


Abstract
Transcriptome analyses have typically disregarded nucleocytoplasmic differences. This approach has ignored some post-transcriptional regulations and their effect on the ultimate protein expression levels. Despite a longstanding interest in the differences between the nuclear and cytosolic transcriptomes, it is only recently that data have become available to study such differences and their associated features on a genome-wide scale. Here, we compared the nuclear and cytosolic transcriptomes of HepG2 and HeLa cells. HepG2 and HeLa cells vary significantly in the differential compartmentalization of their transcript isoforms, indicating that nucleocytoplasmic compartmentalization is a cell-specific characteristic. The differential compartmentalization is manifested at the transcript isoform level instead of the gene level because alternative isoforms of one gene can display different nucleocytoplasmic distributions. The isoforms enriched in the cytosol tend to have more introns and longer introns in their pre-mRNAs. They have more functional RNA folds and unique exons in the 3′ regions. These isoforms are more conserved than the isoforms enriched in the nucleus. Surprisingly, the presence of microRNAs does not have a significant impact on the nucleocytoplasmic distribution of their target isoforms. In contrast, nonsense-mediated decay is significantly more associated with the isoforms enriched in the nucleus than those enriched in the cytosol.
INTRODUCTION
Gene expression regulation comprises multiple levels including transcription, splicing, nuclear export, microRNA (miRNA) inhibition and mRNA decay. Because protein translation occurs in the cytoplasm, the compartmentalization of mature mRNA in the nucleus and the cytosol has an immediate impact on protein expression levels. The compartmentalization is closely related to the export kinetics of mRNA from the nucleus to the cytoplasm and the degradation rate of mRNA in the cytoplasm. Both of these processes are fundamental to gene expression regulation (1). Transcriptome analyses have typically profiled polyadenylated (poly(A)+) transcripts without tracking nucleocytoplasmic differences. This may not interfere with studies on transcriptional regulation but ignores post-transcriptional regulation and its effect on the ultimate output of gene expression regulation, the protein translation level. It is thus necessary to understand the difference between the nuclear and cytoplasmic transcriptomes. It is equally important to investigate whether the nucleocytoplasmic compartmentalization of mRNA transcripts displays any global features and/or reflects any cell-specific characteristics.
The nucleocytoplasmic compartmentalization of mRNA has an immediate impact on the protein expression level. Meanwhile, alternative pre-mRNA splicing is an important gene regulation mechanism for expanding proteomic diversity in higher eukaryotes because multiple transcript isoforms produced from a single gene can lead to protein isoforms with distinct functions (2). It has been estimated that >90% of human genes are alternatively spliced (3,4). Little is known about the nucleocytoplasmic compartmentalization of alternative transcripts on the global scale. Therefore, there is great interest in studying the difference between the nuclear transcriptome and the cytosolic transcriptome at the individual transcript isoform level. Recently, we developed Bayesian Analysis of Splicing IsoformS (BASIS) to compare transcriptomes at the individual transcript isoform level (5). This is achieved by a hierarchical Bayesian model based on the high-throughput sequencing data or the high-resolution tiling array data and transcript isoform splicing patterns assembled from databases.
Here, we applied BASIS to a high-density tiling array data set to compare individual transcript isoforms between the nucleus and the cytosol of HepG2 and HeLa cells. The array data are from the Kapranov et al.'s (6) in which they profiled the steady-state nuclear and cytosolic poly(A)+ RNAs >200 nt using the whole-genome 5 bp resolution tiling arrays. Based on the results from BASIS, we identified transcript isoforms enriched in the nucleus or in the cytosol. In the rest of the article, we will call these enriched classes nuclear isoforms and cytosolic isoforms, respectively (although ‘nuclear isoforms’ do not necessarily mean that they are not found in the cytosol, and vice versa). For the first time on a genome-wide scale, we discovered some global features of nuclear isoforms and cytosolic isoforms.
Introns and their splicing have been reported to affect gene expression at many different levels, including transcription (7–12), polyadenylation (13,14), mRNA export (15–21), translational efficiency (22–24) and mRNA decay (25). In Xenopus oocyte injection experiments, mRNAs generated through splicing are exported to the cytoplasm more rapidly than identical mRNAs transcribed from intron-free cDNA constructs (15–17). In mammalian cells, studies on the SV40 gene (19) and the human ceruloplasmin gene (20) show that splicing efficiently exports mRNAs to the cytoplasm. Tokunaga et al. (21) microinjected fluorescently labeled ftz pre-mRNA into mammalian cell nuclei, and found that the spliced mRNAs were efficiently exported but the intron-less mRNA was diffusely distributed in the nucleus. More recently, Valencia et al. (18) used a similar approach combining FISH and transfection or nuclear microinjection to re-examine the relationship between splicing and mRNA export in mammalian cells. They concluded that splicing promotes rapid and efficient mRNA export. As a result of pre-mRNA splicing, a set of proteins called the exon junction complex (EJC) are deposited on the mRNA upstream of exon–exon junctions. The EJC can provide a binding platform for the mRNA export machinery and link the excision of introns with mRNA export (1,15,26). In addition, introns can regulate mRNA degradation rates (25). To explore the effect of introns on nucleocytoplasmic mRNA distribution on a genomic scale, we examined various attributes of the introns for the nuclear and cytosolic isoforms.
We then compared the enrichment of functional RNA folds between the nuclear and cytosolic isoforms. The untranslated regions (UTRs) of mRNAs contain multiple cis-acting elements that regulate mRNA export, stability, and sub-cellular localization (27). Many of these cis-acting elements contain secondary structures such as stem loops but lack recognizable primary sequence features (28,29). We therefore performed the functional RNA fold studies for the nuclear and cytosolic isoforms. Next, we examined the relationship of miRNAs and differentially distributed isoforms. miRNAs inhibit protein translation by base pairing imperfectly to the 3′ UTR of their target genes in animals (30,31). In some instances, this induces mRNA degradation (32,33) and thus may affect the dynamics of mRNA nucleocytoplasmic localization. We therefore investigated the global impact of miRNAs on the mRNA transcript nucleocytoplasmic distribution. We also examined the relationship between nonsense-mediated decay (NMD) and the differential compartmentalization of transcript isoforms. NMD degrades mRNAs that carry premature translation termination codons (34–36). It has been shown to be coupled with alternative splicing to control protein expression levels or malicious protein expression (termed AS-NMD). The studies on the global prevalence of functional AS-NMD have conflicting views (37,38). EST and cDNA sequence-based analyses predicted that ∼35% of alternative splicing events have the potential to be regulated by NMD (37), but microarray-based studies show that these transcript isoforms are generally produced at low levels and are independent of the action of NMD (38). Because NMD occurs in the cytosol, it may impact the nucleocytoplasmic distribution of a transcript isoform. Finally, we compared the conservation levels of the nuclear isoforms with those of the cytosolic isoforms.
MATERIALS AND METHODS
Tiling array data pre-processing
The non-redundant transcript isoform information for human genes was downloaded from the Alternative Splicing and Transcript Diversity database (39) (http://www.ebi.ac.uk/astd/, release 1.1, names begin with ‘TRAN’) and the Ensembl Genome Browser (http://www.ensembl.org/index.html, release 50, names begin with ‘ENST’). The expression levels of these transcripts were from the whole-genome tiling arrays in which the human genome is split into 91 chips with 5 bp resolution (6). We studied the expression data for the HeLa and HepG2 cell lines in the cytosol and the nucleus. Each cell line had about three replicates. The profiled RNAs were polyadenylated and >200 nt. The preprocessed probe signal and the transcribed regions (transfrags) were downloaded from the GEO database with the accession number GSE7576 (http://www.ncbi.nlm.nih.gov/geo). The signal thresholds for the transfrags correspond to a 4% false discovery rate. The probe intensity was further quantile-normalized for the cytosol and nucleus. The probe coordinates were from the NCBI version 35. The UCSC liftOver tool (http://genome.ucsc.edu/) was used to convert the coordinates between version 35 and version 36. We treated transcripts as ‘present’ if ≥50% of their exons had ≥50% of their positions overlapping with transfrags. Among the 20 892 protein-coding genes [note that some transcript isoforms do not have ‘coding sequence (CDS)’ features available], 9953 genes for HepG2 and 9607 genes for HeLa were removed because none of their transcripts was present in either sub-cellular location. In our pre-screening procedures, we removed genes with incompletely spliced transcripts. If an intron had ≥50% positions overlapping with transfrags, we treated it as an unspliced intron. Only 3–8% of introns were un-spliced. This indicates that polyadenylation occurs after splicing is mostly finished. If a transcript contained un-spliced introns, we treated it as an incompletely spliced transcript and removed the gene (1196 genes for HepG2 and 1015 genes for HeLa). According to the procedures, we also removed genes with retained introns, which are considered a special category of alternative splicing. However, it is generally agreed that it is difficult to distinguish intron retention from experimental artifacts (e.g. incomplete splicing). Other pre-screening procedures were performed as described previously (5), and an additional 1192 genes for HepG2 and 1189 genes for HeLa were removed. A total of 8551 genes with 67 680 possible transcript isoforms were considered in BASIS for HepG2, and a total of 9081 genes with 70 902 possible transcript isoforms were considered in BASIS for HeLa.
Hierarchical Bayesian model (BASIS)
For each exonic probe i of gene g, consider the linear model:
where Δygi is the intensity difference between two sub-cellular locations for probe i of gene g (ygi1 − ygi2), Δβgj is the expression difference between two locations for the jth transcript isoform of gene g, xgij is the binary indicator whether probe i belongs to isoform j’s exon region, and Δεgi is the error term of probe i of gene g. For a dataset, g is from 1 to G where G is the total number of genes, i is from 1 to ng where ng is the total number of probes for gene g, and j is from 1 to sg where sg is the total number of transcript isoforms for gene g. The total Δεgi’s (g = 1, … , G and I = 1, … , ng) are divided into 100 bins. Each bin contains thousands of probes with similar ygi1 + ygi2 values. Because the intensity variance is dependent on the intensity mean, probes in the same bin will have similar variances.
A hierarchical Bayesian model is built as:
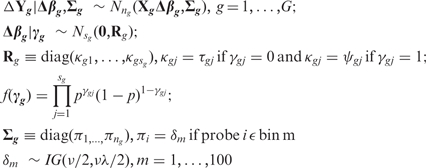 |
where γg is a latent variable, Nng and Nsg stand for multivariate normal distributions, IG stands for inverse gamma distribution. When γgj = 0, Δβgj ∼ N(0, τ) and when γgj = 1, Δβgj ∼ N(0, ψ). We set τ to be small so that when γgj = 0, Δβgj is small enough to be estimated as 0. And ψ is set to be large so that when γgj = 1, Δβgj is big enough to be included in the final model. Therefore, the latent variable γ can perform variable selection for the regression model. The errors of probes belonging to the same gene can be heteroscedastic through the bin assignment. Through BASIS, we can identify transcript isoforms that are differentially distributed between two sub-cellular locations. The details about BASIS can be found in Zheng et al. (5). Hyperparameters of the model include (τ, ψ, p, ν, λ) and we chose the default parameters. We used 100 as the initial value for each δm. A total of 10 000 burn-in iterations followed by 40 000 iterations were generated to estimate the posterior probabilities.
Transcript isoforms with prob (γ = 1 | data) > 0.5 were declared differentially distributed. Then, for each gene, the R2-value was used to assess the overall fit of the regression model, which includes the declared differentially distributed isoforms only. The regressions with R2 < 0.5 were excluded from further analyses. If the Δβ estimate for the differentially expressed transcript was positive, the isoform was declared to be enriched in the cytosol (or cytosolic isoforms). Otherwise, the isoform was declared to be enriched in the nucleus (or nuclear isoforms). The predictions are accessible athttp://www-rcf.usc.edu/∼liangche/software.html. Other statistical tests were performed using R software.
Other data sources
The PhastCons conservation score (40) was downloaded from the UCSC Genome Browser (http://genome.ucsc.edu/, hg18). The score of each site is the posterior probability that the site is in the conserved state of the phylogenetic hidden Markov model for 17 vertebrates. The average PhastCons score for the positions of introns was used as the conservation measure for introns. The average PhastCons score across every exonic position of transcript isoforms was used to represent the conservation level of transcripts.
The predicted EvoFold structures (41) were downloaded from the UCSC Genome Browser. These are the predicted functional RNA secondary structures based on probabilistic models of RNA secondary structure and primary sequence evolution across eight vertebrates.
The miRNA target sites were downloaded from the miRBase Targets database (http://microrna.sanger.ac.uk/targets/v5/). These are the predicted miRNA targets that are highly complementary sites to the given miRNA sequences, and the sites are conserved across species. The miRNA expression data was from the Landgraf et al. (42). Landgraf and colleagues cloned and sequenced more than 330 000 small RNA sequences from 256 small RNA libraries. The cloning frequencies of the miRNAs were used as a measure of the miRNA expression for more than 300 distinct mature human miRNAs. In total, there were 96 miRNAs with a least one clone in the HepG2 cell line and 104 miRNAs present in the HeLa cell line.
Isotope-labeled RT–PCR
We identified the unique exon of the CAPN1 transcript TRAN00000084965 and the BCL6 transcript ENST00000232014. Thus, the expression level of this exclusively included exon can represent the expression level of the transcript isoform that includes it. A different exon that does not belong to the tested isoform but belongs to a non-differentially localized isoform was used as a control. The nuclear and cytosolic poly(A)+ RNAs of HepG2 cells were provided by Dr Gingeras’ group. The nuclear and cytosolic RNAs were DNase I (Roche) treated and reverse transcribed with SuperScript III (Invitrogen) according to the manufacturer’s instructions. The PCR reactions contained 200 000–500 000 counts per minute of the 32P-labeled common primer. Multiplex PCR of the test transcript and the control transcript used one common primer and two specific primers. The PCR reactions were run in an MJ Research PTC-200 thermocycler for 22–26 cycles, depending on the gene expression levels with an annealing temperature of 60°C. The PCR reactions were mixed 1:1 with 95% formamide and loaded onto 8% polyacrylamide, 7.5 M urea gels and electrophoresed. Subsequently, the gels were dried and imaged on a Typhoon PhosphorImager (Molecular Dynamics). The primer sequences are as follows: CAPN1: (F) CGGATGACCCGGACGACTAC, (R1) GGGGTGTGTGACTGCAGGTG, (R2) TGCTGACCTCTCGCAGGTTG; BLC6: (F1) AAGTCCTCCCCTGCCACGTA, (F2) TCCCTTCCCCACTTCCTTCC, (R) TGCCTTGCTTCACAGTCCAA.
RESULTS
The hierarchical Bayesian model BASIS was developed to infer differentially distributed transcript isoforms on the individual isoform level. In BASIS, we introduced a latent variable γ = (γ1, …, γs)T where γj = 1 means that the j-th isoform is differentially distributed and γj = 0 means that it is not differentially distributed. The probes for the same gene can be assigned to different bins and each bin has a unique variance parameter. Therefore, BASIS can handle the heteroscedastic errors of probe intensity. A homogeneous ergodic Markov chain was generated by our Gibbs Sampler. The details can be found in our previous work with a discussion about the convergence and the robustness of the model (5).
HepG2 and HeLa cells contain different nuclear and cytosolic isoforms
Among the 67 680 isoforms that passed the pre-screening procedures and were considered in BASIS for HepG2 cells, 7590 of them were nuclear isoforms and 6901 of them were cytosolic isoforms (Figure 1A). Among the 70 902 isoforms considered for HeLa cells, 11 529 were nuclear isoforms and 10 139 were cytosolic isoforms. To understand whether the differential nucleocytosolic compartmentalization was at the gene level or at the transcript isoform level, we separated genes into four categories according to whether they contained nuclear isoforms and/or cytosolic isoforms. In the HepG2 cell line (Figure 1B), 25.0% of the considered genes had both nuclear and cytosolic isoforms (category 1), 7.3% of them had cytosolic isoforms but no nuclear isoforms (category 2), and 2.7% of them had nuclear isoforms but no cytosolic isoforms (category 3). The remaining genes (65%) did not have nuclear isoforms or cytosolic isoforms (category 4). The percentages for HeLa cells were 35.9% for category 1, 11.9% for category 2, 5.6% for category 3 and 46.6% for category 4 (Figure 1C). Apparently, a majority of genes with differentially distributed isoforms (categories 1 to 3) contained both nuclear isoforms and cytosolic isoforms (category 1). In addition, many genes in categories 1–3 contained isoforms that were not differentially distributed between the nucleus and the cytosol, further indicating the multiplicity in the mRNA nucleocytosolic distribution for a given gene. Therefore, the differential compartmentalization of mRNA is not gene-specific but instead is alternative isoform-specific. Meanwhile, we also observed the cell-specific nucleocytosolic compartmentalization of transcript isoforms. Despite some overlap, HepG2 and HeLa contained different nuclear and cytosolic isoforms (Figure 1A). Large numbers of transcript isoforms were differentially distributed in one cell line but not in the other cell line. Such a difference was not completely due to cell-specific transcription. Among the differentially distributed transcript isoforms unique to HepG2 (3729 + 4255 = 7984), 80.5% of them were present in HeLa. Among the differentially distributed transcript isoforms unique to HeLa (7107 + 8054 = 15 161), 77.2% of them were present in HepG2. The results indicate that the nucleocytosolic localization of a transcript isoform can be cell type-specific. It also implies another type of cell-specific post-transcriptional regulation. Figure 1D and E shows examples of differentially distributed isoforms, which were confirmed by isotope-labeled RT-PCR. The CAPN1 transcript TRAN00000084965 contains an alternative exon 12a and was a nuclear isoform. Another transcript isoform, ENST00000279247, contains exon 12 and was not differentially distributed. The BCL6 transcript ENST00000232014 has a promoter at exon 1a and was enriched in the nucleus. Transcript isoform ENST00000406870 has an alternative promoter at exon 1 and was not differentially distributed. Additional validation experiments for the BASIS model itself have been described by Zheng et al. (5).
Figure 1.

The differentially distributed isoforms predicted by BASIS. (A) A Venn diagram of the cytosolic and nuclear isoforms for the HepG2 and HeLa cell lines. The genes considered in the BASIS analysis for HepG2 (B) or HeLa (C) can be divided into four categories. Category 1: genes having both cytosolic isoforms and nuclear isoforms (2136 and 3258 for HepG2 and HeLa, respectively). Category 2: genes having cytosolic isoforms but no nuclear isoforms (626 and 1083). Category 3: genes having nuclear isoforms but no cytosolic isoforms (235 and 506). Category 4: genes having no differentially distributed transcript isoforms (5554 and 4234). (D)–(E) The isotope-labeled RT-PCR of the CAPN1 and BCL6 transcripts in HepG2 cells. The forward and reverse primer positions are labeled with F or R. (D) CAPN1: Exon 12a is uniquely present in the test isoform, TRAN00000084965, which was predicted to be enriched in the nucleus. Exon 12 is present in a non-differentially distributed transcript isoform, ENST00000279247. (E) BCL6: The test isoform, ENST00000232014, has a promoter at exon 1a and was predicted to be enriched in the nucleus. Another isoform, ENST00000406870, has an alternative promoter at exon 1 and was used as a control. N, nuclear samples; C, cytosolic samples.
Introns and the nucleocytosolic distribution of mRNA
The differential compartmentalization of mRNA is a manifestation of various regulatory mechanisms from nuclear export to cytosolic mRNA decay. For example, transcript isoforms enriched in the cytosol may result from an efficient nuclear export and slow mRNA decay. Although the detailed mechanisms of mRNA export remain to be clarified, it appears that pre-mRNA splicing substantially enhances the nuclear export efficiency (16). We were interested to know if the differential compartmentalization of mRNA transcript isoforms was related with pre-mRNA splicing on the genome-wide scale. We first looked at the intron numbers (i.e. the numbers of exon–exon junctions). The median number of introns was five for HepG2 cytosolic transcripts and three for HepG2 nuclear transcripts. In the HeLa cell line, the median intron number was four for the cytosolic transcripts and three for the nuclear transcripts. For the transcript isoforms enriched in the cytosol of both cell lines, the median intron number was five. For the transcript isoforms enriched in the nucleus of both cell lines, the median number decreased to three. Such a difference was significant (the P-values based on the one-sided Wilcoxon test <2.2 × 10−16). Because the genes with retained introns were not considered in our analysis (see ‘Materials and Methods’ section), the alternative splicing of retained introns cannot explain the difference. The difference is also not due to the cassette exon inclusion in which an intron is broken into one cassette exon and two shorter introns so that the intron number is increased by one. Specifically, the isoforms including cassette exons were not particularly associated with the nuclear isoforms or the cytosolic isoforms compared with the isoforms excluding cassette exons (the P-value based on the chi-squared test was 0.27 for HepG2 and 0.14 for HeLa).
We further investigated the length of the introns specific to the cytosolic or nuclear isoforms. To our surprise, the introns specific to the cytosolic isoforms were significantly longer than those specific to the nuclear isoforms (the P-values based on the one-sided Wilcoxon test <2.2 × 10−16). The median length of the introns specific to the cytosolic isoforms was about 1.7–2.9 times longer than that of the introns specific to the nuclear isoforms. In addition, the median length of the introns specific to the non-differentially distributed transcript isoforms was between those of the cytosolic isoforms and the nuclear isoforms. On the contrary, the exon lengths among the three groups were almost the same. Table 1 lists the median length of the introns and exons specific to the nuclear isoforms, cytosolic isoforms or non-differentially distributed isoforms. Because the cytosolic isoforms had more introns and their unique introns were longer, they had a much larger fraction of intronic regions at the pre-mRNA level than the nuclear isoforms (the P-values based on the one-sided Wilcoxon test <2.2 × 10−16). Figure 2 shows the boxplot of the intronic proportion. These analyses were carried out by counting the individual isoforms. We performed similar analyses by counting the individual gene loci to exclude the possible bias introduced by the different numbers of transcript isoforms for the different genes. Specifically, we averaged the intronic proportions of the nuclear isoforms (or cytosolic isoforms) from the same gene and used the mean values for the boxplots. The results were similar (Supplementary Data 1). If we excluded category 1 genes (genes with both nuclear and cytosolic isoforms) or considered category 1 genes alone, the patterns were still similar to Figure 2 (Supplementary Data 2 and 3). We have also studied the intron conservation. Based on the PhastCons (40) conservation scores, the introns specific to the cytosolic isoforms tended to be more conserved than those specific to the nuclear isoforms. The median conservation level was 0.063 versus 0.046 for HepG2 and 0.064 versus 0.051 for HeLa. The P-values based on the one-sided Wilcoxon test were <2.2 × 10−16.
Table 1.
The lengths of the introns and exons specific to the cytosolic isoforms, nuclear isoforms or non-differentially distributed isoforms
|
Median intron length (in bp) |
Median exon length (in bp) |
|||||
|---|---|---|---|---|---|---|
| Cytosol | Nucleus | Non-differentially distributed | Cytosol | Nucleus | Non-differentially distributed | |
| HepG2 | 2165 | 1088 | 1739 | 139 | 154 | 144 |
| HeLa | 2319 | 1369 | 1804 | 142 | 150 | 145 |
| Both | 2307 | 802 | 1806 | 141 | 158 | 145 |
The non-redundant introns and exons specific to the cytosolic isoforms, nuclear isoforms or non-differentially distributed isoforms were collected to calculate the median length. The isoforms are for the HepG2, HeLa or both cell lines. Some of the transcript isoforms contain no intron and did not contribute to the calculation. The introns and exons shared by the cytosolic isoforms, nuclear isoforms and non-differentially distributed isoforms were excluded from the analysis.
Figure 2.

The intronic proportions of cytosolic and nuclear isoforms. The intronic proportion was calculated as [intronic region (in bp)]/[intronic region (in bp) + exonic region (in bp)]. Note that the cytosolic isoforms have larger intronic proportions. The boxplots are for the transcripts enriched in the cytosol or the nucleus of HepG2, HeLa or both cell lines.
Functional RNA secondary structures and the nucleocytosolic distribution
RNA secondary structure is involved in mRNA nuclear export and mRNA stability. We thus studied whether the cytosolic and nuclear isoforms have distinct patterns of RNA secondary structures. The average coverage of functional RNA folds at each position of the 5′ region and the 3′ region was calculated for both the nuclear and cytosolic isoforms (Figure 3). In general, the 3′ regions were more likely to contain a functional RNA fold than the 5′ regions (median average coverage 0.0063–0.0084 versus 0.0033–0.0042). Interestingly, the cytosolic isoforms were more likely to contain a functional RNA fold in the 3′ region than the nuclear isoforms (median average coverage 0.0077–0.0084 versus 0.0063). The P-value based on the one-sided Wilcoxon test was <2.2 × 10−16 for either HepG2 cells or HeLa cells.
Figure 3.

The average functional RNA fold-coverage for the cytosolic and nuclear isoforms. For every site in a mature mRNA, x was defined as the position relative to the nearest terminal. It is positive for distances from the 5′ terminal and negative for distances from the 3′ terminal [excluding poly(A)]. The y-axis is the average RNA fold-coverage at each position for the cytosolic isoforms of HepG2 (red solid line), the nuclear isoforms of HepG2 (blue solid line), the cytosolic isoforms of HeLa (red dashed line), and the nuclear isoforms of HeLa (blue dashed line).
Potential miRNA targets and the nucleocytosolic distribution
miRNAs are important regulators of translation and mRNA degradation (33). The miRNA-mediated silencing of mRNAs occurs in the cytosol. If the miRNA-mediated mRNA degradation is superior to the nuclear export of mRNAs, we expect miRNA target transcripts to be enriched in the nucleus. Hypergeometric test was performed to test whether the nuclear or cytosolic isoforms tended to be the targets of miRNA. The results are summarized in Table 2. To our greatest surprise, the P-values for the enrichment of the cytosolic isoforms with miRNA targets were very significant (<1.0 × 10−14), whereas the P-values for the nuclear isoforms were close to one. Thus, there were many more cytosolic isoforms with miRNA target sites than expected by chance. On the contrary, there were far fewer nuclear isoforms with miRNA target sites than expected by chance. Such an analysis only takes into account whether the cis-elements of an isoform could be the miRNA target sites regardless of the miRNA expression.
Table 2.
The nucleocytosolic locations of transcripts and the presence of miRNA target sites
| With miRNA target site(s) | Without miRNA target site(s) | P-value | |
|---|---|---|---|
| Total | 65 398 | 60 862 | |
| HepG2: cytosol | 3883 | 3018 | 8.62 × 10−15 |
| HepG2: nucleus | 2991 | 4599 | 1.00 |
| HeLa: cytosol | 6121 | 4018 | 1.49 × 10−73 |
| HeLa: nucleus | 4378 | 7151 | 1.00 |
| Both: cytosol | 1747 | 897 | 2.59 × 10−51 |
| Both: nucleus | 1053 | 1894 | 1.00 |
Among the 126 260 transcript isoforms assembled from databases, 65 398 of them contain a possible miRNA target site. ‘HepG2: cytosol’ means the cytosolic isoforms in HepG2 cells. The same applies to the other groups. The counts of the transcript isoforms with or without miRNA target sites are listed. The P-value is the probability of randomly drawing the same or higher number of transcript isoforms with miRNA target sites. It was calculated based on the hypergeometric test.
It is interesting to investigate whether the physical presence of miRNA would change the results. We obtained the miRNA expression data of the HepG2 and HeLa cell lines from the Landgraf et al. (42). Both the cytosolic and nuclear isoforms were further separated into three groups: (i) isoforms with at least one target site of expressed miRNAs; (ii) isoforms with the target site(s) of only non-expressed miRNAs; and (iii) isoforms without any miRNA target sites. As shown in Figure 4, for transcript isoforms without any miRNA target sites (white bars or group 3), the ratio between the number of cytosolic isoforms and the number of nuclear isoforms was 0.66, 0.56 and 0.47 for the HepG2 cell line, the HeLa cell line and both cell lines, respectively. For the transcript isoforms that have miRNA target sites but none of the corresponding miRNAs present in the cell (grey bars or group 2), these ratios increased to 1.28, 1.39 and 1.65, respectively. If any of the corresponding miRNA(s) was present in the cell (black bars or group 1), the ratios were 1.30, 1.40 and 1.65, respectively, which are not significantly different from those of group 2. The fact that the miRNA expression does not impact these ratios indicates that the miRNA-mediated mRNA decay in the two human cell lines is not a significant contributor to the differential nucleocytosolic compartmentalization of the transcript isoforms.
Figure 4.

The expression of miRNAs does not significantly impact their target isoforms’ nucleocytosolic locations. The y-axis is the counts of the transcripts. The cytosolic isoforms (C) and the nuclear isoforms (N) of individual cell lines or both cell lines were considered. They were further divided into the transcript isoforms with at least one target site of an expressed miRNA (+, black bars), the transcript isoforms with target site(s) of non-expressed miRNA(s) (−, grey bars), and the transcript isoforms without any miRNA target site (na, white bars). The ratios of the cytosolic isoform counts to the nuclear isoform counts (C/N) are listed. Note that the C/N ratios are not significantly different between the (+) and (−) groups.
NMD and the nucleocytosolic distribution
NMD degrades abnormal mRNAs that prematurely terminate translation. Some alternative splicing events also exploit NMD to achieve quantitative post-transcriptional regulation (AS-NMD). In our studies, we identified NMD targets if a transcript has a premature termination codon (PTC) that occurs >50 nt upstream of the final splice junction. The criterion was the same as found in the work by Nagy et al. (43). About 6.8% of the cytosolic isoforms with CDS features in HepG2 were predicted to have a PTC and may trigger NMD. The percentage increased to 11.3% for the nuclear isoforms. The percentages for HeLa were 5.6 versus 10.0%, respectively. Thus, the nuclear isoforms had higher proportions of NMD targets than the cytosolic isoforms. The P-value based on the chi-squared test was 4.6 × 10−12 for HepG2 and <2.2 × 10−16 for HeLa. These results seem to be consistent with the hypothesis because NMD targets are degraded in the cytosol, they tend to be enriched in the nucleus. However, the presence of NMD targets in some cytosolic isoforms indicates that the NMD effect may be selective to certain isoforms or NMD rate is slower than the export rate for certain isoforms.
The conservation score and the nucleocytosolic distribution
We have mentioned that the introns specific to the cytosolic isoforms were more conserved than the introns specific to the nuclear isoforms. For the exonic regions, the quantile–quantile plot of the conservation levels (Figure 5A) also shows that the cytosolic isoforms were much more conserved than the nuclear isoforms. Nevertheless, the transcript isoforms belonging to one gene locus contain both shared and unique exonic regions. To further dissect the difference in these regions between the cytosolic and nuclear isoforms, we then focused on the genes that had both the cytosolic and nuclear isoform(s) (category 1 in Figure 1B and C). Such a gene has three different types of exonic regions: exonic regions shared by both the cytosolic and nuclear isoform(s) exonic regions unique to the cytosolic isoform(s) and exonic regions unique to the nuclear isoform(s). The shared exonic positions were significantly more conserved (median conservation level 0.992) than the unique exonic positions (the P-values based on the one-sided Wilcoxon test <2.2 × 10−16, Figure 5B). This most likely reflects the higher conservation in constitutive exons than in alternative exons globally (44). Interestingly, the exonic positions specific to the cytosolic isoforms were more conserved than those specific to the nuclear isoforms (median conservation level: 0.224 versus 0.002 for HepG2, 0.062 versus 0.005 for HeLa, the P-values based on the one-sided Wilcoxon test <2.2 × 10−16). In addition, we observed a bi-modal distribution of the conservation scores (see the density curves in Figure 5B). Even for the exonic positions specific to the nuclear isoforms, a small number of the positions showed high conservation levels, although the majority of them had low conservation levels. When we looked at the relative positions of these exonic positions, those specific to the cytosolic isoforms tended to be at the 3′ termini of genes (Figure 6A and D). However, the exons specific to the nuclear isoforms tended to be in the middle of genes (Figure 6B and E).
Figure 5.

The conservation scores and nucleocytosolic locations. (A) A quantile–quantile plot for the conservation scores of the cytosolic isoforms (x-axis) and those of the nuclear isoforms (y-axis). The average PhastCons score across every exonic position of a transcript isoform was used to represent the conservation level of that transcript. (B) The violin plots of the conservation scores for the exonic sites specific to the cytosol or nucleus or the exonic sites shared by the cytosolic and nuclear isoforms. Only the genes having both the cytosolic and nuclear isoforms were considered. A violin plot is similar to a boxplot except that it adds the kernel density plot of the data.
Figure 6.

The relative positions of the exonic sites. Only the genes having both cytosolic and nuclear isoforms were considered. The non-redundant exonic sites from the different transcript isoforms of the same gene were pooled together and sorted according to their genomic coordinates (from 5′–3′). The relative position of the i-th exonic site was calculated as (i − 1)/(n − 1) where n is the total number of non-redundant exonic sites. The y-axis represents the counts of exonic sites with a specific relative position. (A) and (D) are for the exonic sites specific to the cytosol. (B) and (E) are for the exonic sites specific to the nucleus. (C) and (F) are for the exonic sites shared by the nuclear and cytosolic transcripts. (A), (B) and (C) are for the HepG2 cell line and (D), (E) and (F) are for the HeLa cell line.
DISCUSSION
In this article, we have applied a hierarchical Bayesian model (BASIS) to identify differentially distributed splicing isoforms between the nucleus and cytosol in the HepG2 and HeLa cell lines. The differential distribution of transcript isoforms was cell type specific (see Figure 1A). A total of 29 652 transcript isoforms were identified to be differentially distributed in the HepG2 and HeLa cell lines. Only 19% of them were enriched in the same sub-cellular location in both cell lines. About 3% of them were enriched in one location for HepG2 but the other location for HeLa. The remaining transcript isoforms (78%) were specific to either cell line. This was not totally due to cell type-specific transcription. For those transcript isoforms differentially distributed in only one cell line, 77.2–80.5% of them were present in the other cell line. In summary, in addition to ‘being expressed’ or ‘not being expressed’, ‘being in the nucleus’ or ‘being in the cytosol’ may be another type of cell-specific gene expression regulation. The mRNA nucleocytoplasmic compartmentalization can be another fingerprint of cell identity.
Despite the cell-specific inventories of nuclear and cytosolic isoforms, the global features of the nuclear and cytosolic isoforms were similar between the two cell lines. This indicates that the two cell lines may have similar mechanisms to control the nucleocytoplasmic compartmentalization. Our genome-wide analyses revealed many differences between the nuclear and cytosolic isoforms. For example, the cytosolic isoforms had more introns than the nuclear isoforms. The intron number reflects exon–exon junction number. Thus, the results can be at least partially explained by the observation that splicing and consequently the EJC on exon–exon junction are important for an efficient nuclear export. Nevertheless, two studies reported little effect of splicing on nuclear export (45,46), so there is probably a large variation in the magnitude of the intron effect on individual transcript isoforms. Our results also show that the introns specific to the cytosolic isoforms tended to be much longer (Table 1) and more conserved than those specific to the nuclear isoforms. The total intronic proportions were also higher in the cytosolic isoforms (Figure 2). These results indicate that the involvement of introns in the nucleocytoplasmic compartmentalization involves more than just the exon–exon junction number. The intron length may be another variable leading to the large variation in the magnitude of the intron effect. But the mechanisms of how an intron affects the nucleocytoplasmic compartmentalization remain to be clarified experimentally.
The effects of mRNA decay on the nucleocytoplasmic distribution also vary depending on the trigger of the mRNA degradation in the cytosol. We have particularly explored the miRNA- and NMD-mediated mRNA decay. In either case, we had originally expected that their targets would be enriched in the nucleus instead of the cytosol. To our surprise, the cytosolic isoforms tended to have miRNA targets and the miRNA expression did not have a significant impact on the isoforms’ nucleocytosolic location. Initial studies show that the mRNA levels of miRNA target genes remain mostly unchanged in animals, but more recent studies show that miRNAs can induce some target mRNA degradation (32,33). Our results do not necessarily argue against either observation. However, they do suggest that the effect of miRNA-mediated mRNA decay is, if not modest, then at least less substantial than the effects of nuclear export and other mechanisms in determining the nucleocytoplasmic compartmentalization of the transcript isoforms. On the other hand, the process of miRNA target recognition takes place in the cytosol. One can speculate that if a transcript is a potential miRNA target, it will tend to be enriched in the cytosol so that the miRNA can act on it efficiently upon induction. The above results also indicate that the nucleocytoplasmic location may be another feature we can integrate into miRNA target prediction. In contrast to the miRNA effect, NMD-mediated decay was significantly more associated with the nuclear isoforms, although some NMD targets were also cytosolic isoforms.
The comparison of the evolutionary conservation shows that the cytosolic isoforms tended to be more conserved than the nuclear isoforms (Figure 5). Parmley et al. (47) found that the evolutionary rates are reduced near intron–exon boundaries and are reduced in intron-rich genes more generally. Thus, the cytosolic isoforms are consequently more conserved because they contain more introns. Marais et al. (48) found a negative correlation between intron size and exon evolution in Drosophila. Our discoveries that cytosolic transcripts have longer introns and more conserved exons in human cells are consistent with their findings. The results also imply that nuclear isoforms may contain more relatively newly evolved transcript isoforms or ‘junk’ transcript isoforms resulting from random splicing events. We also note that some of the exonic positions specific to the nuclear isoforms were highly conserved (Figure 5B). Most likely there is a functional significance for the enrichment of these isoforms in the nucleus. In order to prevent inappropriate or malicious protein expression, these transcript isoforms may not be able to pass scrutiny during nuclear export. This eventually results in their enrichment in the nucleus. The enriched functional RNA folds, and miRNA targets in the cytosolic isoforms further indicate the selection pressure on the cytosolic isoforms.
Several of our results indicate that the 3′ UTRs are important for the transcript enrichment in the cytosol. First, the exonic positions specific to the cytosolic isoforms were enriched in the 3′ regions (Figure 6A and D). However, the exonic positions specific to the nuclear isoforms were relatively more evenly distributed and with a slight excess in the middle regions of genes (Figure 6B and E). Second, the cytosolic isoforms tended to have more functional RNA folds in their 3′ region than the nuclear isoforms, perhaps because those RNA folds contain mRNA localization signals, facilitate mRNA export, or stabilize the mRNA in the cytosol. The detailed mechanisms need to be explored through experimental approaches.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (P50 HG 002790); the American Federation for Aging Research Grant; start-up fund from USC. Funding for open access charge: A start-up fund from University of Southern California.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
I thank Michael Waterman (USC) and Hongyu Zhao (Yale) for scientific critiques during the manuscript preparation. I would also like to acknowledge Thomas Gingeras’ group (Affymetrix) for providing the nuclear and the cytosolic mRNA of the HepG2 cell line. I would like to specially thank Doug Black (UCLA) for his scientific advice on the study and generous support for the validation experiments. I also thank Sika Zheng (UCLA) for his help for the validation experiments.
REFERENCES
- 1.Moore MJ. From birth to death: the complex lives of eukaryotic mRNAs. Science. 2005;309:1514–1518. doi: 10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- 2.Black DL. Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000;103:367–370. doi: 10.1016/s0092-8674(00)00128-8. [DOI] [PubMed] [Google Scholar]
- 3.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 5.Zheng S, Chen L. A hierarchical Bayesian model for comparing transcriptomes at the individual transcript isoform level. Nucleic Acids Res. 2009;37:e75. doi: 10.1093/nar/gkp282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermuller J, Hofacker IL, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–1488. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- 7.Fong YW, Zhou Q. Stimulatory effect of splicing factors on transcriptional elongation. Nature. 2001;414:929–933. doi: 10.1038/414929a. [DOI] [PubMed] [Google Scholar]
- 8.Furger A, O'S;ullivan JM, Binnie A, Lee BA, Proudfoot NJ. Promoter proximal splice sites enhance transcription. Genes Dev. 2002;16:2792–2799. doi: 10.1101/gad.983602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwek KY, Murphy S, Furger A, Thomas B, O'G;orman W, Kimura H, Proudfoot NJ, Akoulitchev A. U1 snRNA associates with TFIIH and regulates transcriptional initiation. Nat. Struct. Mol. Biol. 2002;9:800–805. doi: 10.1038/nsb862. [DOI] [PubMed] [Google Scholar]
- 10.Rigo F, Martinson HG. Functional coupling of last-intron splicing and 3′-end processing to transcription in vitro: the poly(A) signal couples to splicing before committing to cleavage. Mol. Cell. Biol. 2008;28:849–862. doi: 10.1128/MCB.01410-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rose AB, Elfersi T, Parra G, Korf I. Promoter-proximal introns in Arabidopsis thaliana are enriched in dispersed signals that elevate gene expression. Plant Cell. 2008;20:543–551. doi: 10.1105/tpc.107.057190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosonina E, Ip JY, Calarco JA, Bakowski MA, Emili A, McCracken S, Tucker P, Ingles CJ, Blencowe BJ. Role for PSF in mediating transcriptional activator-dependent stimulation of pre-mRNA processing in vivo. Mol. Cell. Biol. 2005;25:6734–6746. doi: 10.1128/MCB.25.15.6734-6746.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Millevoi S, Decorsiere A, Loulergue C, Iacovoni J, Bernat S, Antoniou M, Vagner S. A physical and functional link between splicing factors promotes pre-mRNA 3′ end processing. Nucleic Acids Res. 2009;37:4672–4683. doi: 10.1093/nar/gkp470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Proudfoot NJ, Furger A, Dye MJ. Integrating mRNA processing with transcription. Cell. 2002;108:501–512. doi: 10.1016/s0092-8674(02)00617-7. [DOI] [PubMed] [Google Scholar]
- 15.Le Hir H, Gatfield D, Izaurralde E, Moore MJ. The exon–exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J. 2001;20:4987–4997. doi: 10.1093/emboj/20.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo MJ, Reed R. Splicing is required for rapid and efficient mRNA export in metazoans. Proc. Natl Acad. Sci. USA. 1999;96:14937–14942. doi: 10.1073/pnas.96.26.14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Z, Luo MJ, Straesser K, Katahira J, Hurt E, Reed R. The protein Aly links pre-messenger-RNA splicing to nuclear export in metazoans. Nature. 2000;407:401–405. doi: 10.1038/35030160. [DOI] [PubMed] [Google Scholar]
- 18.Valencia P, Dias AP, Reed R. Splicing promotes rapid and efficient mRNA export in mammalian cells. Proc. Natl Acad. Sci USA. 2008;105:3386–3391. doi: 10.1073/pnas.0800250105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryu WS, Mertz JE. Simian virus 40 late transcripts lacking excisable intervening sequences are defective in both stability in the nucleus and transport to the cytoplasm. J. Virol. 1989;63:4386–4394. doi: 10.1128/jvi.63.10.4386-4394.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rafiq M, Suen CK, Choudhury N, Joannou CL, White KN, Evans RW. Expression of recombinant human ceruloplasmin—an absolute requirement for splicing signals in the expression cassette. FEBS Lett. 1997;407:132–136. doi: 10.1016/s0014-5793(97)00325-6. [DOI] [PubMed] [Google Scholar]
- 21.Tokunaga K, Shibuya T, Ishihama Y, Tadakuma H, Ide M, Yoshida M, Funatsu T, Ohshima Y, Tani T. Nucleocytoplasmic transport of fluorescent mRNA in living mammalian cells: nuclear mRNA export is coupled to ongoing gene transcription. Genes Cells. 2006;11:305–317. doi: 10.1111/j.1365-2443.2006.00936.x. [DOI] [PubMed] [Google Scholar]
- 22.Braddock M, Muckenthaler M, White MR, Thorburn AM, Sommerville J, Kingsman AJ, Kingsman SM. Intron-less RNA injected into the nucleus of Xenopus oocytes accesses a regulated translation control pathway. Nucleic Acids Res. 1994;22:5255–5264. doi: 10.1093/nar/22.24.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsumoto K, Wassarman KM, Wolffe AP. Nuclear history of a pre-mRNA determines the translational activity of cytoplasmic mRNA. EMBO J. 1998;17:2107–2121. doi: 10.1093/emboj/17.7.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nott A, Le Hir H, Moore MJ. Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes Dev. 2004;18:210–222. doi: 10.1101/gad.1163204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao C, Hamilton T. Introns regulate the rate of unstable mRNA decay. J. Biol. Chem. 2007;282:20230–20237. doi: 10.1074/jbc.M700180200. [DOI] [PubMed] [Google Scholar]
- 26.Kataoka N, Diem MD, Kim VN, Yong J, Dreyfuss G. Magoh, a human homolog of Drosophila mago nashi protein, is a component of the splicing-dependent exon–exon junction complex. EMBO J. 2001;20:6424–6433. doi: 10.1093/emboj/20.22.6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuersten S, Goodwin EB. The power of the 3′ UTR: translational control and development. Nat. Rev. Genet. 2003;4:626–637. doi: 10.1038/nrg1125. [DOI] [PubMed] [Google Scholar]
- 28.Van de Bor V, Davis I. mRNA localisation gets more complex. Curr. Opin. Cell Biol. 2004;16:300–307. doi: 10.1016/j.ceb.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Jansen RP. mRNA localization: message on the move. Nat. Rev. Mol. Cell Biol. 2001;2:247–256. doi: 10.1038/35067016. [DOI] [PubMed] [Google Scholar]
- 30.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 31.Pillai RS, Bhattacharyya SN, Filipowicz W. Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 2007;17:118–126. doi: 10.1016/j.tcb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 32.Wu L, Fan J, Belasco JG. MicroRNAs direct rapid deadenylation of mRNA. Proc. Natl Acad. Sci. USA. 2006;103:4034–4039. doi: 10.1073/pnas.0510928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006;20:515–524. doi: 10.1101/gad.1399806. [DOI] [PubMed] [Google Scholar]
- 34.Baker KE, Parker R. Nonsense-mediated mRNA decay: terminating erroneous gene expression. Curr. Opin. Cell Biol. 2004;16:293–299. doi: 10.1016/j.ceb.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 35.Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 2004;36:1073–1078. doi: 10.1038/ng1429. [DOI] [PubMed] [Google Scholar]
- 36.Rebbapragada I, Lykke-Andersen J. Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr. Opin. Cell Biol. 2009;21:394–402. doi: 10.1016/j.ceb.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 37.Lewis BP, Green RE, Brenner SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc. Natl Acad. Sci. USA. 2003;100:189–192. doi: 10.1073/pnas.0136770100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan Q, Saltzman AL, Kim YK, Misquitta C, Shai O, Maquat LE, Frey BJ, Blencowe BJ. Quantitative microarray profiling provides evidence against widespread coupling of alternative splicing with nonsense-mediated mRNA decay to control gene expression. Genes Dev. 2006;20:153–158. doi: 10.1101/gad.1382806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stamm S, Riethoven JJ, Le Texier V, Gopalakrishnan C, Kumanduri V, Tang Y, Barbosa-Morais NL, Thanaraj TA. ASD: a bioinformatics resource on alternative splicing. Nucleic Acids Res. 2006;34:D46–D55. doi: 10.1093/nar/gkj031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Felsenstein J, Churchill GA. A Hidden Markov Model approach to variation among sites in rate of evolution. Mol. Biol. Evol. 1996;13:93–104. doi: 10.1093/oxfordjournals.molbev.a025575. [DOI] [PubMed] [Google Scholar]
- 41.Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, Lander ES, Kent J, Miller W, Haussler D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput. Biol. 2006;2:e33. doi: 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci. 1998;23:198–199. doi: 10.1016/s0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- 44.Chen L, Zheng S. Identify alternative splicing events based on position-specific evolutionary conservation. PLoS ONE. 2008;3:e2806. doi: 10.1371/journal.pone.0002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nott A, Meislin SH, Moore MJ. A quantitative analysis of intron effects on mammalian gene expression. RNA. 2003;9:607–617. doi: 10.1261/rna.5250403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu S, Cullen BR. Analysis of the stimulatory effect of splicing on mRNA production and utilization in mammalian cells. RNA. 2003;9:618–630. doi: 10.1261/rna.5260303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parmley JL, Urrutia AO, Potrzebowski L, Kaessmann H, Hurst LD. Splicing and the evolution of proteins in mammals. PLoS Biol. 2007;5:e14. doi: 10.1371/journal.pbio.0050014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marais G, Nouvellet P, Keightley PD, Charlesworth B. Intron size and exon evolution in Drosophila. Genetics. 2005;170:481–485. doi: 10.1534/genetics.104.037333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.