline), once a day for 3 consecutive days using a previously validated protocol (Panter et al., 1985). Saline was used as the vehicle control.
Administration of sulforaphane to induce haptoglobin expression.
For the in vivo experiments, 5 mg/kg sulforaphane (SF) (LKT Labs; S8044) (dissolved in 10% corn oil) was injected intraperitoneally, at 30 min and 24 h after M-ICH. Corn oil (10% in saline) was used as the vehicle control. For the cell culture experiments, SF was directly applied into the culture medium at 0.1∼2.0 μm, 30 min before induction of injury. DMSO (0.1% in PBS) was used as the vehicle control.
Animal perfusion and tissue collection.
All animals were fatally anesthetized with chloral hydrate (0.5 g/kg, i.p.) and intracardially perfused with ice-cold PBS. For histology or biochemical analyses, the whole brains or the subdissected tissues representing hematoma-affected striatum were immediately frozen in −80°C 2-methylbutane and stored in −80°C freezer before cryosectioning, RNA isolation, or protein extraction.
Neurological deficits score.
The neurological deficits score (NDS) was determined by a battery of behavioral tests, including postural flexing, corner test, foot fault, and forelimb placing, as we described in rat and mouse (Zhao et al., 2007b, 2009).
Brain edema.
The brain edema was measured using the wet-weight/dry-weight method (Tang et al., 2004). Briefly, after a short (∼30 s) intracranial perfusion with PBS, the brains were removed and snap-frozen in −80°C 2-methylbutane. The brain tissue was collected from three coronal brain sections (40 μm thick), representing levels of needle track (blood injection site) and 0.25 mm rostrally and caudally to the needle track. The tissue weight of these sections was determined before and after drying in an 80°C oven overnight using microbalance (model ATI CAHN C-33) capable of measuring weight with 1.0 μg precision. The data were calculated as percentage of water content: (wet weight − dry weight)/wet weight × 100. This nonconventional approach was validated with respect to the tissue size and PBS perfusion.
Western blot.
The protein levels were determined using Western blot as we described previously (Zhao et al., 2007b, 2009). Rabbit anti-mouse/rat Hp antibodies (1:2000; ICL), mouse anti-rat/mouse neurofilament (NF-M) (1:500; Abcam), goat anti-mouse Tau (1:500; Santa Cruz), and chicken anti-mouse myelin basic protein (MBP) (1:1000; Abnov) were used. After incubation with the second antibodies conjugated to HRP (1:5000; Zymed), the immunopositive bands were visualized with ECL (Pierce). Semiquantification of luminescence signal intensity on x-ray film was determined by analyses of optical density using a computer-assisted Kodak Analysis (EDAS) 290 system.
Oxidative stress.
Oxidative stress in the brain after M-ICH was determined by measuring the oxidative damage to proteins through 3′-nitrotyrosine formation (anti-3′-NT; Millipore) and to lipids through 4-hydroxynonenal formation (anti-4-HNE; Calbiochem), using the dot immunoblots as we reported previously (Zhao et al., 2007b). The optical densities were quantified with the Kodak EDAS 290 system and normalized to the naive control.
Hp dot blot.
The relative changes in blood Hp level were determined with dot immunoblot. Briefly, 5 μl of 1:10 diluted sera was spotted on a nitrocellulose membrane and immunolabeled with rabbit anti-rat Hp antibody (1:1000; ICL). After application of secondary antibody, the visualization of the resulting antibody–antigen complex was achieved using ECL, and the OD was quantified by the Kodak EDAS 290 system.
Hp ELISA.
The Hp amount in blood sera and cell culture media was also measured using a rat or human Hp ELISA kit (ICL), according to the instruction provided by the manufacturer.
Primary neuronal cultures.
The primary neuronal cultures from the cortices of embryonic day 18 (E18) fetuses were prepared as we described previously (Zhao et al., 2006a). The cells were plated on poly-l-lysine-coated culture plates in Neurobasal medium with B27 (Invitrogen), and maintained in 5% CO2 incubator for 12–15 d in vitro.
Primary neuron–glial cocultures.
The primary neuron–glial cocultures from E18 prenatal rat/mouse embryos were prepared as we described previously with minor modifications (Zhao et al., 1999). Briefly, the forebrains were dissected under a dissection microscope after removing the pia meninges and olfactory bulbs. The dissected brains were triturated with flame-restricted glass pipettes, and the dissociated cells were suspended in the DMEM with 4.5 mg/ml glucose, DM1 and DM2, 5% bovine calf serum (BCS), 2 mm l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (DMEM-DM-BCS), and seeded on poly-l-lysine-coated culture flasks or German glass (inserted in the culture well) at a cell density of 4 × 105 cells/mm2. The cultures were maintained for 12–15 d until the astroglia become confluent. Using anti-MAP2, anti-glial fibrillary acidic protein (GFAP), OX42, and anti-MBP antibodies to immunolabel neurons, astroglia, microglia, and oligodendrocyte, respectively, we established that our neuron–glial coculture consists of 52% astrocytes, 19% neurons, 12% oligodendroglia, and 7% microglia at 15 d in culture.
Oligodendrocyte-enriched culture.
Oligodendrocytes were purified and cultured from mixed glial cell cultures as described previously (Giulian and Baker, 1986; Negishi et al., 2003). Briefly, the mixed glial cells prepared from postnatal day 1 (P1)∼P2 rat/mouse postnatal pups were plated onto the poly-l-lysine-coated flasks and cultured in the DMEM plus 10% FBS plus penicillin/streptomycin (DMEM-FBS) for 15–20 d. After removing the microglia using the shaking method, the cultures were further subjected to a shaking step at 220 rpm for 16 h. Harvested cells were centrifuged at 350 × g for 5 min and reseeded on poly-d-lysine-coated culture plates and cultured in DMEM plus B27 (DMEM-B27) for another 7–10 d. We changed one-half of the media every 3 d.
ICH-like injury model and RBC lysate toxicity in vitro.
Because there are no standard in vitro cell injury models that could mimic the ICH injury, we exposed the 12- to 15-d-old primary brain cell cultures to freshly prepared RBC lysate with or without 1 h of hypoxia. The RBC isolated using density gradient centrifugation (BD Vacutainer CPT) were lysed in distilled H2O at a density of 1 × 109 RBC/ml, and the RBC lysate was added into the culture medium at a ratio of 1 μl/100 μl medium. For “ICH-like” injury, the cells were additionally subjected to a mild hypoxic injury by exposing them to 1 h of oxygen deprivation, using an approach we described previously (Kim et al., 2004).
Media transfer experiment.
The 15-d-old neurons [prepared from wild-type (WT) mice] (see the methods for primary neuronal culture above) cultured in Neurobasal medium with B27 were evaluated for their susceptibility to injury produced by ICH-like injury (RBC lysates plus hypoxia) (see above). Twenty-day-old oligodendrocyte-enriched cultures (OECs) from Hp-Tg and Hp-KO mice cultured in DMEM-B27 were used as media donor. Both Hp-Tg and Hp-KO oligodendroglia showed similar morphology, and the media for growing these cells showed low and similar content of lactate dehydrogenase (LDH), indicating no difference in the natural viability/senescence. In our hands, Neurobasal and DMEM media containing B27 are compatible with both cultured neurons and oligodendroglia. Thus, the transfer of the media does not cause additional stress to neurons. Fifteen minutes before ICH-like injury, two-thirds of the media from neuronal culture was removed and immediately replaced with an equal volume of media from OEC. LDH level was determined right before the injury and 24 h later. LDH changes at 24 h after injury in the neuronal culture media were calculated as fold increase over the control (right before subjecting to injury).
Excitotoxic damage.
The neuronal cultures were exposed to 500 μm NMDA in the presence of 4.5 μm Hp or 10 μm dizocilpine maleate (MK-801) (Zhao et al., 2006a). The LDH released into the culture media was measured using a LDH assay kit (Promega) at 24 h after the insult.
RNA isolation and reverse transcription-PCR.
The ipsilateral corpus striatum (containing the hematoma and perihematoma areas) was dissected on ice, snap-frozen, and processed for mRNA extraction. Alternatively, the primary brain cell culture was washed with RNase/DNase-free PBS and lysed in Trizol reagents for RNA isolation, and used for reverse transcription-PCR (RT-PCR) analysis as we described previously (Zhao et al., 2006b, 2009). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an internal control. The sequences of primers for rat or mouse Hp and GAPDH are listed in Table 1. Measurements of the gene products were normalized to the optical density of GAPDH bands. The results were calculated as percentage change over the control (naive animal, in vivo) or the media control in vitro.
Table 1.
Primer sequence for RT-PCR
Determination of LDH.
The cell injury was assessed by determining the amount of LDH (Zoldhelyi et al., 1996) released into the culture medium, using an LDH assay kit (Promega) (Zhao et al., 1999, 2006a, 2007a).
Immunofluorescence.
The immunohistochemistry for Hp detection was performed using the procedure we described previously (Zhao et al., 2006a, 2007a). Briefly, the brain coronal cryosection (10 μm) or cells grown on German glass was treated with 95% methanol containing 5% acetic acid for 10 min at −20°C and incubated in chicken (1:100; Novus), rabbit (1:1000; ICL), or mouse (1:500; Sigma-Aldrich) anti-Hp antibody overnight at 4°C. Rabbit anti-chicken, goat anti-rabbit, or goat anti-mouse IgG-Alexa Fluor 488 (Invitrogen) was used to visualize Hp signals.
Double immunohistochemistry was used to localize Hp (detected as above) to a specific cell type by using the cell-specific antibody followed by the second antibody conjugated to Alexa Fluor 546 (Invitrogen). Hoechst 33258 was applied to label the nuclei. The specific brain cell markers included the following: NeuN (Millipore Bioscience Research Reagents) or MAP2 (Sigma-Aldrich) for neurons, mouse or rabbit GFAP (Sigma-Aldrich) for astrocytes, mouse anti-rat or rat anti-mouse CD11b (AbD Serotec) for microglia, and oligodendrocyte-specific protein (OSP) (Abcam) and MBP (Abcam) for oligodendrocytes.
Image capture and cell counting.
A Zeiss Axioskop-2 microscope equipped with a CCD camera and operated by MetaMorph 6.2 software was used for image acquisition. The fluorescence-labeled cells were visualized using Ex/Em of 490/520 for Alexa 488, Ex/Em of 550/575 nm for Alexa 546, and Ex/Em of 365/480 nm for Hoechst 33258. This computer-controlled image acquisition system permits automatic and unbiased cell counting. To determine the cell type composition of the brain neuron–glial cocultures, we took advantage of a motorized stage that allows image stitching, thus permitting cell counting throughout the larger surface area. Three 12-mm-diameter German glass coverslips were used for analysis. Sixteen images from each coverslip were captured under 20× objective for a total area of 2.4 mm2 (each image covers 448 × 335 μm). The ratio of each cell type was calculated as fraction of the total cell number labeled by Hoechst 33258.
Haptoglobin transgenic and haptoglobin knock-out mice.
The haptoglobin transgenic (Hp-Tg) mice (carrying a 9 kb human Hp genomic DNA, which contains the entire Hp2 gene plus 1 kb of the 5′- and 1.5 kb of the 3′-flanking regions) (Yang et al., 2003) and haptoglobin knock-out (Hp-KO) (Lim et al., 1998) mice were provided by Dr. Franklin G. Berger (Department of Biological Sciences, University of South Carolina, Columbia, SC). Hp-Tg and Hp-KO mice were backcrossed into the C57BJ/6 background for >10 generations, and C57BJ/6 mice were used as the wild-type control. The experiments were conducted on randomized mice (8∼16 weeks of age) including both genders (groups were equally balanced for both sexes) of offspring from different parents and different generations. In agreement with the previous studies, both Hp-KO and Hp-Tg mice appeared to be normal with respect to gross cerebrovascular anatomy, behavior, growth, rectal temperature, and prolificacy.
Statistical analysis.
All data are expressed as mean ± SEM unless otherwise stated in the text. All the in vitro experiments were repeated at least three times. The statistical analyses were performed using the GraphPad and InStat programs. The data were evaluated using one-way ANOVAs and Newman–Keuls post test for multiple-group comparison. Nonpaired t test was used when two groups were compared.
Results
HHp aggravates brain injury after ICH
Although it is known that blood plasma Hp can bind and neutralize Hb after intravascular hemolysis, the relevance of Hp this process in the brain after ICH is not clear.
First, we established a modified rodent ICH model (M-ICH) in which whole-blood lysate was injected into the basal ganglia to mimic the environment/events occurring after RBC lysis in the brain hematoma after ICH (Fig. 1 A). M-ICH results in histological damage and visible perihematoma edema (Fig. 1 B).
Figure 1.

HHp aggravates ICH-mediated damage. A, Photograph of representative rat brain coronal section illustrating location of hematoma produced with intrastriatal injection of lysed blood (M-ICH). B, Representative photomicrograph of H&E-stained cryosection showing edema surrounding hematoma (outlined with dotted lines), as captured at 24 h after M-ICH. C, Bar graph illustrating the densitometrical analysis of immunoreactivity (immuno-dot blots technique) for 3′-NT and 4-HNE in the hematoma-affected tissue homogenates of normohaptoglobinemic control (ICH) and HHp (ICH+HHp) rats 24 h after M-ICH. The data are expressed as mean ± SEM (n = 5). *p ≤ 0.05 from the control (ICH). D, Bar graph illustrating brain edema measured by wet-to-dry weight ratio (percentage of water) at 24 h after M-ICH in the control (ICH) and HHp (ICH+HHp) rats. The data are expressed as mean ± SEM (n = 7). *p ≤ 0.05 from the control (ICH).
To test the role of Hp in this M-ICH injury model, we reduced the blood Hp level with daily intraperitoneal injection of purified Hb, using a previously validated protocol (Panter et al., 1985). Repeated injection of Hb caused a 52% reduction in blood Hp (203 ± 24 vs 419 ± 21 mg/dl) compared with the saline control. Subsequently, we subjected these hypohaptoglobinemic and control (normohaptoglobinemic) rats to M-ICH and then recorded the oxidative damage and brain edema as the indices of brain injury. The HHp rats experienced significantly higher level of oxidative brain damage (Fig. 1 B) and worsened brain edema (Fig. 1 C,D), compared with the control at 24 h after M-ICH, suggesting that Hp may play important role in protecting the brain from the toxicity of hemolytic products.
The level of haptoglobin expression is associated with susceptibility to ICH-mediated brain damage
To better define the relationship between the abundance of Hp and ICH pathogenesis, we used genetically engineered mice that are deficient in Hp (Hp-KO) and mice that overexpress the human Hp2 gene (Hp-Tg). Specifically, we subjected the WT, Hp-KO, and Hp-Tg mice to M-ICH injury. At 3 d after M-ICH, we assessed the level of neurological deficits (NDS) and the severity of brain damage in the ICH-affected basal ganglia by measuring loss of neuronal-specific Tau and 160 kDa neurofilament (NF-M) proteins as surrogates of gray matter injury, and loss of oligodendroglia-specific MBP as surrogate of white matter injury.
In agreement with the outcome of the experiment with the Hb-induced HHp rats, Hp-KO mice subjected to M-ICH, based on intrastriatal injection of 12 μl of autologous hemolysates, showed more severe neurological deficits than WT mice (Fig. 2 A). The severity of neurological dysfunction was associated with more extensive loss of NF-M, Tau, and MBP in Hp-KO mice, compared with WT mice (Fig. 2 B,C). In additional support of the beneficial role of Hp in ICH, Hp-Tg mice showed less severe neurological deficits (Fig. 2 A) and reduced loss of NF-M, Tau, and MBP proteins (Fig. 2 B,C), compared with WT and Hp-KO mice. Overall, the Hp expression level is negatively associated with the susceptibility of mice to ICH-mediated brain injury.
Figure 2.

Hp protects brain from injury produced by M-ICH. A, Bar graph illustrating the composite neurological deficit scores (NDS; left panel) and deficit scores for the individual (postural reflex, corner, right forelimb placing, and right foot fault) tests at 7 d after M-ICH in WT, Hp-KO, and Hp-Tg mice. The data are expressed as mean ± SEM (n = 9). *p ≤ 0.05 from all the remaining groups. B, Photograph of immunoblot showing bands of NF-M, Tau, and MBP in hematoma-affected striatum of WT, Hp-KO, and Hp-Tg mice with (+) and without (−) M-ICH injury at 7 d after the insult. C, Bar graph of densitometrical quantification of NF-M, Tau, and MBP on the immunoblots. The data are calculated as percentage over naive animals and expressed as mean ± SEM (n = 5). *p ≤ 0.05 from all the remaining groups.
Haptoglobin is expressed in oligodendrocytes and can be upregulated by sulforaphane
When analyzing ICH-affected mouse brain for expression of Hp mRNA (Fig. 3 A) and protein (Fig. 3 B), we found that Hp mRNA and Hp protein levels in the perihematoma brain tissue demonstrated a consistent increase that was evident as early as 3 h after the onset of ICH (Fig. 3 A). Subsequently, the levels of Hp mRNA and protein increased over time in the first 3 d and remained at a higher level for at least 7 d (Fig. 3 A,B). Similar to the increase of Hp in hematoma-affected brains, ICH triggered a robust increase of Hp in the peripheral blood (Fig. 3 C). These results suggest that not only peripheral organs but also the brain is capable of generating Hp in response to ICH, thus helping to protect the ICH-affected brain from cytotoxic Hb.
Figure 3.
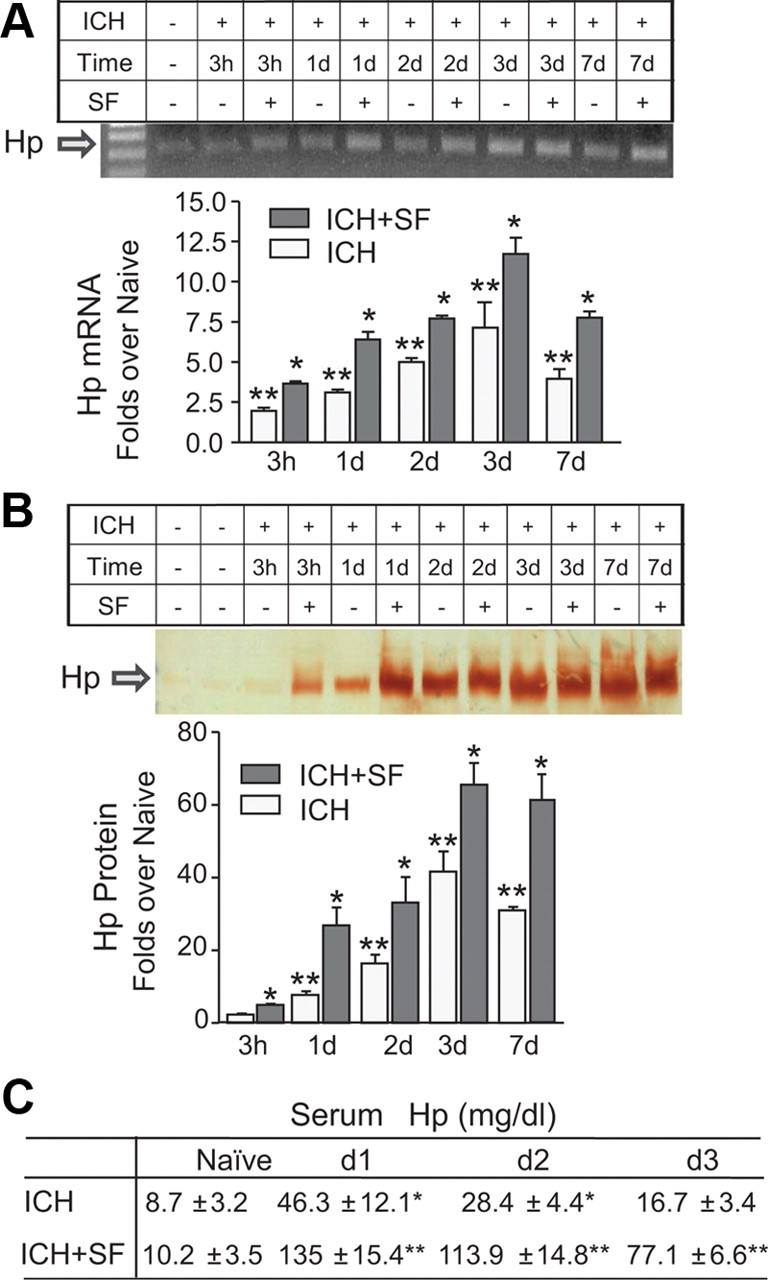
Hp in the hemorrhage-affected striatum is increased after ICH and is augmented by SF. The SD rats were injected intraperitoneally with either saline (ICH) or SF (ICH+SF; 5 mg/kg) at 30 min and 24 h after ICH. A, Photograph of RT-PCR products for Hp on representative agarose gel, and bar graph illustrating the densitometrical quantification of Hp mRNA at 3 h, 1, 2, 3, and 7 d after ICH. The data are calculated as mean ± SEM (n = 4) and expressed as fold increase over the naive groups. *p < 0.05 from naive and the saline control at the same time point; **p < 0.05 from naive. B, Photograph of the representative immunoblot for Hp and bar graph illustrating the densitometrical quantification of Hp protein at 3 h, 1, 2, 3, and 7 d after ICH in rats. The data are calculated as mean ± SEM (n = 4) and expressed as fold increase over the naive groups. *p < 0.05 from naive and ICH control at the same time point; **p < 0.05 from naive. C, Table indicating blood plasma Hp level in naive mice (first column) and in mice with (ICH+SF) and without (ICH) treatment with SF as determined at 1, 2, and 3 d after ICH. The data are expressed as mean ± SEM (n = 5). *p ≤ 0.05 from naive; **p < 0.05 from naive and ICH at the same time point.
The Hp gene is upregulated in response to activation of nuclear factor-erythroid 2-related factor 2 (Nrf2) (Thimmulappa et al., 2002; Kensler et al., 2007), a basic leucine zipper transcription factor that regulates redox balance and stress responses. We have recently demonstrated that the Nrf2 activator, SF, reduces brain injury in a rat model of ICH (Zhao et al., 2007b). To explore whether SF treatment can induce expression of Hp in the brain, we treated animals with SF after ICH and measured Hp expression in the ICH-affected brains. As anticipated, SF administered robustly induced the expression of Hp mRNA (Fig. 3 A) and protein (Fig. 3 B) in the brain within ∼3 h. This induction lasted for at least 7 d. Similar to its effect in the brain, SF also increased peripheral blood Hp (Fig. 3 C, table), thus suggesting that the protective effect of SF in ICH could in part be attributable to induction of Hp.
Our next goal was to identify what cells in the ICH-affected brain produce Hp. Using immunohistochemistry; we first found that virtually no Hp expression is present in naive rat brains. However, Hp immunoreactivity after ICH was very strong and encompassed the whole hematoma territory including adjacent corpus striatum (Fig. 4, left) and corpus callosum (Fig. 4, right). Using double immunohistochemistry to localize Hp to brain cell type, we determined that Hp colocalizes primarily to MBP (Fig. 4)- and OSP (data not included)-immunopositive cells, but not NeuN-, GFAP-, or OX42-positive cells (data not included), suggesting that Hp is expressed primarily by oligodendroglia (Fig. 4) and not by neurons, astroglia, or microglia. This experiment suggests that oligodendroglia represents a main source of Hp in the ICH-affected brain.
Figure 4.
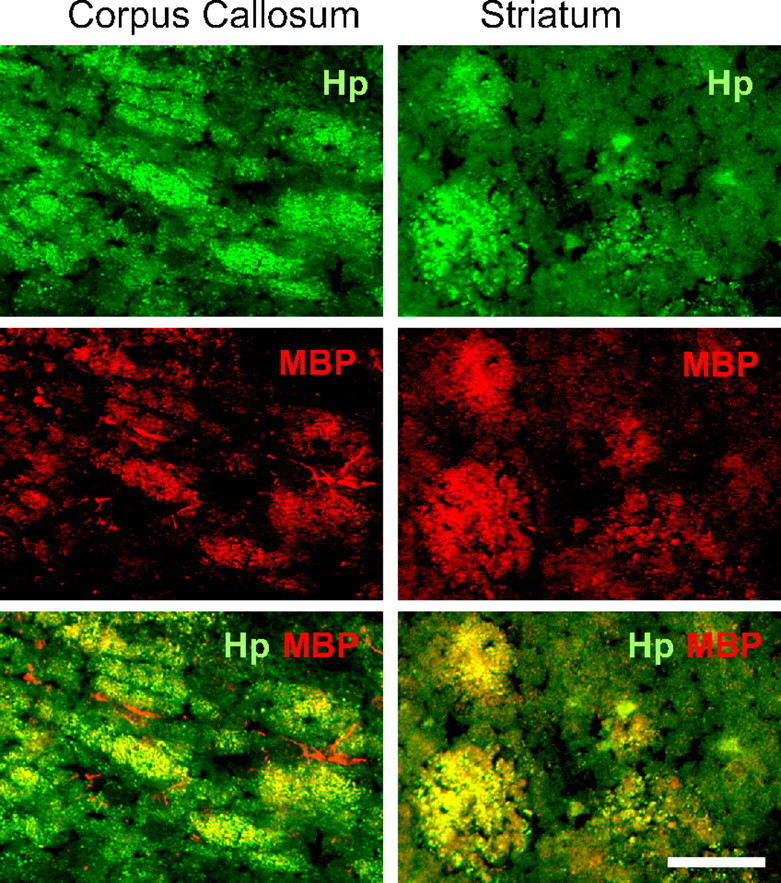
The representative Hp expression pattern in the ICH-affected brain after ICH. The rats at 24 h after M-ICH were analyzed for presence of Hp using immunohistochemistry. Hp-immunopositive cells (green) were detected in corpus callosum and in the basal ganglia around the hematoma. The double-immunofluorescence labeling with anti-MBP (red) and anti-Hp (green) antibodies indicates that Hp colocalizes with oligodendrocytes (MBP-immunopositive cells). Scale bar, 50 μm. ICH, Hematoma.
Hp can neutralize Hb toxicity, but not excitotoxicity in the primary neuronal culture
Hb represents a main component of the lysed erythrocytes that imposes toxicity to the brain after ICH (Sadrzadeh et al., 1987; Koeppen et al., 1995; Xi et al., 1998, 2006), and in particular to neurons (Regan and Panter, 1993, 1996). Thus, Hb neutralization by Hp could play an important role in protecting neurons after ICH.
We first showed that, in our primary neuronal cultures, Hb in a dose-dependent manner induces neuronal damage (Fig. 5 A). Subsequently, to establish the role of Hp in Hb neurotoxicity, we incubated Hb with purified human Hp before adding it to neurons to induce injury. Consistent with the beneficial role of Hp in the animal model of ICH, Hb in presence of Hp demonstrated significantly reduced neurotoxicity when compared with Hb alone, as established by mitigated loss of morphological integrity (Fig. 5 B), reduced degradation of neurofilament protein (Fig. 5 C), and reduced LDH release (Fig. 5 D). These data suggest that Hp in fact can effectively neutralize the cytotoxic effects of Hb.
Figure 5.
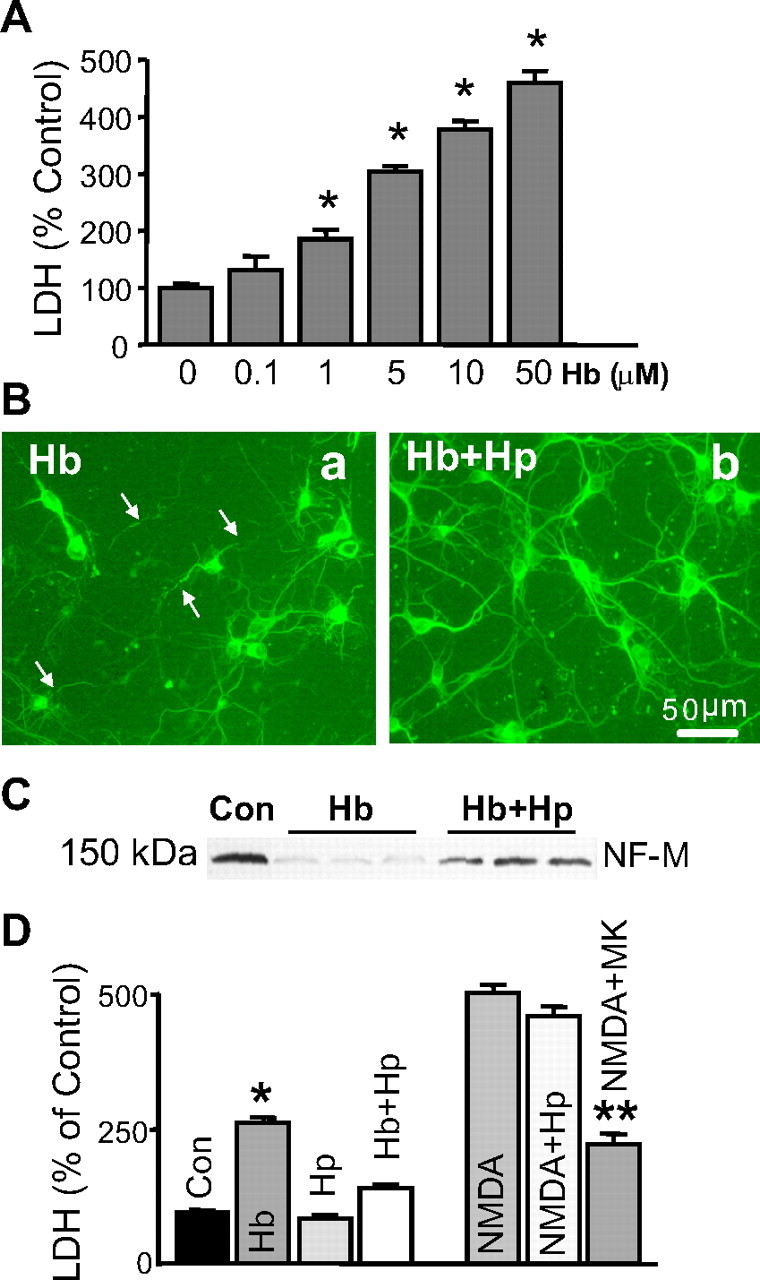
A, Bar graph of LDH released into culture media by neurons in culture exposed to 0.1∼50 μm Hb for 24 h. The data are expressed as percentage over the media control and displayed as mean ± SEM (n = 3). *p ≤ 0.05 from the media control. B, Photomicrograph of MAP2 immunofluorescence in the neuronal cultures after exposure to Hb, demonstrating morphological damage. The 15-d-old neurons in culture were incubated with the following: 3 μm Hb (a) or the complexes of Hp-Hb (b) (4.5 μm Hp and 3.0 μm Hb were coincubated for 15 min before the use) for 24 h, and the morphological integrity was assessed using immunostaining for MAP2. The arrows show the breaks of neurites. C, Photograph of the immunoblot analyzing NF-M in lysates of neurons in culture subjected to Hb alone or as complexes with Hp (Hb+Hp), under experimental conditions described for B. D, Bar graph of LDH released into culture media by neurons in response to: saline (Con), 3 μm Hb, 4.5 μm Hp, Hp plus Hb, 0.5 mm NMDA, 0.5 mm NMDA plus 4.5 μm Hp, or 0.5 mm NMDA plus 10 μm MK-801 (MK). The values of LDH are expressed as percentage change over the control (Con). The data are displayed as mean ± SEM (n = 6). *p ≤ 0.05 from Con, Hp, and Hb-Hp; **p ≤ 0.05 from NMDA and NMDA plus Hp.
Finally, we found that, in contrast to NMDA antagonist, MK-801, Hp is ineffective in protecting neurons from NMDA-mediated excitotoxic damage (Fig. 5 C), thus suggesting that the Hp-mediated neuroprotection is more likely through neutralization of Hb rather than through generalized effect on neuronal resistance to noxious stimuli.
Hp is synthesized and secreted by oligodendrocytes
Animal studies suggested that Hp is generated primarily by oligodendroglia. To further explore the relationship between oligodendroglia and Hp, we first analyzed Hp expression by immunohistochemistry in rat primary neuron–glial coculture. This coculture is normally composed of 52% astrocytes, 19% neurons, 12% oligodendroglia, and 7% microglia. Consistent with the location of Hp in the brains after ICH, Hp in the coculture localized to the oligodendrocytes and was distributed throughout oligodendrocyte soma and fine processes (Fig. 6 A). No Hp immunostaining was detected in cells other than oligodendrocytes.
Figure 6.

Hp is expressed by oligodendroglia and its expression is stimulated by SF. A, Photomicrograph of MBP-immunolabeled oligodendrocyte expressing Hp. The mixed neuronal–glial cocultures were incubated with 1 μm SF for 24 h and then immunolabeled for MBP (a) (green) and Hp (b) (red). c, Merged image of MBP and Hp. The nuclei were visualized with Hoechst 33258 (blue). Note that, among all cells in the field, only MBP-positive cell is immunopositive for Hp. Hp proteins are detected in the soma and fine processes of MBP-positive cells. Scale bar, 100 μm. B, Photograph of RT-PCR product for Hp on representative agarose gel (mRNA, top panel) and Hp immunoblot (protein, bottom panel), illustrating levels of Hp expression in the oligodendroglia-enriched cultures in absence (CON) or presence of SF (1 μm for 24 h). C, Bar graph quantifying Hp protein in the oligodendrocyte-enriched culture media, at 48 h after treatment with 0.1∼2 μm SF, determined by ELISA. *p < 0.05 from vehicle control.
To further characterize Hp in oligodendrocytes, we used an OEC system. By using this system, we showed that oligodendroglia normally express low levels of Hp message and also protein (Fig. 6 B). However, in response to SF, oligodendroglia robustly increased the expression of Hp mRNA (470%) and protein (235%) (Fig. 6 B). Interestingly, SF increased not only intracellular Hp expression but also in a dose-dependent manner augmented Hp in the culture media (Fig. 6 C). The Hp concentration in the culture media increased 3.4-fold compared with the vehicle-treated control at 48 h after exposing oligodendroglia to 1 μm SF, indicating that Hp synthesized in oligodendrocytes can be secreted into the extracellular space.
Conditioned media from Hp-overexpressing, but not Hp-deficient, oligodendroglia renders neuroprotection
Since oligodendroglia can release Hp into the culture media, we sought to determine whether this process could benefit neurons during hemolytic products-mediated injury. To test this idea, we used oligodendroglia-enriched cultures (OEC; Oligo) from Hp-deficient (Hp-KO) and Hp-overexpressing (Hp-Tg) mice. After 12 d in culture, the amount of Hp in the conditioned media of OECs from Hp-Tg mice was ∼490 ng/ml, whereas essentially no Hp was detected in the media from Hp-deficient mice. To examine how these conditioned media affect neuronal vulnerability, we harvested media from the Hp-KO and Hp-Tg OECs and transferred it into a culture dishes containing primary neurons before subjecting them to an “ICH-like” injury. Blank media was used as control. Note that the process of media transfer between the culture dishes did not cause additional cytotoxicity (Fig. 7 A; compare 0 vs 24 h without injury). In agreement with the beneficial role of Hp, neurons in the presence of conditioned media from Hp-Tg OEC showed 30.9% less injury than the neurons containing media from Hp-deficient (Hp-KO) oligodendrocytes (Fig. 7 A).
Figure 7.

A, Bar graph illustrating LDH release by 15-d-old mouse neurons in culture subjected to “ICH-like” injury (lysed RBC plus hypoxia) in the presence of conditioned media from OEC prepared from either Hp-KO or Hp-Tg mice or blank (control) media. The conditioned or blank medium was transferred into the neuronal culture (replacing two-thirds volume of the media used to grow neurons) and incubated for 15 min before induction of ICH-like injury. The cytotoxicity level after media transfer (before injury; 0 h) and at 24 h with and without induction of injury was assessed by the analysis of LDH released into the culture media. The LDH release is expressed as optical density. The data are calculated as mean ± SEM (n = 3). *p ≤ 0.05 from Hp-KO. B, Bar graph of LDH release by Hp-deficient (Hp-KO) and Hp-overexpressing (Hp-Tg) OEC on injury induced by exposure to RBC lysates. The levels of LDH released into culture media during first 16 h after induction of the insult are expressed as fold increase over the control (without RBC lysate). The data are calculated as mean ± SEM (n = 6). *p ≤ 0.05 from Hp-KO. C, Representative photomicrograph of OEC from Hp-KO or Hp-Tg mice that were exposed to RBC lysates for 16 h. Oligodendroglia were visualized by immunostaining for MBP. Note that the oligodendrocytes overexpressing Hp (Hp-Tg) show better-preserved morphology, compared with Hp-deficient (Hp-KO) ones. Scale bar, 50 μm. D, Representative Western blot for MBP as determined in homogenates from OEC exposed to 1 or 3 μm Hb in the presence or absence of 4.5 μm Hp for 24 h. *p ≤ 0.05 from control (Cont).
Finally, to investigate whether Hp could also protect oligodendrocytes (self-protection) from hemolytic products-mediated injury, we added RBC lysates directly into the OECs prepared from Hp-Tg and Hp-KO mice. As anticipated, the Hp-deficient oligodendroglia were more susceptible to injury caused by RBC lysates than the Hp-overexpressing oligodendroglia, as determined using LDH assay (Fig. 7 B) and immunohistochemistry for MBP (Fig. 7 C). Since Hp alone or in combination with modestly toxic concentration of Hb (1 μm) did not directly increase MBP expression (Fig. 7 D, Western blot), we propose that the MBP immunoreactivity loss, as seen with 3 μm Hb, represents an appropriate indicator of oligodendroglia viability. Our data suggest that Hp is protective not only to neurons but also to oligodendrocytes, the cells that produce Hp.
Discussion
Hemoglobin is the most abundant protein of RBCs. And because of the presence of iron within its moiety, Hb may act as a Fenton's reagent generating highly reactive hydroxyl radicals capable of imposing robust oxidative stress (Xi et al., 1998; Wang et al., 2002; Wagner et al., 2003). Thus, after RBC lyses, the cell-free Hb released from the hematoma into brain parenchyma could produce oxidative brain injury and devastating edema (Huang et al., 2002; Wagner et al., 2003; Thiex and Tsirka, 2007; Keep et al., 2008). Hence, approaches aimed at neutralizing Hb could represent a powerful strategy in limiting brain damage caused by ICH.
Haptoglobin is a plasma protein and a key component of Hb detoxification system known for its role in conditions associated with intravascular hemolysis. Hp is primarily synthesized by hepatocytes and secreted into the circulating blood (Peters and Alper, 1966; Bowman and Kurosky, 1982). Hp binds cell-free Hb to form essentially irreversible (K d = 1 pm) Hp–Hb complexes, thereby blocking Hb-induced oxidative toxicity, enabling clearance of cell-free Hb and aiding in recycling of iron (Allison and Rees, 1957; Wang et al., 2001; Tolosano et al., 2002). The clearance of Hb–Hp complexes from the hemolysis-affected environment is effectively mediated by circulating blood monocytes and tissue macrophages expressing the Hb scavenger receptor CD163 (Kristiansen et al., 2001; Schaer et al., 2005; Zuwala-Jagiello, 2006).
Hp reserves in blood plasma are normally relatively high to allow for efficient buffering of Hb in case of minor acute hemolytic events; however, serum levels of Hp can be dramatically increased in response to acute stress, inflammation, and other pathological states (Baumann and Muller-Eberhard, 1987), which improve efficiency of neutralization of free Hb in case of severe hemolytic events. The timely binding of Hp to cell-free Hb plays an important role not only by blocking the pro-oxidative properties of Hb toward cellular constituents, but also toward Hb itself. The latter is particularly important since oxidatively modified Hb loses its binding affinity to Hp and CD163. Normally, when Hb is bound by Hp, the Hb–Hp complex supports structural stabilization of Hb and protects it from structural/oxidative modification. Otherwise, once the free Hb is oxidatively modified, Hp is no longer able to control its anti-oxidative properties and clearance through CD163-mediated endocytosis via macrophages (Buehler et al., 2009).
Although the role of Hp in intravascular hemolysis is well defined, its role in the brain and its expression by brain cells is essentially unknown. A recent study together with our present findings indicate that only very low Hp levels are present in the normal brain, thus contrasting with the high levels of Hp detected in the circulating blood and some peripheral organs. Interestingly, previous experiments suggested that Hp expression could be induced in peripheral organs and not in the brain. For example, intraperitoneal injection of bacterial endotoxin was shown to cause robust increase in Hp expression in peripheral organs and blood serum, but not in the brain (D'Armiento et al., 1997). This selective expression could be explained either by the distinct regulation of Hp expression in brain, or more likely by the fact that the signal triggering Hp expression in the periphery has no access to the normal brain. Although induction of the peripheral Hp may occur without upregulation of the brain Hp, our present study indicates that selective brain damage by intracerebral injection of blood induces not only local (perihematoma) increase in the Hp mRNA and protein of the brain, but it also leads to a robust increase in Hp in peripheral blood. Interestingly, the expression profile of Hp in the brain follows the time course of hemolysis-mediated cytotoxicity, perihematoma edema, and tissue damage after ICH (Wagner et al., 2003; Wagner and Dwyer, 2004), suggesting that Hp may represent an endogenous factor normally involved in neutralization of hemolytic products toxicity in ICH-affected brain.
The role of oligodendroglia in Hp synthesis has not been suggested to date. Using in vivo and in vitro models of ICH, we have demonstrated here that the Hp of the brain is almost exclusively synthesized by oligodendrocytes. Although some previous studies suggested presence of Hp in the astroglia in hippocampus after global ischemia (Lee et al., 2002), blood clot in subarachnoid (Borsody et al., 2006) and in neutrophils (Theilgaard-Mönch et al., 2006), our present study failed to detect Hp in brain cells other than oligodendroglia after ICH. We consequently propose that oligodendrocytes, which are known for their role in shielding and defending axons from excitotoxins and oxidative stress (Dewar et al., 2003), could also protect axons from the damage imposed by hemolytic products via releasing Hb-neutralizing Hp into the periaxonal space. During intracerebral bleeding extravasated blood tends to spread along white matter tracts (Qureshi et al., 2001), in which it may initiate direct toxicity to axons within the nerve tracts and therefore adversely affect neurological status (MacWalter et al., 2001; Qureshi et al., 2001). In agreement with this concept, mice overexpressing Hp showed better preservation of axonal integrity than Hp-null mice when subjected to ICH, as we determined by the reduced damage to axon-specific proteins, neurofilament, and Tau in the hematoma-affected brain. Similarly, a reduced damage to neurofilament was observed in animals subjected to ICH and treated with SF (data not included), the intervention associated with increased Hp expression (see below), further supporting the evidence for an important role of Hp in protecting axons after ICH. Finally, oligodendroglia are also present in the gray matter in which they could play a protective role in context of lobar hemorrhage.
Oligodendrocytes themselves are highly vulnerable to oxidative stress (Juurlink et al., 1998; Dewar et al., 2003; McTigue and Tripathi, 2008) and could easily be damaged by hemolytic products, an event that we propose could be prevented by Hp. In agreement with this notion, the Hp-overexpressing oligodendrocytes in culture showed decreased loss of MBP on exposure to hemolytic products, compared with oligodendroglia from Hp-deficient mice. This suggests that Hp is cytoprotective not only to neurons but also to oligodendrocytes, cells that produce Hp.
We and others have recently shown that Nrf2, a ubiquitous pleiotropic transcription factor and a key genomic homeostatic regulator of intracellular stress (Moi et al., 1994), protects the brain from damage caused by ICH in rodents (Wang et al., 2007; Zhao et al., 2007b). The original interpretation of these results was that Nrf2-mediated mobilization of self-defense enzymes such as catalase, superoxide dysmutase-1, or glutathione transferase protects the brain by preventing oxidative damage caused by ICH. Our present study identifies Hp as an additional factor contributing to the protective effects of the Nrf2. Nrf2 activator, SF, used under conditions that reduced ICH-mediated brain damage (Zhao et al., 2007b), is uniquely effective in mobilizing expression of Hp in the ICH-affected brain and peripheral blood. Similarly, SF applied to primary neuron–glial cocultures under conditions demonstrated to increase Hp expression (Fig. 6) was effective in reducing damage to cultured cells subjected to ICH-like injury (data not included). These findings are overall consistent with the previous studies identifying Hp as target of Nrf2, based on the transcriptional pattern of gene expression, in the small intestines of Nrf2 wild-type and deficient mice (Thimmulappa et al., 2002).
The exact mechanism underlying the beneficial role of Hp in ICH is not entirely clear. Even though neutralization of Hb appears to offer logical explanation for Hp benefit in ICH, Hp has also been proposed to act as extracellular chaperon (Yerbury et al., 2005), inhibitor of cathepsin B activity (Snellman and Sylven, 1967), or ligand for CD11b/CD18 and CD22 (El Ghmati et al., 1996, 2002). As such, it raises the possibility that Hb could protect cells by improving generalized resistance of neurons to Hb-mediated injury. To address this issue, we subjected neurons in culture to excitotoxic injury and tested whether Hp could ameliorate this damage as it does toward Hb toxicity. The results of this experiment showed that Hp is ineffective in blocking NMDA-induced excitotoxicity, suggesting that it is unlikely that Hp protects neurons via a mechanism producing generalized resistance to deleterious stimuli.
In conclusion, our present study showed for the first time that (1) Hp is important for protecting brain from damage caused by ICH, (2) Hp can be synthesized locally in the brain by oligodendroglia and released into the extracellular space in which it can protect brain cells from damage mediated by hemolytic products, and (3) that activation of transcription factor Nrf2 with SF stimulates production of circulating and brain Hp, thus suggesting its therapeutic usefulness for diseases associated with hemolytic product toxicity, including ICH.
Footnotes
This work was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grants R01 NS060768 and R21 NS057284 and American Heart Association Grant 0665200Y. We thank Dr. Franklin G. Berger and Karen Barbour (Department of Biological Sciences, University of South Carolina) for providing Hp-Tg and Hp-KO mice.
References
- Allison AC, Rees WA. The binding of haemoglobin by plasma proteins (haptoglobins); its bearing on the renal threshold for haemoglobin and the aetiology of haemoglobinuria. Br Med J. 1957;2:1137–1143. doi: 10.1136/bmj.2.5054.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronowski J, Hall CE. New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol Res. 2005;27:268–279. doi: 10.1179/016164105X25225. [DOI] [PubMed] [Google Scholar]
- Baumann H, Muller-Eberhard U. Synthesis of hemopexin and cysteine protease inhibitor is coordinately regulated by HSF-II and interferon-beta 2 in rat hepatoma cells. Biochem Biophys Res Commun. 1987;146:1218–1228. doi: 10.1016/0006-291x(87)90778-9. [DOI] [PubMed] [Google Scholar]
- Bhasin RR, Xi G, Hua Y, Keep RF, Hoff JT. Experimental intracerebral hemorrhage: effect of lysed erythrocytes on brain edema and blood-brain barrier permeability. Acta Neurochir Suppl. 2002;81:249–251. doi: 10.1007/978-3-7091-6738-0_65. [DOI] [PubMed] [Google Scholar]
- Borsody M, Burke A, Coplin W, Miller-Lotan R, Levy A. Haptoglobin and the development of cerebral artery vasospasm after subarachnoid hemorrhage. Neurology. 2006;66:634–640. doi: 10.1212/01.wnl.0000200781.62172.1d. [DOI] [PubMed] [Google Scholar]
- Bowman BH, Kurosky A. Haptoglobin: the evolutionary product of duplication, unequal crossing over, and point mutation. Adv Hum Genet. 1982;12:189–261. 453–454. doi: 10.1007/978-1-4615-8315-8_3. [DOI] [PubMed] [Google Scholar]
- Buehler PW, Abraham B, Vallelian F, Linnemayr C, Pereira CP, Cipollo JF, Jia Y, Mikolajczyk M, Boretti FS, Schoedon G, Alayash AI, Schaer DJ. Haptoglobin preserves the CD163 hemoglobin scavenger pathway by shielding hemoglobin from peroxidative modification. Blood. 2009;113:2578–2586. doi: 10.1182/blood-2008-08-174466. [DOI] [PubMed] [Google Scholar]
- Chen W, Lu H, Dutt K, Smith A, Hunt DM, Hunt RC. Expression of the protective proteins hemopexin and haptoglobin by cells of the neural retina. Exp Eye Res. 1998;67:83–93. doi: 10.1006/exer.1998.0494. [DOI] [PubMed] [Google Scholar]
- D'Armiento J, Dalal SS, Chada K. Tissue, temporal and inducible expression pattern of haptoglobin in mice. Gene. 1997;195:19–27. doi: 10.1016/s0378-1119(97)00123-6. [DOI] [PubMed] [Google Scholar]
- Dewar D, Underhill SM, Goldberg MP. Oligodendrocytes and ischemic brain injury. J Cereb Blood Flow Metab. 2003;23:263–274. doi: 10.1097/01.WCB.0000053472.41007.F9. [DOI] [PubMed] [Google Scholar]
- Dobrotina NA, Ezhova GP. Glycoprotein components of the skin under normal conditions and in several types of pathology (in Russian) Vopr Med Khim. 1977;23:215–219. [PubMed] [Google Scholar]
- Dobryszycka W. Biological functions of haptoglobin—new pieces to an old puzzle. Eur J Clin Chem Clin Biochem. 1997;35:647–654. [PubMed] [Google Scholar]
- El Ghmati SM, Van Hoeyveld EM, Van Strijp JG, Ceuppens JL, Stevens EA. Identification of haptoglobin as an alternative ligand for CD11b/CD18. J Immunol. 1996;156:2542–2552. [PubMed] [Google Scholar]
- El-Ghmati SM, Arredouani M, Van Hoeyveld EM, Ceuppens JL, Stevens EA. Haptoglobin interacts with the human mast cell line HMC-1 and inhibits its spontaneous proliferation. Scand J Immunol. 2002;55:352–358. doi: 10.1046/j.1365-3083.2002.01067.x. [DOI] [PubMed] [Google Scholar]
- Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoj L, Binder V, Espersen F, Greibe J, Rasmussen SN, Rask-Madsen J. Secretion rates of immunoglobulins, albumin, haptoglobin and complement factors C3 and C4 in the perfused jejunum and ileum of human Salmonella carriers. Acta Pathol Microbiol Immunol Scand C. 1984;92:129–132. doi: 10.1111/j.1699-0463.1984.tb00063.x. [DOI] [PubMed] [Google Scholar]
- Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- Juurlink BH, Thorburne SK, Hertz L. Peroxide-scavenging deficit underlies oligodendrocyte susceptibility to oxidative stress. Glia. 1998;22:371–378. doi: 10.1002/(sici)1098-1136(199804)22:4<371::aid-glia6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Keep RF, Xiang J, Ennis SR, Andjelkovic A, Hua Y, Xi G, Hoff JT. Blood-brain barrier function in intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:73–77. doi: 10.1007/978-3-211-09469-3_15. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Kim DH, Zhao X, Tu CH, Casaccia-Bonnefil P, Chao MV. Prevention of apoptotic but not necrotic cell death following neuronal injury by neurotrophins signaling through the tyrosine kinase receptor. J Neurosurg. 2004;100:79–87. doi: 10.3171/jns.2004.100.1.0079. [DOI] [PubMed] [Google Scholar]
- Koeppen AH, Dickson AC, McEvoy JA. The cellular reactions to experimental intracerebral hemorrhage. J Neurol Sci. 1995;134(Suppl):102–112. doi: 10.1016/0022-510x(95)00215-n. [DOI] [PubMed] [Google Scholar]
- Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, Moestrup SK. Identification of the haemoglobin scavenger receptor. Nature. 2001;409:198–201. doi: 10.1038/35051594. [DOI] [PubMed] [Google Scholar]
- Lee MY, Kim SY, Choi JS, Lee IH, Choi YS, Jin JY, Park SJ, Sung KW, Chun MH, Kim IS. Upregulation of haptoglobin in reactive astrocytes after transient forebrain ischemia in rats. J Cereb Blood Flow Metab. 2002;22:1176–1180. doi: 10.1097/01.wcb.0000037989.07114.d1. [DOI] [PubMed] [Google Scholar]
- Lim SK, Kim H, Lim SK, bin Ali A, Lim YK, Wang Y, Chong SM, Costantini F, Baumman H. Increased susceptibility in Hp knockout mice during acute hemolysis. Blood. 1998;92:1870–1877. [PubMed] [Google Scholar]
- MacWalter RS, Ersoy Y, Wolfson DR. Cerebral haemorrhage. Parenchymal intracranial haemorrhage. Gerontology. 2001;47:119–130. doi: 10.1159/000052785. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Tripathi RB. The life, death, and replacement of oligodendrocytes in the adult CNS. J Neurochem. 2008;107:1–19. doi: 10.1111/j.1471-4159.2008.05570.x. [DOI] [PubMed] [Google Scholar]
- Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Keep RF, Hua Y, Hoff JT, Xi G. Oxidative DNA injury after experimental intracerebral hemorrhage. Brain Res. 2005;1039:30–36. doi: 10.1016/j.brainres.2005.01.036. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Keep RF, Hua Y, Nagao S, Hoff JT, Xi G. Iron-induced oxidative brain injury after experimental intracerebral hemorrhage. Acta Neurochir Suppl. 2006;96:194–198. doi: 10.1007/3-211-30714-1_42. [DOI] [PubMed] [Google Scholar]
- Negishi T, Ishii Y, Kyuwa S, Kuroda Y, Yoshikawa Y. Primary culture of cortical neurons, type-1 astrocytes, and microglial cells from cynomolgus monkey (Macaca fascicularis) fetuses. J Neurosci Methods. 2003;131:133–140. doi: 10.1016/j.jneumeth.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Panter SS, Sadrzadeh SM, Hallaway PE, Haines JL, Anderson VE, Eaton JW. Hypohaptoglobinemia associated with familial epilepsy. J Exp Med. 1985;161:748–754. doi: 10.1084/jem.161.4.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, Alper CA. Haptoglobin synthesis. II. Cellular localization studies. J Clin Invest. 1966;45:314–320. doi: 10.1172/JCI105345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi AI, Tuhrim S, Broderick JP, Batjer HH, Hondo H, Hanley DF. Spontaneous intracerebral hemorrhage. N Engl J Med. 2001;344:1450–1460. doi: 10.1056/NEJM200105103441907. [DOI] [PubMed] [Google Scholar]
- Regan RF, Guo Y. Toxic effect of hemoglobin on spinal cord neurons in culture. J Neurotrauma. 1998;15:645–653. doi: 10.1089/neu.1998.15.645. [DOI] [PubMed] [Google Scholar]
- Regan RF, Panter SS. Neurotoxicity of hemoglobin in cortical cell culture. Neurosci Lett. 1993;153:219–222. doi: 10.1016/0304-3940(93)90326-g. [DOI] [PubMed] [Google Scholar]
- Regan RF, Panter SS. Hemoglobin potentiates excitotoxic injury in cortical cell culture. J Neurotrauma. 1996;13:223–231. doi: 10.1089/neu.1996.13.223. [DOI] [PubMed] [Google Scholar]
- Sadrzadeh SM, Anderson DK, Panter SS, Hallaway PE, Eaton JW. Hemoglobin potentiates central nervous system damage. J Clin Invest. 1987;79:662–664. doi: 10.1172/JCI112865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaer DJ, Schleiffenbaum B, Kurrer M, Imhof A, Bächli E, Fehr J, Moller HJ, Moestrup SK, Schaffner A. Soluble hemoglobin-haptoglobin scavenger receptor CD163 as a lineage-specific marker in the reactive hemophagocytic syndrome. Eur J Haematol. 2005;74:6–10. doi: 10.1111/j.1600-0609.2004.00318.x. [DOI] [PubMed] [Google Scholar]
- Snellman O, Sylven B. Haptoglobin acting as a natural inhibitor of cathepsin B activity. Nature. 1967;216:1033. doi: 10.1038/2161033a0. [DOI] [PubMed] [Google Scholar]
- Tang J, Liu J, Zhou C, Alexander JS, Nanda A, Granger DN, Zhang JH. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004;24:1133–1145. doi: 10.1097/01.WCB.0000135593.05952.DE. [DOI] [PubMed] [Google Scholar]
- Theilgaard-Mönch K, Jacobsen LC, Nielsen MJ, Rasmussen T, Udby L, Gharib M, Arkwright PD, Gombart AF, Calafat J, Moestrup SK, Porse BT, Borregaard N. Haptoglobin is synthesized during granulocyte differentiation, stored in specific granules, and released by neutrophils in response to activation. Blood. 2006;108:353–361. doi: 10.1182/blood-2005-09-3890. [DOI] [PubMed] [Google Scholar]
- Thiex R, Tsirka SE. Brain edema after intracerebral hemorrhage: mechanisms, treatment options, management strategies, and operative indications. Neurosurg Focus. 2007;22:E6. doi: 10.3171/foc.2007.22.5.7. [DOI] [PubMed] [Google Scholar]
- Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- Tolosano E, Fagoonee S, Hirsch E, Berger FG, Baumann H, Silengo L, Altruda F. Enhanced splenomegaly and severe liver inflammation in haptoglobin/hemopexin double-null mice after acute hemolysis. Blood. 2002;100:4201–4208. doi: 10.1182/blood-2002-04-1270. [DOI] [PubMed] [Google Scholar]
- Wada T, Oara H, Watanabe K, Kinoshita H, Yachi A. Autoradiographic study on the site of uptake of the haptoglobin-hemoglobin complex. J Reticuloendothel Soc. 1970;8:185–193. [PubMed] [Google Scholar]
- Wagner KR, Dwyer BE. Hematoma removal, heme, and heme oxygenase following hemorrhagic stroke. Ann N Y Acad Sci. 2004;1012:237–251. doi: 10.1196/annals.1306.020. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23:629–652. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Doré S. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. 2007;43:408–414. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Mori T, Sumii T, Lo EH. Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke. 2002;33:1882–1888. doi: 10.1161/01.str.0000020121.41527.5d. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kinzie E, Berger FG, Lim SK, Baumann H. Haptoglobin, an inflammation-inducible plasma protein. Redox Rep. 2001;6:379–385. doi: 10.1179/135100001101536580. [DOI] [PubMed] [Google Scholar]
- Woo D, Sauerbeck LR, Kissela BM, Khoury JC, Szaflarski JP, Gebel J, Shukla R, Pancioli AM, Jauch EC, Menon AG, Deka R, Carrozzella JA, Moomaw CJ, Fontaine RN, Broderick JP. Genetic and environmental risk factors for intracerebral hemorrhage: preliminary results of a population-based study. Stroke. 2002;33:1190–1195. doi: 10.1161/01.str.0000014774.88027.22. [DOI] [PubMed] [Google Scholar]
- Wu J, Hua Y, Keep RF, Nakamura T, Hoff JT, Xi G. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003;34:2964–2969. doi: 10.1161/01.STR.0000103140.52838.45. [DOI] [PubMed] [Google Scholar]
- Xi G, Keep RF, Hoff JT. Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J Neurosurg. 1998;89:991–996. doi: 10.3171/jns.1998.89.6.0991. [DOI] [PubMed] [Google Scholar]
- Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- Yang F, Haile DJ, Berger FG, Herbert DC, Van Beveren E, Ghio AJ. Haptoglobin reduces lung injury associated with exposure to blood. Am J Physiol Lung Cell Mol Physiol. 2003;284:L402–L409. doi: 10.1152/ajplung.00115.2002. [DOI] [PubMed] [Google Scholar]
- Yerbury JJ, Rybchyn MS, Easterbrook-Smith SB, Henriques C, Wilson MR. The acute phase protein haptoglobin is a mammalian extracellular chaperone with an action similar to clusterin. Biochemistry. 2005;44:10914–10925. doi: 10.1021/bi050764x. [DOI] [PubMed] [Google Scholar]
- Zhao X, Pike BR, Newcomb JK, Wang KK, Posmantur RM, Hayes RL. Maitotoxin induces calpain but not caspase-3 activation and necrotic cell death in primary septo-hippocampal cultures. Neurochem Res. 1999;24:371–382. doi: 10.1023/a:1020933616351. [DOI] [PubMed] [Google Scholar]
- Zhao X, Ou Z, Grotta JC, Waxham N, Aronowski J. Peroxisome-proliferator-activated receptor-gamma (PPARgamma) activation protects neurons from NMDA excitotoxicity. Brain Res. 2006a;1073–1074:460–469. doi: 10.1016/j.brainres.2005.12.061. [DOI] [PubMed] [Google Scholar]
- Zhao X, Zhang Y, Strong R, Grotta JC, Aronowski J. 15d-Prostaglandin J(2) activates peroxisome proliferator-activated receptor-gamma, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J Cereb Blood Flow Metab. 2006b;26:811–820. doi: 10.1038/sj.jcbfm.9600233. [DOI] [PubMed] [Google Scholar]
- Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, Grotta JC, Aronowski J. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol. 2007a;61:352–362. doi: 10.1002/ana.21097. [DOI] [PubMed] [Google Scholar]
- Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, Grotta JC, Aronowski J. Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke. 2007b;38:3280–3286. doi: 10.1161/STROKEAHA.107.486506. [DOI] [PubMed] [Google Scholar]
- Zhao X, Grotta J, Gonzales N, Aronowski J. Hematoma resolution as a therapeutic target: the role of microglia/macrophages. Stroke. 2009;40:S92–S94. doi: 10.1161/STROKEAHA.108.533158. [DOI] [PubMed] [Google Scholar]
- Zoldhelyi P, McNatt J, Xu XM, Loose-Mitchell D, Meidell RS, Clubb FJ, Jr, Buja LM, Willerson JT, Wu KK. Prevention of arterial thrombosis by adenovirus-mediated transfer of cyclooxygenase gene. Circulation. 1996;93:10–17. doi: 10.1161/01.cir.93.1.10. [DOI] [PubMed] [Google Scholar]
- Zuwala-Jagiello J. Haemoglobin scavenger receptor: function in relation to disease. Acta Biochim Pol. 2006;53:257–268. [PubMed] [Google Scholar]