

Abstract
Alcoholic patients have an increased risk of respiratory infections, which is partially due to an impaired immune response of alveolar macrophages. The mechanisms by which alcohol impairs alveolar macrophage function are poorly understood. In this study, we demonstrated in a guinea pig model that chronic ethanol ingestion significantly impaired alveolar macrophage differentiation and function. Isolated alveolar macrophages were separated into 4 different subpopulations with varying densities and levels of maturation. Compared to control values, chronic ethanol ingestion decreased the percentage of alveolar macrophages in the mature fractions by ~60%. Alveolar macrophage function in each subpopulation was determined by measuring phagocytosis of FITC-labeled S. aureus. Alveolar macrophages from ethanol-fed animals had ~80% decrease in the phagocytic index. Western blot and immunohistochemical analysis of the differential markers GM-CSF receptor α (GM-CSFR-α), PU.1, CD11c, and CD11b verified that alcoholic macrophages displayed impaired terminal differentiation. While oral supplementation with the glutathione precursor S-adenosyl-methionine (SAM) did not alter the maturational status of control animals, SAM s supplementation shifted the distribution of macrophages to more mature fractions, normalized the phagocytic index; as well as normalized expression of CD11c, CD11b, PU.1, and GM-CSFR-α. Chronic ethanol ingestion also impaired the differentiation status of interstitial macrophages which was normalized by SAM supplementation. This improvement in the maturational status suggested that ethanol-induced oxidant stress is a central feature in impaired terminal differentiation of macrophages in the interstitial and alveolar space. Therefore, strategies targeting pulmonary oxidant stress may restore macrophage differentiation and function even after chronic ethanol ingestion.
Keywords: lung, acute respiratory distress syndrome, oxidative stress, glutathione
Introduction
Patients with a known history of alcohol abuse have a 43% chance of developing acute respiratory distress syndrome (ARDS) (Joshi and Guidot, 2007)., an acute, severe injury to the lungs associated with a variety of risk factors (Ware and Matthay, 2000). ARDS is characterized by lung inflammation that leads to an impairment of gas exchange and the release of pro-inflammatory cytokines (Aytacoglu et al., 2006). The incidence and severity of community-acquired pneumonia, the most common cause of ARDS in alcoholic patients, is associated with a 30% increase in the in-hospital mortality rate (Jong et al., 1995, Esper A., 2006). This increased increased risk of bacterial pneumonia is partially due to impaired alveolar macrophage function (Rosseau et al., 2000).
Peripheral blood monocytes pass through the pulmonary circulation and become sequestered within pulmonary capillaries and then migrate into interstitial and alveolar spaces where they mature into pulmonary macrophages (Shibata et al., 2001, Akagawa et al., 1988, Lloberas et al., 1999). Monocytes are rich in adhesion molecules, such as members of the CD11 family (CD11a, CD11b, and CD11c), but possess diminished phagocytotic ability. Since monocytes must migrate through the pulmonary interstitium en route to the alveolus, the interstitial macrophage is thought to represent an intermediate stage of differentiation between monocytes and alveolar macrophages (Laskin et al., 2001). As macrophages mature in the alveolar space, they increase in size and decrease in density. Compared to alveolar macrophages, interstitial macrophages are much smaller in size, have blunt pseudopodia, and closely resemble monocytes. Mature alveolar macrophages have minimal levels of such adhesion molecules but have significantly greater phagocytic capacity (Lundahl et al., 1996) and play a vital role in innate and acquired immunity (Trapnell and Whitsett, 2002).
Granulocyte/macrophage colony-stimulating factor (GM-CSF), a glycoprotein responsible for controlling the terminal differentiation of peripheral blood monocyte into terminally differentiated alveolar macrophages, promotes the proliferation and differentiation of myeloid progenitors (Burgess and Metcalf, 1980, Baldwin, 1992),(DeKoter et al., 1998, Berclaz et al., 2007, Shibata et al., 2001). In the GM-CSF knockout mouse, alveolar macrophages displayed impaired surfactant expression, clearance, phagocytosis, as well as altered receptor expression (Berclaz et al., 2002, Trapnell and Whitsett, 2002). PU.1 is a regulatory transcription factor required for normal hemopoietic cell development, proliferation and differentiation of myeloid progenitors, and the generation of both the innate and the adaptive immune system (Scott et al., 1994, Lloberas et al., 1999). Previous studies have shown GM-CSF regulates alveolar macrophage differentiation through PU.1. In a rat model, chronic ethanol ingestion in decreased expression of PU.1 and the GM-CSF receptor (GM-CSFR), suggesting decreased terminal differentiation (Joshi et al., 2005).
Glutathione (GSH), the most abundant thiol in living organisms, is essential for protein synthesis, DNA synthesis and detoxification of oxygen radicals. In the lung, GSH is concentrated in the epithelia lining fluid through transport by alveolar type II cells (Brown et al., 2007). In clinical and animal studies, chronic ethanol ingestion depletes GSH within the alveolar space resulting in chronic oxidative stress and a loss of alveolar macrophage functions (Brown et al., 2004, Yeh et al., 2007, F. Holguin 1998, Brown et al., 2007). In animal studies, chronic ethanol-induced depletion of GSH in the lung impairs alveolar type II cell function (Guidot et al., 2000), impairs clearance of microbes and decreases alveolar macrophage cell viability (Velasquez A, 2002, Aytacoglu et al., 2006).
Previous studies by this laboratory and others demonstrated chronic ethanol ingestion impairs alveolar macrophage binding and internalization of inactivated Staphylococcus aureus (S. aureus) (Brown et al., 2004, Standiford TJ, 1997). Moreover, in vivo treatment with GSH precursors improved alveolar macrophage phagocytosis during chronic ethanol ingestion (Brown et al., 2007). These results suggested the decreased functions of alveolar macrophages were due to decreased GSH availability and subsequent oxidant stress in the alveolar space. However, the precise mechanisms by which ethanol impairs alveolar macrophage function remain poorly understood. Therefore, we hypothesized that chronic ethanol ingestion alters macrophage terminal differentiation and function and that these alterations were secondary to ethanol-induced decreases in antioxidant availability. To address this hypothesis, we examined the role of chronic ethanol consumption on the maturation and terminal differentiation of peripheral blood monocytes into pulmonary macrophages and their phagocytic capacity. Previous studies in a guinea pig model demonstrated that alveolar macrophages can be separated into subpopulations with distinct densities, as well as morphologic and functional properties (Chandler et al., 1986, Dauber JH, 1983, Laskin et al., 2001). In these experiments, we capitalized on this change in density to separate the alveolar macrophages by a percoll density gradient into four fractions with a range in terminal maturation. Assessment of these different fractions suggested that chronic ethanol ingestion impaired terminal differentiation of macrophages and that these effects were secondary to ethanol-induced oxidant stress.
Material and Methods
Guinea pig model of chronic ethanol ingestion
Since the adult guinea pig has a well-defined subpopulation of resident alveolar macrophage that correlate directly with their function (Dauber JH, 1983), we chose to examine the effects of chronic ethanol exposure on the maturation of the alveolar macrophage in this model. Pathogen-free 6–8 week old female Hartley guinea pigs weighing 400–500 grams were shipped from the vendor (Charles River). Three days after delivery to the Emory Animal Facility, animals were randomly assigned to drinking water with or without ethanol. The ethanol content of the drinking water was incrementally increased to 4% (Mato et al., 1999). For some of the control and ethanol-treated animals, the GSH precursor S-adenosyl-methionine (SAM) (0.04%) was also added to the drinking water. During the 25 days of treatment, the only access to drinking water was the experimental mixture. Solid diet was provided ad libitum to all four groups. All animals were used in accordance with NIH guidelines (Guide for the Care and Use of Laboratory Animals) as described in protocols reviewed and approved by the Emory University Institutional Animal Care Committee.
Alveolar macrophage isolation
After pentobarbital anesthesia, the trachea was cannulated and the guinea pig lung underwent 4 bronchoalveolar lavages, consisting of 10 ml each, for a total of 40 ml of sterile phosphate-buffered saline (37°C, pH 7.4). The pooled lavage fluid was centrifuged at 500 g for 8 min and the cell pellet was resuspended in 2 ml of sterile Hank’s Buffered Salt Solution (HBSS). After staining with Diff Quik (Dade Behring, Newark, DE) and counting on a hemacytometer, the cell population was determined to be ~95% macrophages.
Alveolar macrophage separation
Alveolar macrophages were subfractionated by centrifugation on a continuous percoll density gradient as described by others (O’Neill et al., 1984, Chandler et al., 1986). After the iso-osmotic percoll was diluted to 49.2% with phosphate buffered saline, the pH was adjusted to 7.2 and 12 mL of the diluted Percoll was then added to a 15 ml glass centrifuge tube (Corning Glass, Corning, NY). Two identical density-based gradients of Percoll were generated for each animal. One tube was used for the separation of alveolar macrophages and the second tube was used for density calibration using marker beads of known density (Sigma-Aldrich, Saint Louis, MO). All of the tubes were centrifuged at 30,000 g for 90 min at 4°C to form a continuous gradient. After 2 ml of the resuspended alveolar macrophages was added to the Percoll tube and 2 ml of HBSS was added to the calibration tube, the tubes were centrifuged at 400 g for 20 min at 4°C. The gradient separated into 4 fractions with the following densities (fraction 1, <1.055 g/ml; fraction 2, 1.056–1.061 g/ml; fraction 3, 1.061–1.065 g/ml; and fraction 4, >1.065 g/ml). Starting with the top layer (layer 1), each gradient fraction was recovered by aspirating with an 18-guage spinal needle. Cells were then resuspended with 5 volumes of HBSS, centrifuged at 200 g for 20 min at 4°C and the resulting cell pellet from each layer was resuspended in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 1% penicillin-streptomycin solution and 10% fetal bovine serum. The cell number in each layer was determined via counting on a hemacytometer.
Phagocytosis of fluorescein isothiocyanate - (FITC)-labeled S. aureus in vitro
Alveolar macrophages from each fraction were plated at a density of 106 cell/mL in DMEM supplemented with 1% penicillin-streptomycin solution plus 10% fetal bovine serum and cultured at 37°C, 5% CO2 for 1 hr. Fluorescein-labeled inactivated S. aureus (Molecular Probes, Eugene, OR) was then added in a 1:10 ratio (macrophage to bacteria). After a 4-hr culture period, alveolar macrophages were washed with DMEM and fixed with 3.7% paraformaldehyde. Phagocytosis of S. aureus was determined via quantitative digital analysis of fluorescence using confocal fluorescent microscopy (Olympus, Melville, NY) as previously reported (Ping et al., 2007, Brown et al., 2007). Using three-dimensional analysis, we evaluated the alveolar macrophages at 50% cell depth in the z plane to ensure that the S. aureus was internalized and not bound to the cell periphery (not within the outer 10 mm of the plasma membrane) (Brown et al., 2007). Fluorescence was determined via quantitative digital analysis using FluoView (Olympus, Melville, NY). Values are presented as mean relative fluorescent units (RFU)/cell ± SEM and the mean percentage of cells fluorescently positive for internalization (per field) ± SEM as tallied from at least 10 experimental fields/set.
Interstitial macrophage isolation
To ensure removal of alveolar macrophages from the lung, two additional bronchoalveolar lavages with 10 mL of saline solution each were performed. After the lavages, the lungs were minced into 2–4 mm pieces with scissors and excess blood removed by rinsing the lung pieces with phosphate buffered saline and filtering through a 70-μm nylon mesh cell strainer membranes (BD Falcon, Bedford, Mass.). The tissue pieces were then incubated with RPMI-1640 medium containing trypsin (166 mg/mL) and EDTA (66 mg/mL) for 40 min at 37°C. To remove particulate matter, the cell suspension was filtered through gauze and then centrifuged for 10 min at 1,500 g at 4°C (Sansores et al., 1997). The cell pellet containing interstitial macrophages was resuspended in RPMI-1640 medium, washed twice, and then centrifuged. The cell pellet containing interstitial macrophages were then resuspended in RPMI-1640 medium, counted, and adjusted to a concentration of 5 × 105 cells/ml before it was cultured at 37°C, 5% CO2 for 1 hr. After this culture period, nonadherent cells were removed and the adherent cells were washed twice with phosphate buffered saline (PBS). The cells were then fixed with 3.7% paraformaldehyde before assessment by immunofluorescence techniques.
Western blot analysis
Alveolar macrophage and interstitial macrophage cell lysates were prepared by adding cell lysis buffer containing protease inhibitor cocktail (Roche) to isolated macrophages. Fifty micrograms of total protein from each sample was separated by SDS-PAGE, and the gel was transferred on to polyvinylidene difluoride membranes. Membranes were blocked at room temperature for 1 h in TBS with 0.2% Tween 20 (TBS-T) containing 5% nonfat dry milk in TBS-T. Primary antibodies at 1:50 dilution in 5% milk in TBS-T were added to the membranes and kept at 4°C overnight. After three washes with T-TBS for 10 min, membranes were incubated at room temperature with secondary antibodies coupled to a horse-radish peroxidase (Santa Cruz Biotechnologies, Santa Cruz, CA) for 2 h. After adding ECL chemiluminescence reagent (Amersham Biosciences) to the membranes, bands were detected and quantified (via densitometry) using a Bio-Rad Imaging System.
Immunofluorescence characterization
Alveolar macrophages and interstitial macrophages were isolated and cultured as described above. After the culture period, the macrophages were fixed with 3.7% paraformaldehyde and nonspecific binding blocked with 1% bovine serum albumin. Cells were then washed twice in PBS, and incubated for 2 hr at room temperature with the following primary antibodies: CD11b (Abcam, Cambridge, MA), CD11c (Antigenix America, Franklin Square, NY), the α subunit of the GM-CSF receptor (GM-CSFR α) (Santa Cruz Biotechnology, Santa Cruz, CA) or PU.1 (Santa Cruz Biotechnology, Santa Cruz, CA). The slides were washed three times with PBS and incubated with the appropriate secondary antibodies (all FITC- and Tetramethyl Rhodamine Iso-Thiocyanate (TRITC)-conjugated antibodies; Sigma-Aldrich) for 1 hr, followed by 2 washes with PBS. Immunofluorescence was determined via quantitative digital analysis of fluorescence (QImaging, Burnaby, BC, Canada) and data analysis performed using Image-Pro Plus for Windows (Jandel Scientific). Background fluorescence was determined and subtracted from the mean relative fluorescent units. Values are presented as the mean relative fluorescent units per cell (± SE) as tallied from at least 10 experimental fields per set.
Statistical analysis
Statistical analysis was performed with Sigma Stat for Windows. The data is presented as means ± SE. Results were analyzed using one-way analysis of variance (ANOVA), followed by Student-Newman-Keuls test comparisons. A p-value of < 0.05 was considered significant.
Results
Chronic ethanol ingestion impaired alveolar macrophage maturation
Pulmonary macrophages increase in size, gain cytoplasm and decrease in density as they mature. As a result, we were able to separate alveolar macrophages into various subpopulations based on densities that contained cells at different maturational stages. The most dense (high nuclear-to-cytoplasmic ratio) alveolar macrophages represent the most immature cells and the least dense (low nuclear-to-cytoplasmic ratio) cells represent the most mature alveolar macrophages (Dauber JH, 1983). We determined 1) the total number of alveolar macrophages isolated by the bronchoalveolar lavage, 2) the number of alveolar macrophages in each of the four subpopulations, and 3) the distribution of alveolar macrophages across the four subpopulations. Across the four experimental groups, there were no significant differences in the total number of alveolar macrophages retrieved by the bronchoalveolar lavage (control 2.2 ± 0.5 ×106/animal; control+ SAM 2.9 ± 0.3 ×106; ethanol 1.6 ± 0.3 ×106/animal; ethanol + SAM 1.9 ± 0.4 ×106/animal). However, chronic ethanol ingestion altered the distribution of alveolar macrophages across the four layers of maturation. Over 75% of the control alveolar macrophages were located within the two most mature fractions (layer 1 and 2), while less than 30% of the ethanol-exposed alveolar macrophages were present in these fractions (Figure 1, a,bp < 0.01 vs. control and control + SAM). When the GSH precursor SAM was added to the drinking water containing ethanol, the percentage of alveolar macrophages in the immature fraction decreased by ~ 20% (Layer 4; Figure 1, cp < 0.001 vs. ethanol), while the percentage of macrophages within layer 2 increased 2-fold (Figure 1, cp < 0.01 vs. ethanol). Therefore, chronic ethanol ingestion resulted in a 60% decrease in the percentage of macrophages present in the two mature fractions but SAM supplementations normalized the alveolar macrophages to a more mature, control-like population.
Figure 1. Alveolar macrophage isolation and distribution.

Pathogen-free guinea pigs were randomly assigned to drinking water, ethanol in the drinking water (4%) or, ethanol + SAM in the drinking water. During the 25 days of treatment, the only access to drinking water was the experimental mixture. Solid diet was provided ad libitum to all three groups. After the appropriate period of experimental diet, the animals were lavaged and the cell pellet subfractionated on a Percoll gradient. A hemacytometer was used to count the percentage of cells obtained from each layer (A). Bars represent means ± SE. a p≤ 0.05 compared to control, b p≤ 0.05 compared to control+SAM, cp≤ 0.05 compared to EtOH+SAM 1 p≤ 0.05 compared with layer 1 (within that group) and 2 p ≤ 0.05 compared with layer 2 (within that group), 3 p ≤ 0.05 compared with layer 3 (within that group). n=5 or more guinea pigs in each group.
Chronic ethanol ingestion impaired alveolar macrophage function in defined subpopulations
Since phagocytic function varies with the different subpopulations of guinea pig alveolar macrophages (Holian A, 1983), we evaluated the different subpopulations from the experimental groups to determine their phagocytic capacity. Alveolar macrophages from each subpopulation were incubated with inactivated FITC-labeled S. aureus in a 1:10 ratio (macrophage: bacteria) for 4 hr. Phagocytosis was evaluated using quantitative digital analysis of fluorescence on the confocal microscopy. Representative photomicrographs at 60X magnification at 50% cell depth in the Z plane of the alveolar macrophages are shown in Figure 2A. As suggested in the confocal images (Figure 2A), the control alveolar macrophages in the two mature fractions (layers 1 and 2) had the greatest percentage of cells positive for internalization, ~ 50% and 70%, respectively (Figure 2B). When the ingested RFU/cell was determined, macrophages isolated from layer 2 phagocytized the greatest amount of S. aureus (Figure 2C). When the percentage of cells positive for internalization was multiplied by the RFU/cell to obtain the phagocytic index, control and control + SAM cells localized to layer 2 were the most functional (Figure 2D).
Figure 2. Chronic ethanol ingestion impaired internalization of S. aureus.


Alveolar macrophage subpopulations were incubated with inactivated FITC-labeled S. aureus in a 1:10 ratio (macrophage: bacteria) for 4 hr. Internalization by the alveolar macrophages in each subpopulation was evaluated using quantitative digital analysis of fluorescence on the confocal microscopy. Representative photomicrographs at 60X magnification at 50% cell depth in the Z plane of the macrophage are shown in Figure A. Bar heights represent the % positive for fluorescence ± SE (B), the mean relative fluorescent units (RFU/cell) ± SE (C), and the Phagocytic Index ± SE (D). Bars in B-D represent means ± SE. Bars represent means ± SE. a p≤ 0.05 compared to control, b p ≤ 0.05 compared to control+SAM, cp≤ 0.05 compared to EtOH+SAM 1 p≤ 0.05 compared with layer 1 (within that group) and 2 p ≤ 0.05 compared with layer 2 (within that group), 3 p ≤ 0.05 compared with layer 3 (within that group),and 4 p≤ 0.05 compared to layer 4 (within that group). n=8 or more fields per layer in each group.
As the confocal images suggested (Figure 2A), phagocytosis was compromised when the alveolar macrophages were derived from ethanol-fed guinea pigs. For alveolar macrophages in layers 1 and 2, there was a ~60% decrease in the percentage of cells positive for internalization compared to control cells (Figure 2B, ap <0.05). Correspondingly, the amount of S. aureus phagocytized by cells in layer 2 was severely diminished as seen by more than a 40% decrease in RFU/cell (Figure 2C, a p <0.01) when compared to control alveolar macrophages located in layer 2. Therefore, the phagocytic index for control alveolar macrophages was ~100,000 and ~170,000 in layers 1 and layer 2 respectively, compared to only ~25,000 in layers 1 and layer 2 respectively for ethanol-exposed alveolar macrophages (Figure 2D, a p <0.05). This translated to an 80% decrease in the phagocytic index for the entire pool of ethanol-exposed alveolar macrophages, when compared to controls.
As suggested in the confocal images (Figure 2A), SAM treatments during ethanol ingestion improved the percentage of cells present in layer 2 that were positive for internalization (Figure 2B, c p <0.001) to levels comparable to that observed in the control group. Importantly, the RFU/cell for the macrophages from the ethanol plus SAM group were comparable to the control cells present in layer 2 (Figure 2C, a,b p >0.05). SAM significantly improved overall phagocytosis of S. aureus (Figure 2D, c p <0.01) as seen by a partial, yet significant, recovery of the phagocytic index in layer 2. In all three treatment groups, the phagocytic index suggested that the cells present in layers 3 and 4 were relatively inactive.
Chronic ingestion severely altered expression of PU.1 in defined subpopulations
As shown above, chronic ethanol ingestion resulted in a shift of the alveolar macrophages to a population with decreased terminal differentiation. To further examine our hypothesis, alveolar macrophage expression of the differentiation marker PU.1 was assessed. PU.1 was chosen because it is required for the terminal maturation of alveolar macrophages and increases as the cell becomes more terminally differentiated. As a result, an increased level of PU.1 is associated with a more terminally differentiated alveolar macrophage.
In control alveolar macrophages, PU.1 expression was highest in the mature layers (layer 1 and layer 2) (Figure 3B). Representative photomicrographs at 60× magnification are shown in Figure 3A. Chronic ethanol ingestion significantly decreased PU.1 expression in layer 1, layer 2, and layer 3, further supporting our hypothesis that ethanol exposure impaired alveolar macrophage maturation. In contrast, SAM supplementation during ethanol ingestion prevented the ethanol-induced alterations of PU.1 expression across the 4 layers (Figure 3B, ap >0.05 vs. control). PU.1 expression in alveolar macrophages from control animals supplemented with SAM did not differ from control values.
Figure 3. Quantification of PU.1 in alveolar macrophage subpopulations.

Representative photomicrographs at 60X magnification are shown in Figure A. PU.1 expressions were determined by staining alveolar macrophages with primary antibodies directed against PU.1 and then secondary antibodies directed against IgG labeled with FITC. The RFU/field was determined by computer analysis of the confocal microscopic images (B). Bar height represents RFU/field ± SE and was tallied from at least 20 experimental fields. Bars represent means ± SE. a p≤ 0.05 compared to control, b p ≤ 0.05 compared to control +SAM, and cp≤ 0.05 compared to EtOH + SAM. n=5 or more fields per group.
Chronic ethanol ingestion severely altered expression of maturational factors
Based on the alveolar macrophage subpopulation data, chronic ethanol ingestion resulted in a shift of the alveolar macrophages to a population with decreased terminal differentiation. To further examine the effects of ethanol on terminal differentiation of alveolar macrophages, we determined expression of markers associated with alveolar macrophage maturation. GM-CSFR-α and PU.1 were chosen because they are both required for the terminal maturation of alveolar macrophages, with increased levels of GM-CSFR-α associated with a more terminally differentiated cell. Previous studies demonstrated in a rat model of chronic ethanol ingestion that expression of the α and β subunits of the GM-CSF receptor (GM-CSFR) were severely decreased in the plasma membrane of alveolar macrophages (Joshi et al., 2005), suggesting a decrease in terminal differentiation. In the current study, total GM-CSFR-α expression was determined across the three treatment groups. When compared to control and control + SAM alveolar macrophages, alveolar macrophages from ethanol-fed animals displayed a 18% and 32% decrease in GM-CSFR-α expression, respectively (Figures 4A and 5A, a,bp < 0.05). SAM supplementation during ethanol ingestion normalized GM-CSFR-α expression (Figures 4A and 5A, ap > 0.05). PU.1 expression was also determined by western blot and immunohistochemical analysis. Compared to control alveolar macrophages, alveolar macrophages from ethanol-fed animals had a 31% decrease in PU.1 expression; a marker of maturity (Figures 4B and 5B, a,bp > 0.05). SAM supplementation prevented the ethanol-induced depression of PU.1 expression (Figures 4B and 5B, ap > 0.05).).
Figure 4. Quantification of GM-CSFR-α, PU.1, CD11b, and CD11c expression in alveolar macrophages by immunohistochemistry.



GM-CSFR-α, PU.1, CD11c, and CD11b expressions were determined by staining alveolar macrophages with primary antibodies directed against GM-CSFR-α (A), PU.1 (B), CD11b (C), and CD11c (D) and then secondary antibodies directed against IgG labeled with FITC or TRITC. The ratio of GM-CSFR-α: CD11b (E) and PU.1:CD11c (F) expression were also evaluated in each layer. The RFU/field was determined by computer analysis of the confocal microscopic images. Bar height represents RFU/field ± SE and was tallied from at least 20 experimental fields. Bars represent means ± SE. a p≤ 0.05 compared to control, b p≤ 0.05 compared to control+SAM, and cp≤ 0.05 compared to EtOH + SAM. n=10 or more fields per group.
Figure 5. Quantification of GM-CSFR-α, PU.1, CD11b, and CD11c expression in alveolar macrophages by western blot analysis.


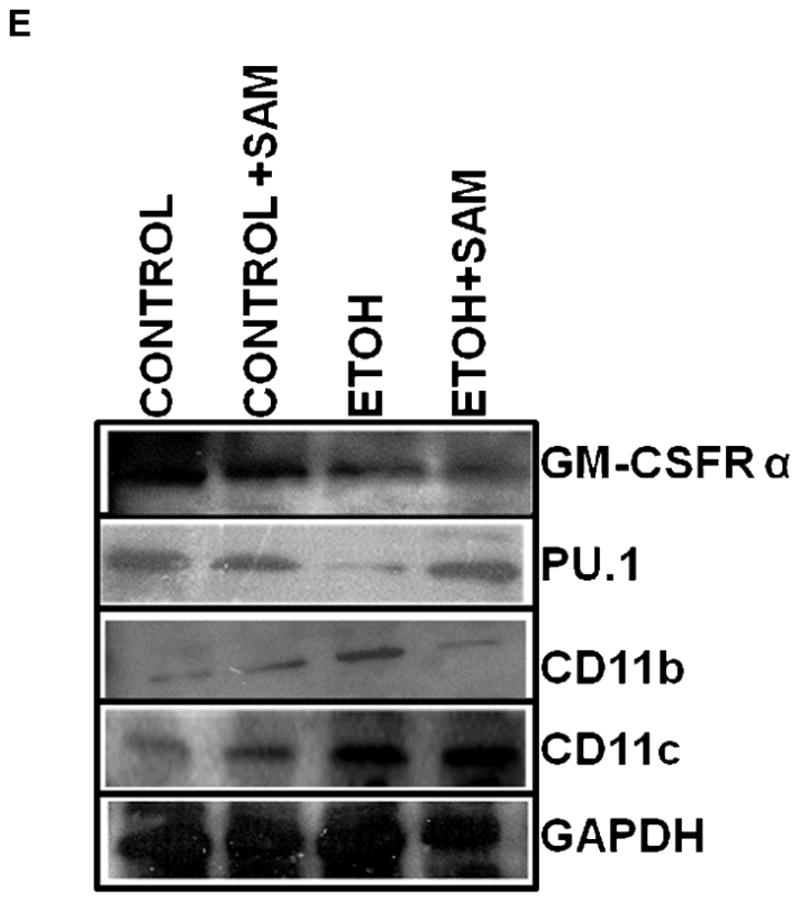
GM-CSFR-α, PU.1, CD11c, and CD11b expressions were determined via western blot analysis. GAPDH, GM-CSFR-α (A), PU.1 (B), CD11b (C), and CD11c (D) protein levels were detected by immunoblotting. Bars represent means ± SE. ap≤ 0.05 compared to control, b p≤ 0.05 compared to control + SAM, and cp≤ 0.05 compared to EtOH + SAM. n=10 or more fields per group. Representative gels of GAPDH, GM-CSFR-α, PU.1, CD11c, and CD11b protein expressions in alveolar macrophages (E).
We also assessed markers associated with immaturity including CD11b, and CD11c which are highly expressed in monocytes. For CD11b expression control and control + SAM alveolar macrophages displayed more than 35% less CD11b expression when compared to alveolar macrophages from ethanol-fed animals (Figures 4C and 5C, a,bp < 0.05). SAM supplementation significantly improved Cd11b expression, as seen as a normalization to control values (Figures 4C and 5C, ap > 0.05). Similar to that observed for CD11b, chronic ethanol ingestion significantly increased expression of the monocytic marker CD11c expression when compared to control alveolar macrophages (Figures 4D and 5D, ap < 0.05). SAM supplementation normalized CD11c levels and were not significantly different from controls. When the ratio of GM-CSFR-α: CD11b expression was assessed, there were comparable ratios for the control, control + SAM, and ethanol +SAM alveolar macrophages (Figure 4E). In the ethanol-fed animals, the ratio of GM-CSFR-α: CD11b was also decreased, compared to the control groups. Furthermore, SAM supplementation prevented this decrease induced by ethanol (Figure 4E, a,b,c p <0.05). Similar to what was observed with the ratio of GM-CSFR-α: CD11b, the ratio of PU.1:CD11c did not differ between the control, control + SAM, and ethanol +SAM alveolar macrophages. Similarly, ethanol-exposed alveolar macrophages had more than a 50% decrease in PU.1:CD11c expression (Figure 4F, a,bp < 0.05).
Chronic ethanol -induced defects were not limited to the alveolar space
To determine if the ethanol-induced derangements in pulmonary macrophage maturation were limited to the alveolar space, the expressions of GM-CSFR-α, PU.1, CD11b, and CD11c, were determined in lung interstitial macrophages. For the interstitial macrophages, chronic ethanol exposure significantly decreased GM-CSFR-α expression when compared to control cells but SAM supplements failed to normalize GM-CSFR-α expression Figures 6A and 7A, a,bp < 0.05). PU.1 expression by the pool of interstitial macrophages was also significantly altered by chronic ingestion of ethanol (Figures 6B and 7B ap < 0.05). When we examined the monocytic markers, chronic ethanol ingestion increased expression of CD11b and CD11c by ~65% and ~15%, respectively, when compared to controls (Figures 6C and 7C, a,bp < 0.05) (Figures 6D and 7D). As observed with alveolar macrophages, these effects of ethanol on monocytic markers were blunted by SAM supplements. When the ratio of GM-CSFR-α: CD11b expression was assessed, the control and control + SAM interstitial macrophages had comparable ratios. In the ethanol-fed animals, the ratio of GM-CSFR-α: CD11b also decreased when compared to control (Figure 4E, a,bp <0.05). When the ratio of PU.1:CD11c expression was assessed, the interstitial macrophages from the control, control + SAM, and ethanol +SAM groups had comparable ratios (Figure 4F). In contrast, ethanol-exposed alveolar macrophages had an approximate 40% decrease in PU.1:CD11c expression (Figure 4F, a,bp <0.05).
Figure 6. Quantification of GM-CSFR-α, PU.1, CD11b, and CD11c expression in interstitial macrophages by immunohistochemistry.



GM-CSFR-α, PU.1, CD11c, and CD11b expressions were determined by staining interstitial macrophages with primary antibodies directed against GM-CSFR-α (A), PU.1 (B), CD11b (C), and CD11c (D) and then secondary antibodies directed against IgG labeled with FITC or TRITC. The ratio of GM-CSFR-α: CD11b (E) and PU.1:CD11c (F) expression were also evaluated in each layer. The RFU/field was determined by computer analysis of the confocal microscopic images. Bar height represents RFU/field ± SE and was tallied from at least 20 experimental fields. Bars represent means ± SE. a p≤ 0.05 compared to control, b p ≤ 0.05 compared to control + SAM, and cp≤ 0.05 compared to EtOH + SAM. n=10 or more fields per group.
Figure 7. Quantification of GM-CSFR-α, PU.1, CD11b, and CD11c expression in interstitial macrophages by western blot analysis.


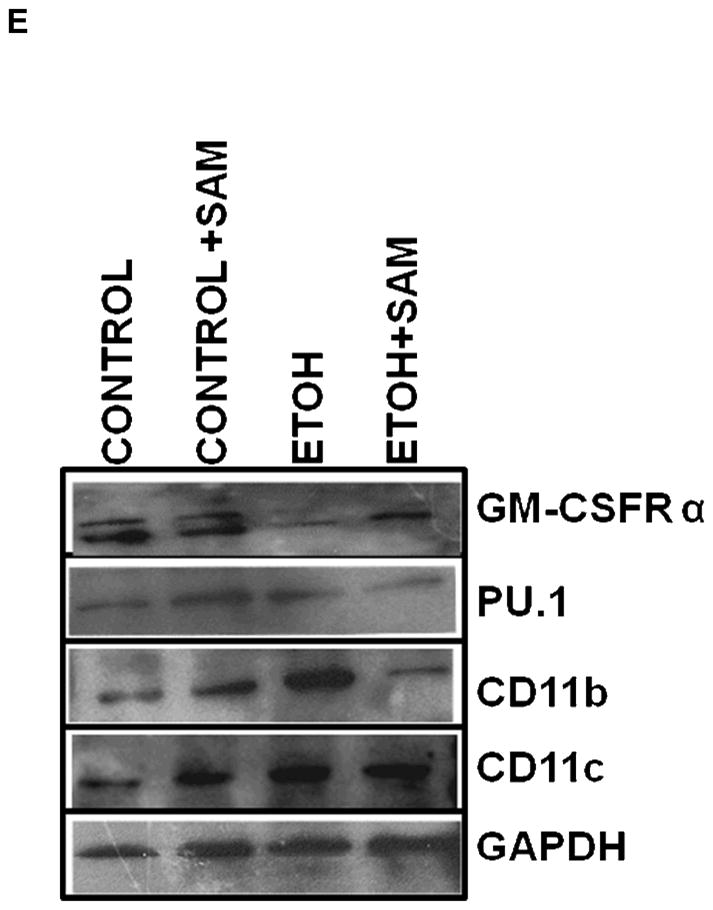
GM-CSFR-α, PU.1, CD11c, and CD11b expressions were determined via western blot analysis. GAPDH, GM-CSFR-α (A), PU.1 (B), CD11b (C), and CD11c (D) protein levels were detected by immunoblotting. Bars represent means ± SE. ap≤ 0.05 compared to control, b p ≤ 0.05 compared to control + SAM, and cp≤ 0.05 compared to EtOH + SAM. n=10 or more fields per group. Representative gels of GAPDH, GM-CSFR-α, PU.1, CD11c, and CD11b protein expressions in interstitial macrophages (E).
Discussion
The deleterious effects of chronic ethanol ingestion on lung physiology and function have been well characterized (Moss et al., 2000, Polikandriotis et al., 2006, Joshi et al., 2005, Aytacoglu et al., 2006). Previous studies by this laboratory demonstrated chronic ethanol ingestion and subsequent chronic oxidant stress impaired phagocytic functions of alveolar macrophages (Brown et al., 2007). In the current study, we examined the possibility that impaired phagocytosis after chronic ethanol ingestion was a result of impaired terminal differentiation. To address this question, we examined the effects of chronic ethanol ingestion on several different markers of alveolar macrophage differentiation including cell density, phagocytosis, and expression of maturational markers.
When cell density was used as a marker of differentiation, approximately 80% of the alveolar macrophages from control and control + SAM animals were equally distributed between the mature layers (layer 1 and layer 2). These two layers also displayed the highest phagocytic capacity based on the percentage of cells positive for phagocytosis and the quantification of bacteria ingested per cell. Thus, the majority of the control and control + SAM alveolar macrophages were terminally differentiated as assessed by cell density, phagocytic capacity, and maturational markers such as GM-CSFRα and PU.1.
As observed previously in an ethanol-fed rat model (Guidot et al., 2000), chronic ethanol consumption in a guinea pig model also severely decreased alveolar macrophage phagocytosis as evidenced by a drastic decrease in the percentage of alveolar macrophages positive for S. aureus internalization. Since the majority of the alcoholic macrophages were localized to the immature fractions, poor phagocytic capacity in these subpopulations was not surprising. However, a small percentage of ethanol-exposed alveolar macrophages had the cell size and density comparable to that observed for the majority of the control cells. Although these ethanol-exposed alveolar macrophages were located in the mature fractions, their phagocytic capacity was compromised. In addition, the cells located in the mature fractions had decreased expression of the maturational marker PU.1. These two diverse markers of maturation, cell density and PU.1 expression, suggest that chronic ethanol ingestion impaired terminal differentiation of the alveolar macrophages. However, we cannot rule out the possibility that chronic ethanol ingestion also alters the profile for receptor surface expression. Additional studies of receptor cell trafficking are needed to determine why these ethanol-exposed cells present in the fractions and deemed to be mature based on cell density demonstrated altered expression of surface receptors and whether this altered receptor expression results in severely compromised phagocytic capacity.
Chronic ethanol ingestion has been shown to severely diminish GSH availability within the alveolar space (Foreman et al., 2002, Moss et al., 2000, Velasquez A, 2002) as well as within the resident cells of the alveolar space, including alveolar type II cells (F. Holguin 1998) and alveolar macrophages (Brown et al., 2007). Previous studies in this laboratory have shown in the rat model of chronic ethanol ingestion that supplementation with GSH precursors procysteine or N-acetyl cysteine during ethanol ingestion maintained alveolar macrophage function, as well as normalized GSH levels (Brown et al., 2007). SAM supplementation has been shown to attenuate alcohol-induced liver injury in an animal model of chronic ethanol consumption and has clinically been shown to improve survival of liver transplantation patients with alcoholic liver cirrhosis (Lieber, 1990, Mato et al., 1999). This is the first study to examine the potential benefits of SAM supplementation to alveolar macrophages in an adult animal model of chronic ethanol ingestion. In the current study, SAM supplementation during chronic ethanol ingestion partially prevented the harmful effects of ethanol on alveolar macrophage maturation and function. More specifically, SAM supplementation led to a shift in the distribution of macrophages to a more mature fraction, as well as a significant increase in phagocytic capacity of the cells present in the mature fractions. However, SAM supplementation in control animals had minimal effects and this further supports the hypothesis that chronic ethanol ingestion leads to increased oxidant stress.
Studies done by Joshi et al. (Joshi et al., 2005) reported in a rat model that chronic ethanol ingestion decreased membrane expression of GM-CSFR-α in alveolar macrophages. In the current study, SAM supplementation during ethanol ingestion increased expression of GM-CSFR-α and PU.1, markers of terminal differentiation. SAM supplementation during chronic ethanol ingestion also decreased expression of CD11b and CD11c, monocytic markers. Therefore, SAM prevented the ethanol-induced alterations in alveolar macrophage density, terminal differentiation, and phagocytic capacity suggesting that ethanol-induced oxidant stress was a central feature in the impaired maturation of alveolar macrophages. Furthermore, SAM supplementation maintained phagocytic function in the small percentage of “mature” ethanol-exposed macrophages when cell density was used as the criteria. Additional studies are necessary to determine whether higher concentrations of SAM are needed to completely protect the alveolar macrophages from the negative effects of chronic ethanol ingestion.
In order to determine if altered maturation and function of pulmonary macrophages in response to chronic ethanol ingestion extended to interstitial macrophages, expressions of GM-CSFR-α, PU.1, CD11b, and CD11c were also determined in interstitial macrophages. Chronic ethanol ingestion led to a significant decrease in GM-CSFR-α expression. Similarly, expressions of CD11b and CD11b were also increased in interstitial macrophages from ethanol-fed animals. This suggested that chronic ethanol ingestion also impaired the differentiation of interstitial macrophages within the lung. SAM supplementation blunted these deleterious effects of ethanol, as seen by normalization of CD11b and CD11c expression. It is also important to note this is the first study to examine the effects of ethanol on pulmonary interstitial macrophages.
In summary, this study demonstrated that chronic ethanol exposure decreased alveolar macrophage maturation, whether defined by decreased cell density, decreased expression of maturational markers, or persistent monocytic characteristics. These alterations were directly correlated to impaired phagocytic function. Since the majority of the ethanol-exposed alveolar macrophages were localized to the immature fractions, our study suggested the poor phagocytic capacity seen with these macrophages was primarily due to impaired terminal differentiation. For the limited percentage of cells that were present in the mature fractions, chronic ethanol ingestion also impaired phagocytic capacity. The capacity of SAM supplementation to normalize most of the detrimental effects of ethanol suggested ethanol-induced oxidant stress played a central role in the impaired terminal differentiation and dysfunction of lung interstitial and alveolar macrophages. Additional studies are needed to determine whether GSH precursors will restore macrophage differentiation and phagocytosis as well as improve resistance to respiratory infections in this selected at-risk patient population.
Acknowledgments
This study was funded by The National Institute of Alcohol Abuse and Addiction Grant R01 AA139879 (LAB), the National Institute of Alcohol Abuse and Addiction Grant R01 AA12197 (LAB), the National Institute of Alcohol Abuse and Addiction Center Grant P50 AA 135757 (LAB and TWG), and the National Institute of Environmental Health Sciences Toxicology Training Grant -T32 ES012870 (SDB).
References
- AKAGAWA KS, KAMOSHITA K, TOKUNAGA T. Effects of granulocyte-macrophage colony-stimulating factor and colony- stimulating factor-1 on the proliferation and differentiation of murine alveolar macrophages. J Immunol. 1988;141:3383–3390. [PubMed] [Google Scholar]
- AYTACOGLU BN, CALIKOGLU M, TAMER L, COSKUN B, SUCU N, KÖSE N, AKTAS S, DIKMENGIL M. Alcohol-Induced Lung Damage and Increased Oxidative Stress. Respiration. 2006;73:100–104. doi: 10.1159/000088680. [DOI] [PubMed] [Google Scholar]
- BALDWIN GC. The biology of granulocyte-macrophage colony-stimulating factor: Effects on hematopoietic and nonhematopoietic cells. Developmental Biology. 1992;151:352–367. doi: 10.1016/0012-1606(92)90175-g. [DOI] [PubMed] [Google Scholar]
- BERCLAZ PY, CAREY B, FILLIPI MD, WERNKE-DOLLRIES K, GERACI N, CUSH S, RICHARDSON T, KITZMILLER J, O’CONNOR M, HERMOYIAN C, KORFHAGEN T, WHITSETT JA, TRAPNELL BC. GM-CSF Regulates a PU.1-Dependent Transcriptional Program Determining the Pulmonary Response to LPS. Am J Respir Cell Mol Biol. 2007;36:114–121. doi: 10.1165/rcmb.2006-0174OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERCLAZ PY, SHIBATA Y, WHITSETT JA, TRAPNELL BC. GM-CSF, via PU.1, regulates alveolar macrophage Fcgamma R-mediated phagocytosis and the IL-18/IFN-gamma -mediated molecular connection between innate and adaptive immunity in the lung. Blood. 2002;100:4193–4200. doi: 10.1182/blood-2002-04-1102. [DOI] [PubMed] [Google Scholar]
- BROWN LAS, HARRIS FL, PING XD, GAUTHIER TW. Chronic ethanol ingestion and the risk of acute lung injury: a role for glutathione availability? Alcohol. 2004;33:191–197. doi: 10.1016/j.alcohol.2004.08.002. [DOI] [PubMed] [Google Scholar]
- BROWN LAS, PING XD, HARRIS FL, GAUTHIER TW. Glutathione availability modulates alveolar macrophage function in the chronic ethanol-fed rat. Am J Physiol Lung Cell Mol Physiol. 2007;292:L824–832. doi: 10.1152/ajplung.00346.2006. [DOI] [PubMed] [Google Scholar]
- BURGESS AW, METCALF D. The nature and action of granulocyte-macrophage colony stimulating factors. Blood. 1980;56:947–958. [PubMed] [Google Scholar]
- CHANDLER DB, FULLER WC, JACKSON RM, FULMER JD. Fractionation of rat alveolar macrophages by isopycnic centrifugation: morphological, cytochemical, biochemical, and functional properties. J Leukoc Biol. 1986;39:371–383. doi: 10.1002/jlb.39.4.371. [DOI] [PubMed] [Google Scholar]
- DAUBER JHHA, ROSEMILLER ME, DANIELE RP. Separation of bronchoalveolar cells from the guinea pig on continuous density gradients of Percoll: morphology and cytochemical properties of fractionated lung macrophages. J Reticuloendothel Soc. 1983;33:119–26. [PubMed] [Google Scholar]
- DEKOTER RP, WALSH JC, SINGH H. PU.1 regulates both cytokine-dependent proliferation and differentiation of granulocyte/macrophage progenitors. EMBO J. 1998;17:4456–4468. doi: 10.1093/emboj/17.15.4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESPER A, BEL, MOSS M. The effect of alcohol abuse on ARDS and multiple organ dysfunction. Anesthesiology. 2006;72:375–381. [PubMed] [Google Scholar]
- HOLGUIN F, IM, BROWN LA, GUIDOT DM. Chronic ethanol ingestion impairs alveolar type II cell glutathione homeostasis and function and predisposes to endotoxin-mediated acute edematous lung injury in rats. J Clin Invest. 1998;101:761–768. doi: 10.1172/JCI1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOREMAN MG, HOOR TT, BROWN LAS, MOSS M. Effects of Chronic Hepatic Dysfunction on Pulmonary Glutathione Homeostasis. Alcoholism: Clinical and Experimental Research. 2002;26:1840–1845. doi: 10.1097/01.ALC.0000042149.71731.B7. [DOI] [PubMed] [Google Scholar]
- GUIDOT DM, MODELSKA K, LOIS M, JAIN L, MOSS IM, PITTET JF, BROWN LAS. Ethanol ingestion via glutathione depletion impairs alveolar epithelial barrier function in rats. Am J Physiol Lung Cell Mol Physiol. 2000;279:L127–135. doi: 10.1152/ajplung.2000.279.1.L127. [DOI] [PubMed] [Google Scholar]
- HOLIAN ADJ, DIAMOND MS, DANIELE RP. Separation of bronchoalveolar cells from the guinea pig on continuous gradients of Percoll: functional properties of fractionated lung macrophages. J Reticuloendothel Soc. 1983;33:157–164. [PubMed] [Google Scholar]
- JONG GM, HSIUE TR, CHEN CR, CHANG HY, CHEN CW. Rapidly fatal outcome of bacteremic Klebsiella pneumoniae pneumonia in alcoholics. Chest. 1995;107:214–217. doi: 10.1378/chest.107.1.214. [DOI] [PubMed] [Google Scholar]
- JOSHI PC, APPLEWHITE L, RITZENTHALER JD, ROMAN J, FERNANDEZ AL, EATON DC, BROWN LAS, GUIDOT DM. Chronic Ethanol Ingestion in Rats Decreases Granulocyte-Macrophage Colony-Stimulating Factor Receptor Expression and Downstream Signaling in the Alveolar Macrophage. J Immunol. 2005;175:6837–6845. doi: 10.4049/jimmunol.175.10.6837. [DOI] [PubMed] [Google Scholar]
- JOSHI PC, GUIDOT DM. The alcoholic lung: epidemiology, pathophysiology, and potential therapies. Am J Physiol Lung Cell Mol Physiol. 2007;292:L813–823. doi: 10.1152/ajplung.00348.2006. [DOI] [PubMed] [Google Scholar]
- LASKIN DL, WEINBERGER B, LASKIN JD. Functional heterogeneity in liver and lung macrophages. J Leukoc Biol. 2001;70:163–170. [PubMed] [Google Scholar]
- LIEBER CSD, ALESSANDRO KIM, CHO-IL SASAKI, NANCY LEO, MARIA A. S-Adenosyl-L-methionine attenuates alcohol-induced liver injury in the baboon. Hepatology. 1990;11:165–172. doi: 10.1002/hep.1840110203. [DOI] [PubMed] [Google Scholar]
- LLOBERAS J, SOLER C, CELADA A. The key role of PU.1/SPI-1 in B cells, myeloid cells and macrophages. Immunology Today. 1999;20:184–189. doi: 10.1016/s0167-5699(99)01442-5. [DOI] [PubMed] [Google Scholar]
- LUNDAHL J, HALLDEN G, SKOLD CM. Human blood monocytes, but not alveolar macrophages, reveal increased CD11b/CD18 expression and adhesion properties upon receptor-dependent activation. Eur Respir J. 1996;9:1188–1194. doi: 10.1183/09031936.96.09061188. [DOI] [PubMed] [Google Scholar]
- MATO JM, CAMARA J, PAZ JFD, CABALLERIA L, COLL S, CABALLERO A, GARCIA-BUEY L, BELTRAN J, BENITA V, CABALLERIA J, SOLA R, MORENO-OTERO R, BARRAO F, MARTIN-DUCE A, CORREA JA, PARES A, BARRAO E, GARCIA-MAGAZ I, PUERTA JL, MORENO J, BOISSARD G, ORTIZ P, RODES J. S-Adenosylmethionine in alcoholic liver cirrhosis: a randomized, placebo-controlled, double-blind, multicenter clinical trial. Journal of Hepatology. 1999;30:1081–1089. doi: 10.1016/s0168-8278(99)80263-3. [DOI] [PubMed] [Google Scholar]
- MOSS M, GUIDOT DM, WONG-LAMBERTINA M, TEN HOOR T, PEREZ RL, BROWN LAS. The Effects of Chronic Alcohol Abuse on Pulmonary Glutathione Homeostasis. Am J Respir Crit Care Med. 2000;161:414–419. doi: 10.1164/ajrccm.161.2.9905002. [DOI] [PubMed] [Google Scholar]
- O’NEILL SJ, HOEHN SK, LESPERANCE E, KLASS DJ. Functional heterogeneity of isopycnic fractions of rat alveolar macrophages. Infect Immun. 1984;46:282–284. doi: 10.1128/iai.46.1.282-284.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PING XD, HARRIS FL, BROWN LAS, GAUTHIER TW. In Vivo Dysfunction of the Term Alveolar Macrophage After in Utero Ethanol Exposure. Alcoholism: Clinical and Experimental Research. 2007;31:308–316. doi: 10.1111/j.1530-0277.2006.00306.x. [DOI] [PubMed] [Google Scholar]
- POLIKANDRIOTIS JA, RUPNOW HL, ELMS SC, CLEMPUS RE, CAMPBELL DJ, SUTLIFF RL, BROWN LAS, GUIDOT DM, HART CM. Chronic Ethanol Ingestion Increases Superoxide Production and NADPH Oxidase Expression in the Lung. Am J Respir Cell Mol Biol. 2006;34:314–319. doi: 10.1165/rcmb.2005-0320OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSSEAU S, HAMMERL P, MAUS U, WALMRATH HD, SCHUTTE H, GRIMMINGER F, SEEGER W, LOHMEYER J. Phenotypic characterization of alveolar monocyte recruitment in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2000;279:L25–35. doi: 10.1152/ajplung.2000.279.1.L25. [DOI] [PubMed] [Google Scholar]
- SANSORES RH, ABBOUD RT, BECERRIL C, MONTANO M, RAMOS C, VANDA B, SELMAN ML. Effect of exposure of guinea pigs to cigarette smoke on elastolytic activity of pulmonary macrophages. Chest. 1997;112:214–219. doi: 10.1378/chest.112.1.214. [DOI] [PubMed] [Google Scholar]
- SCOTT EW, SIMON MC, ANASTASI J, SINGH H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- SHIBATA Y, BERCLAZ PY, CHRONEOS Z, YOSHIDA H, WHITSETT JA, TRAPNELL BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15:557. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- STANDIFORD TJDJ. Ethanol Feeding Inhibits Proinflammatory Cytokine Expression from Murine Alveolar Macrophages Ex Vivo. Alcoholism: Clinical and Experimental Research. 1997;21:1212–1217. [PubMed] [Google Scholar]
- TRAPNELL BC, WHITSETT JA. GM-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu Rev Physiol. 2002;64:775–802. doi: 10.1146/annurev.physiol.64.090601.113847. [DOI] [PubMed] [Google Scholar]
- VELASQUEZ ABR, LEWIS JF, MALLOY J, MCCAIG L, BROWN LA, GUIDOT DM. Glutathione Replacement Preserves the Functional Surfactant Phospholipid Pool Size and Decreases Sepsis-Mediated Lung Dysfunction in Ethanol-Fed Rats. Alcoholism: Clinical and Experimental Research. 2002;26:1245–1251. doi: 10.1097/01.ALC.0000024269.05402.97. [DOI] [PubMed] [Google Scholar]
- WARE LB, MATTHAY MA. The Acute Respiratory Distress Syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- YEH MY, BURNHAM EL, MOSS M, BROWN LAS. Chronic Alcoholism Alters Systemic and Pulmonary Glutathione Redox Status. Am J Respir Crit Care Med. 2007;176:270–276. doi: 10.1164/rccm.200611-1722OC. [DOI] [PMC free article] [PubMed] [Google Scholar]