

Abstract
Application of electric fields tangent to the plane of a confined patch of fluid bilayer membrane can create lateral concentration gradients of the lipids. A thermodynamic model of this steady-state behavior is developed for binary systems and tested with experiments in supported lipid bilayers. The model uses Flory’s approximation for the entropy of mixing and allows for effects arising when the components have different molecular areas. In the special case of equal area molecules the concentration gradient reduces to a Fermi–Dirac distribution. The theory is extended to include effects from charged molecules in the membrane. Calculations show that surface charge on the supporting substrate substantially screens electrostatic interactions within the membrane. It also is shown that concentration profiles can be affected by other intermolecular interactions such as clustering. Qualitative agreement with this prediction is provided by comparing phosphatidylserine- and cardiolipin-containing membranes.
Electric fields can be used to reorganize molecules in fluid lipid bilayer membranes. In 1977 Poo and Robinson (1) observed that application of an electric field caused the membrane-bound protein, Con A, to congregate on one side of a living cell. The distribution of protein at steady-state results from a balance between field-induced drift and diffusion within the confined membrane area (2, 3). The shape of a field-induced concentration profile thus contains information about the forces affecting molecules in the membrane. However, the small size and complexity of native cell membranes hinders analysis of these profiles. Recently, electric fields have been used to reorganize and concentrate lipids and proteins in supported bilayers where quantitative analysis is greatly simplified (4, 5).
Supported bilayers can be created by spontaneous fusion of unilamellar phospholipid vesicles with an appropriate substrate such as silica (6, 7). The resulting membrane is typically separated from the solid substrate by a thin (10 Å) layer of water (8–10) and retains many of the properties of free membranes, including lateral fluidity. The fluidity is macroscopically long-range with mobile components of both leaflets of the bilayer freely diffusing over the entire surface of the support. The diffusive mixing and flow of molecules within the membrane can be confined by imposing barriers on this lateral motion. Manually scratching the membrane-coated surface can effectively create such barriers. Alternatively, more precisely partitioned membranes have been formed by using pre-patterned substrates to impose structure onto the membrane (11). Application of electric fields tangent to the plane of supported membranes partitioned by either method causes molecules to reorganize within the confined membrane corrals. The size and geometry of these corrals can be tailored to allow precise analysis of the field-induced concentration profiles.
In this work, a general description of the steady-state electric field-induced reorganization of molecules in a two-component planar bilayer membrane is developed. The model is based on thermodynamic equilibrium and revolves around the use of area fractions in place of mole fractions following Flory’s approximation for the entropy of mixing in a binary system composed of differently sized molecules. This formulation describes the behavior of integral membrane components and is important for the study of systems where large size differences between components may be present (protein in a lipid membrane, for example). It differs from the steric exclusion model, introduced by Ryan et al. (3), which applies specifically to peripheral membrane proteins.
The present model accurately describes experimental results and provides some useful predictions. In particular, molecular clustering is expected to produce observable effects on the shape of the steady-state concentration profile. Qualitative comparison of phosphatidylserine- and cardiolipin-containing membranes is in agreement with calculations. A corollary of these results is that the membrane systems studied exist in a homogeneous phase, coexisting phases were not observed.
MATERIALS AND METHODS
Planar supported bilayers were formed by fusion of small unilamellar vesicles (SUV) with clean glass coverslips (Corning). The SUVs were made roughly according to the Barenholz procedure (12). A lipid solution in chloroform was evaporated onto the walls of a round bottom flask, which then was evacuated overnight. Lipids were resuspended in distilled water by vortexing moderately for several minutes. The lipid concentration at this point was around 6 mg/ml. The lipid dispersion then was probe-sonicated to clarity on ice. The SUVs were separated from other lipid structures by ultracentrifugation for 3 hr at 192,000 g. The supernatant contained the SUVs with yields of 50–75%. SUVs were stored at 4°C and typically were stable for a few weeks to several months. Before use, the SUV suspensions were exchanged into the desired buffered salt solution, generally 5 mM Tris, pH 8.0, 50 mM NaCl. l-α phosphatidylcholine from egg (egg-PC), 1,2-dioleoyl-sn-glycero-3-[phospho-l-serine], sodium salt (DOPS) and cardiolipin, sodium salt were obtained from Avanti Polar Lipids. The fluorescent probes, N-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (NBD-PE) and N-(Texas Red sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (Texas Red DHPE) were obtained from Molecular Probes.
Glass surfaces were prepared for membrane deposition by boiling in a 1:4 dilution of ICN 7X for 20 min and rinsing extensively with deionized water followed by a 3-min etch in argon plasma. The bilayer was allowed to self-assemble by placing the coverslip over an 80-μl drop of vesicle suspension in a Petri dish for several minutes. The dish then was carefully filled with distilled water, and the excess vesicles were rinsed away by shaking gently. Confined regions were created by scratching the membrane-coated surface with a pair of tweezers. The supported bilayer next was assembled into a sandwich with another coverslip, taking care not to expose the membrane-coated surface to air.
The coverslip sandwich was mounted in a membrane electrophoresis cell and field strengths of 10–30 V/cm were applied. The electrophoresis cell was similar to previously described versions (5) with slight modification to accommodate use of an inverted microscope. Currents were monitored with a Keithly picoammeter (Cleveland, OH) and were typically between 1 and 4 μA for a 25-mm square coverslip sandwich. This corresponds to a total power dissipation of order 10−4 W, which should produce a negligible amount of Joule heating. Membranes were viewed at room temperature with a Zeiss Axiovert 100 epifluorescence microscope equipped with a Princeton Instruments (Trenton, NJ) TK512D back-illuminated charged-coupled device camera. Images were acquired and analyzed by using Image1/Metamorph from Universal Imaging Corporation (West Chester, PA).
Thermodynamic Model.
We construct expressions for the chemical potential of each component in the binary mixture by using an entropy term that takes account of molecular size differences. The equilibrium concentration profiles are obtained by imposing the condition that the gradient of this potential is zero.
Flory has shown that the entropic free energy of mixing in ideal liquids composed of differently sized molecules can be written in terms of the area fractions, ϕi, and the number of molecules, Ni, of each component,
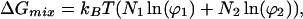 |
1 |
where kB is the Boltzmann constant and T is the temperature (13). Here
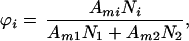 |
2 |
where Ami is the molecular area of component i. The entropic contribution to the chemical potential, μSi, can be obtained by differentiating Eq. 1 with respect to Ni:
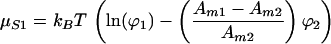 |
3 |
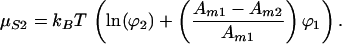 |
4 |
In addition to the logarithmic term, there is a second term depending on the size difference of the two components.
Application of an electric field tangent to the membrane plane imparts a force on each molecule. It has been shown that this electrophoretic force is the result of an electrical force acting directly on the charged molecules and an electroosmotic drag, produced by frictional coupling to the bulk electroosmotic flow (14, 15). Assuming the total force per unit area acting on component i, fi, is independent of lateral position in the membrane, the contribution to the chemical potential from the electric field is Amifir where r denotes the position along the direction of the field. Additionally, lateral pressure in the membrane, Π, contributes AmiΠ to the chemical potential. Summing up the entropic, electrophoretic, and pressure terms we obtain the following expressions for the chemical potential:
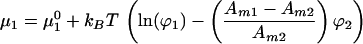 |
5 |
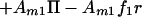 |
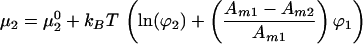 |
6 |
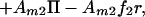 |
where μ10 and μ20 are the chemical potentials of pure membranes of component 1 and 2, respectively.
At equilibrium, the chemical potential gradients are zero:
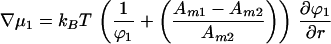 |
7 |
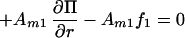 |
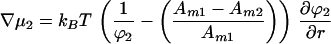 |
8 |
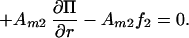 |
Combining Eqs. 7 and 8 to eliminate the pressure term we obtain
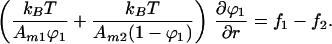 |
9 |
Solutions to this equation can be written in the form
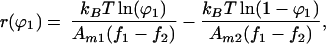 |
10 |
where r(ϕ1) is the relative position in the concentration profile at which the area fraction of component 1 is ϕ1. It is natural to define the concentration profile as ϕ1(r); thus Eq. 10 is the inverse concentration profile function. The boundary condition on Eq. 10 for a confined region, ℛ, requires that the total amount of each molecular component remain constant:
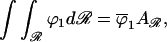 |
11 |
where ϕ̄1 is the average area fraction of component 1 and Aℛ is the area of the confined region.
In the special case when both molecular components are of equal size, Eq. 10 can be inverted:
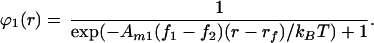 |
12 |
This is the familiar Fermi–Dirac distribution where the energy function, −Am1(f1 − f2)r, is the electrophoretic potential of component 1 relative to component 2. The boundary requirement is met by adjusting the parameter, rf. The quantity, −Am1(f1 − f2)rf, has units of energy and is analogous to the Fermi level in quantum mechanical systems. The similarity between field-induced concentration profiles in a membrane and the Fermi–Dirac distribution provides a convenient way of conceptualizing these systems (Fig. 1). In the limit as the entropic contribution to the chemical potential goes to zero (T → 0), the concentration profile takes on the form of a step function (Fig. 1B). Without any thermal energy to counter the electrophoretic forces, the two components will separate completely with the location of the boundary, rf, determined only by the total area occupied by each component. At increased temperatures some of the molecules will be excited into higher energy positions in the electrophoretic potential gradient, thus producing a smooth distribution (Fig. 1C). The value of rf is temperature dependent and can be obtained by integrating Eq. 12 and imposing the boundary requirement (Eq. 12). Eq. 10 thus describes a general class of functions resembling the Fermi–Dirac distribution in which the effect of differently sized molecules is included.
Figure 1.
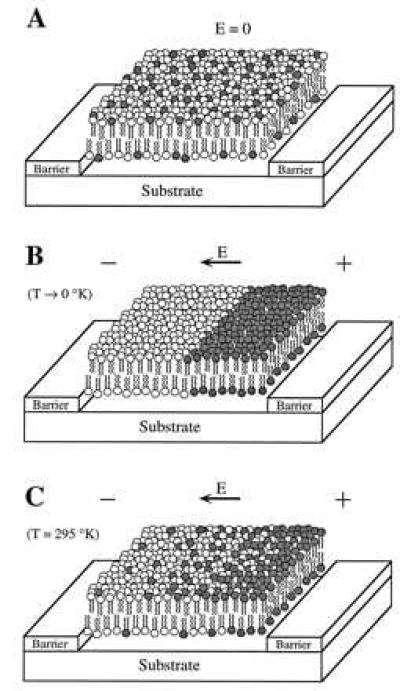
(A) Schematic diagram of a supported bilayer laterally confined by barriers. Before application of a field both components are fully mixed and the membrane is of uniform composition. (B) Hypothetical depiction of the molecular reorganization induced by a field in the limit as thermal energy goes to zero. Complete separation of the two components is shown with the negative lipids (shaded dark) gathering toward the anode. The boundary separating the two components has analogies to the Fermi level in quantum mechanical systems. (C) Under normal conditions thermal energy causes a substantial amount of mixing around the boundary, giving rise to a smooth distribution.
Another convenient parallel can be drawn between Eq. 12 and earlier descriptions of steady-state concentration profiles as a competition between electric field-induced drift and diffusion (4). In the dilute limit, the drift velocity of component 1 relative to component 2, vd, is given by
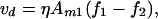 |
13 |
where η is the mobility. Combining Eq. 13 with the Einstein relation for the diffusion coefficient, D = ηkBT, we find that
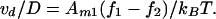 |
14 |
Hence, Eq. 12 can be rewritten in terms of the drift velocity and the diffusion coefficient:
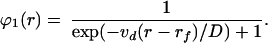 |
15 |
The exponential form of the concentration profile (4), is a dilute limit approximation to Eq. 15.
Electrostatic Effects.
The effect of electrostatic interactions between charged molecules within the membrane can be calculated by including their contribution to the chemical potential. Letting the molecular charge of component 1 be zm1e, Eq. 5 becomes:
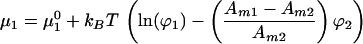 |
16 |
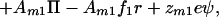 |
where ψ is the surface potential of the membrane. Imposing the equilibrium requirement that the gradients of the chemical potentials are zero and eliminating the pressure terms as done previously, we find
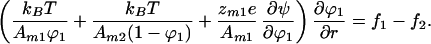 |
17 |
Here we have taken component 2 to be neutral because this is a common experimental situation. Eq. 17 can be solved giving
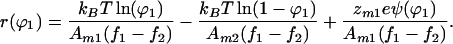 |
18 |
Calculating the effect of electrostatic interactions within the membrane is thus reduced to determining ψ(ϕ1).
At low ionic strengths, Guoy–Chapman theory provides a suitable model of the electrostatic environment near the membrane (16, 17). The surface potential is related to the surface charge density, σ, by the Guoy equation:
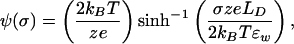 |
19 |
where z is the valence of the symmetrical electrolyte solution, e is the elementary charge, ɛw is the dielectric constant of water, and LD is the Debye length defined as LD = (kBTɛw/2Iz2e2)1/2 with I denoting the ionic strength of the bulk solution. The implicit assumption is that σ can be treated as a uniform charge distribution. This assumption is sensible as long as the Debye length is large compared with the average spacing between charged molecules. For the low ionic strengths typically used in these experiments, Debye lengths are in the range of 10–100 nm, suggesting that the Guoy equation should apply for densities of charged lipids above a few percent. At lower concentrations of charged molecules in the bilayer, it is expected that the apparent screening would be greater than that predicted by Guoy–Chapman theory.
For supported membranes, the surface charge density has contributions from both the membrane and the substrate:
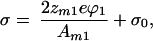 |
20 |
where the factor of 2 corresponds to the two leaflets of the bilayer membrane, zm1 is the valence of the charged component in the membrane, and σ0 represents the charge contribution from the substrate. We will make the simplifying assumption that all charges on the substrate and in the membrane can be treated as though they were in a single plane, thus neglecting any screening that occurs over the 1-nm layer of water separating the membrane from the substrate. Again, this is expected to be a reasonable approximation under low ionic strength (bulk solution < 1 mM) working conditions.
Fig. 2 depicts calculated concentration profiles under various circumstances illustrating the predicted effect of lateral electrostatic repulsion between molecules in the membrane. The top curve represents the case where electrostatic interactions are ignored. The bottom curve was obtained by setting σ0 = 0. This is the predicted concentration profile for a free membrane with no screening from the substrate. The middle two curves illustrate different treatments of the screening effect provided by the substrate. In the second from the top, the substrate is treated as contributing a constant surface charge density, σ0 = −0.25 C/m2, based on the charge density expected on a silicon oxide surface at typical membrane formation conditions (pH = 8.0, ionic strength = 50 mM) (18). Experimentally, there is evidence suggesting that the substrate is not at equilibrium with the bulk solution (9), implying that σ0 can be approximated by a constant determined by the conditions when the membrane was formed. Here we are neglecting charge contributions from ions trapped in the 10 Å layer of water between the membrane and the support. This omission has little consequence because the screening effect from the substrate is not sensitive to the precise value of σ0. Because σ0 is relatively large, thus pushing the interface into a regime where changes in charge density have a diminished effect on the surface potential, that is the primary source of screening. The third curve was generated by explicitly calculating σ0 as a function of ϕ1 and solving Eq. 18 numerically. For this calculation it was assumed that the silicon oxide surface is at equilibrium with the bulk solution and that the membrane’s effect is simply to contribute charge density to the interface. In this case, σ0 is given by
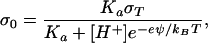 |
21 |
where σT is the density of chargeable groups on the substrate, Ka is their solution equilibrium constant, and [H+] is the proton concentration. For the membranes studied here, it is expected that the σ0 = −0.25 C/m2 calculation is the most applicable because of the apparent tendency of the supported bilayer to isolate the substrate from the bulk solution, thus fixing the surface charge density to that at the time of membrane formation.
Figure 2.
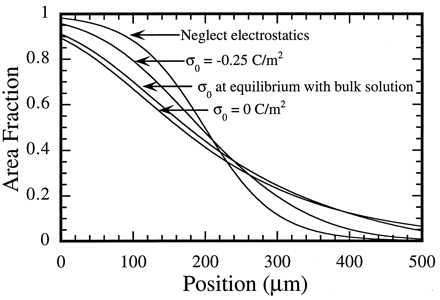
Calculated concentration profiles illustrating the effect of lateral electrostatic interactions within the membrane. Three distributions of substrate charge, σ0, are shown together with the case where electrostatic interactions between molecules are neglected. These curves were generated for a binary mixture of equally sized molecules where one carries a net charge of −1. There is a barrier at the zero position, and this is the anode side of the region. The net force per molecule is 8.9 × 10−17 N, which is comparable to forces realized experimentally. All profiles were normalized to contain the same amount of lipid.
RESULTS AND DISCUSSION
A typical electric field-induced molecular reorganization of a supported bilayer is depicted in Fig. 3. Three corrals of different sizes are seen in these images separated by scratch boundaries. Micropatterned substrates could be used to partition the membrane into more precisely defined arrays of corrals but the scratch remains a simple and effective method. Fig. 3A is an image of the membrane before application of the field. The corrals are all of uniform composition consisting of egg-PC, DOPS, and the fluorescent probe N-(Texas Red sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt in a 90:9:1 ratio. The Texas Red and the DOPS both carry a net negative charge whereas the egg-PC is neutral. When the field is turned on the charged components drift within the membrane and eventually reach an equilibrium distribution such as that shown in Fig. 3B. Equilibrium profiles generally are reached within 1 hr for field strengths of 10–30 V/cm and corrals up to 350 μm in length. The membrane in this configuration is still completely fluid and remains sensitive to changes in the field. The profile becomes steeper when the field is increased and shallower when it is decreased. The field can be reversed, causing the profile to reverse, or it can be turned off, allowing the membrane to relax back to uniformity by diffusive mixing.
Figure 3.

(A) Epifluorescence image of several corrals of fluid membrane separated by scratch boundaries. The membrane is of uniform composition in the absence of an electric field. Fluorescence is from 1 mol% of N-(Texas Red sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt doped into this egg-PC (90%) and DOPS (9%) membrane. (B) Epifluorescence image of a steady-state molecular reorganization induced by an applied field of 25 V/cm. The negatively charged components have built up concentration gradients toward the anode. A trace of the fluorescence intensity across the image is depicted below with the scratch boundaries marked with gray bars. The concentration profiles can be observed to a greater extent in the larger corrals.
Image analysis can be used to trace the fluorescence intensity of the probe, plotted in the lower panel of Fig. 3B, allowing quantitation of the concentration profile. In the far right corral an exponential profile is observed, consistent with the dilute-limit approximation described earlier. In the larger corrals, more of the profile is visible and saturation effects become noticeable. Note the shape of the profile against the left boundary of the larger corrals. It is an important feature of these experiments that the length of a corral, along with the average concentration, determines the maximum concentration and thus the extent of the profile observed. The functional form of the profile, however, is independent of corral size. It is determined entirely by the microscopic balance between forces acting on the molecules and thermal energy.
The accuracy of the thermodynamic model to describe the concentration profile was tested on a membranes containing egg-PC, DOPS, and NBD-PE in an 85:14:1 ratio. NBD-PE was chosen as the fluorescent probe based on its similarity in size and charge to DOPS so that the resulting mixture would behave as a quasi two-component system. Fluorescence from the NBD-PE can be used to quantitate the concentration profile. Because it is only a small fraction of the negatively charged lipid, fluorescence self-quenching is not a significant problem. Measurements on membrane standards containing known concentrations of NBD-PE confirm a linear relationship between fluorescence intensity and NBD-PE concentration up to 6.5 mol%. Probe concentrations remain in this range for quasi two-component systems where the probe is 1/15 or less of the negative lipid.
Quantitative analysis of a concentration profile along with a theoretical fit to the data is illustrated in Fig. 4. The theoretical curve was generated from Eq. 18 and includes electrostatic interactions within the membrane. The substrate was assumed to have a constant surface charge density of −0.25 C/m2, and the two components were assigned the same molecular area (60 Å2). All other parameters in Eq. 18 are known except the differential force (f1 − f2), which was obtained from the fit procedure. The net force acting on the charged component was found to be 8.9 ± 0.4 × 10−17 N per molecule, which is consistent with the applied field strength of 10–15 V/cm and a typical amount of electroosmotic drag (15). The total area fraction of negatively charged lipid in this profile is 13%, which is in relatively good agreement with the 15 mol% initial condition. The difference may be because of some leakage into the scratch region, discrepancy in the molecular area occupied by the various components in the membrane, or variation in the lipid composition during the vesicle preparation. Fig. 4B is a close-up view of the transition region of the profile, illustrating its shape and extension of the calculated curve beyond the boundary.
Figure 4.
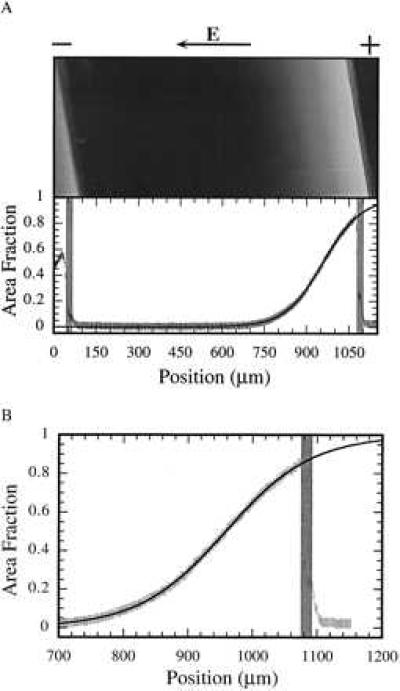
(A) Epifluorescence image of a concentration profile in a membrane containing egg-PC, DOPS, and NBD-PE in an 85:14:1 mol ratio. This system is expected to behave like a two-component system because of the similarity in size and charge of NBD-PE and DOPS. A fluorescence intensity trace across the region is shown below with the location of the scratch boundaries marked with gray bars. A theoretical fit to the data (Eq. 18) is drawn with a solid line. A net force of 8.9 ± 0.4 × 10−17 N per molecule is predicted, which is consistent with the applied field strength of 10–15 V/cm. (B) Close-up view of the transition region of the profile illustrating its shape and extension of the calculated curve beyond the boundary.
An interesting and potentially useful prediction of this model is that clustering of molecules in the membrane will have an observable effect on the shape of the concentration profile. Calculations presented in Fig. 5A demonstrate this effect for a membrane consisting of the same area fraction of charged molecules organized as monomers, dimers, or large clusters. It can be seen that progressively steeper profiles are predicted for the dimerized and clustered arrangement of the molecules. The primary reason for this effect is that the applied force on each particle scales linearly with size whereas the balancing force originates from thermal energy and is less strongly dependent on size, especially at lower concentrations. More subtle differences resulting from pressure as well as entropic effects become noticeable for large clusters. These forces create a distinctive asymmetry in the concentration profile of a binary system with unequal sized components, which could be used to extract their size ratio.
Figure 5.
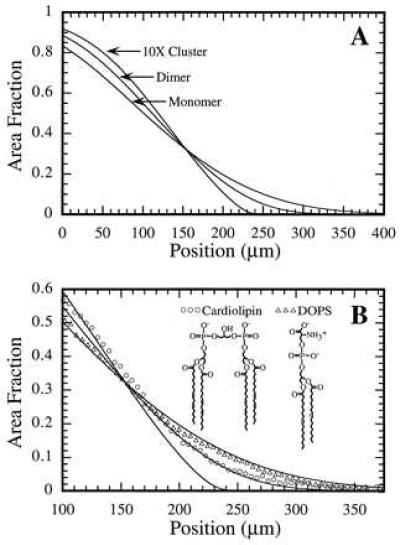
(A) Calculations illustrating the effect of molecular clustering on the concentration profile for membranes consisting of the same area fraction of charged molecules organized as monomers, dimers, or clusters of 10. The monomer was taken to have the same molecular area as the other component and a charge of −1. The net force per monomer was set to 8.9 × 10−17 N. In addition to becoming steeper with more clustering, the profiles show a greater asymmetry resulting from pressure and entropic effects. (B) Observed concentration profiles in egg-PC/DOPS/NBD-PE (85:14:1) and egg-PC/cardiolipin/NBD-PE (85:7:1) membranes under similar experimental conditions. The three solid lines are the same calculated curves shown in A. The steeper profile seen with the cardiolipin (approximating a dimer of DOPS molecules) is in agreement with the calculated profile for a dimer of DOPS molecules.
Qualitative agreement with the predicted effect of clustering can be found by comparing the egg-PC/DOPS/NBD-PE membrane described above to an egg-PC/cardiolipin/NBD-PE (85:7:1) membrane. Cardiolipin carries a net charge of −2 and has four hydrophobic tail groups; it is a reasonable approximation to a dimer of DOPS molecules. The two membranes mentioned have the same area fraction (15%) of charged lipid, but in the case of cardiolipin, the charged component is organized in dimers (each cardiolipin molecule resembling a dimer of DOPS molecules). Fig. 5B illustrates the measured difference in the shape of the concentration profiles for these two systems. Observed cardiolipin profiles were always steeper than DOPS profiles under similar conditions. This comparison is only qualitative because we are not directly observing the cardiolipin concentration profile. The observed fluorescence is from the NBD-PE probe, which is different from cardiolipin, and thus will not exactly trace the cardiolipin profile. An estimate of the cardiolipin concentration was obtained from the probe profile by normalizing the apparent plateau to 1. A full quantitative analysis requires the system be treated as a genuine three-component system.
The theoretical model described here assumes the membrane is in a homogeneous phase. That it accurately describes the observed concentration profiles confirms that the membranes examined are comprised of only a single thermodynamic phase. Preliminary calculations indicate that application of a field to a phase-separated membrane would produce a concentration profile with a discontinuity at the boundary of the two phases. Although no phase separation was observed, we have noted evidence of critical mixing effects on the concentration profile in egg-PC/cardiolipin/NBD-PE membranes.
Recent advances have substantially increased the ease and precision with which supported bilayers can be partitioned into μm-size corrals of isolated fluid membrane. These micropartitioned membranes are well suited to steady-state electric field-induced molecular reorganization experiments. The thermodynamic model presented here provides a framework for interpretation of the resulting concentration profiles, yielding insights into molecular organization within bilayer membranes.
Acknowledgments
We thank James Sabry and Jim Spudich for use of microscope imaging facilities in their lab. This work was supported in part by the National Science Foundation Biophysics Program (to S.G.B.) and by National Science Foundation Grant MCB-9316256 (to H.M.M.).
ABBREVIATIONS
- SUV
small unilamellar vesicle
- egg-PC
l-α phosphatidylcholine from egg
- DOPS
1,2-dioleoyl-sn-glycero-3-[phospho-l-serine], sodium salt
- NBD-PE
N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt
References
- 1.Poo M-M, Robinson K R. Nature (London) 1977;265:602–605. doi: 10.1038/265602a0. [DOI] [PubMed] [Google Scholar]
- 2.Jaffe L F. Nature (London) 1977;265:600–602. doi: 10.1038/265600a0. [DOI] [PubMed] [Google Scholar]
- 3.Ryan T A, Myers J, Holowka D, Baird B, Webb W W. Science. 1988;239:61–64. doi: 10.1126/science.2962287. [DOI] [PubMed] [Google Scholar]
- 4.Groves J T, Boxer S G. Biophys J. 1995;69:1972–1975. doi: 10.1016/S0006-3495(95)80067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Groves J T, Wülfing C, Boxer S G. Biophys J. 1996;71:2716–2723. doi: 10.1016/S0006-3495(96)79462-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brian A A, McConnell H M. Proc Natl Acad Sci USA. 1984;81:6159–6163. doi: 10.1073/pnas.81.19.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sackmann E. Science. 1996;271:43–48. doi: 10.1126/science.271.5245.43. [DOI] [PubMed] [Google Scholar]
- 8.Johnson S J, Bayerl T M, McDermott D C, Adam G W, Rennie A R, Thomas R K, Sackmann E. Biophys J. 1991;59:289–294. doi: 10.1016/S0006-3495(91)82222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayerl T M, Bloom M. Biophys J. 1990;58:357–362. doi: 10.1016/S0006-3495(90)82382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koenig B W, Krueger S, Orts W J, Majkrzak C F, Berk N F, Silverton J V, Gawrisch K. Langmuir. 1996;12:1343–1350. [Google Scholar]
- 11.Groves J T, Ulman N, Boxer S G. Science. 1997;275:651–653. doi: 10.1126/science.275.5300.651. [DOI] [PubMed] [Google Scholar]
- 12.Barenholz Y, Gibbes D, Litman B J, Goll J, Thompson T E, Carlson F D. Biochemistry. 1977;16:2806–2810. doi: 10.1021/bi00631a035. [DOI] [PubMed] [Google Scholar]
- 13.Guggenheim E A. Mixtures. London: Oxford Univ. Press; 1952. pp. 204–205. [Google Scholar]
- 14.McLaughlin S, Poo M-M. Biophys J. 1981;34:85–93. doi: 10.1016/S0006-3495(81)84838-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stelzle N, Miehlich R, Sackmann E. Biophys J. 1992;63:1346–1354. doi: 10.1016/S0006-3495(92)81712-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peitzsch R M, Eisenberg M, Sharp K A, McLaughlin S. Biophys J. 1995;68:729–738. doi: 10.1016/S0006-3495(95)80253-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McLaughlin S. Curr Top Membr Transp. 1977;9:71–144. [Google Scholar]
- 18.Bergna H, editor. The Colloid Chemistry of Silica. Vol. 234. Washington, DC: Am. Chem. Soc.; 1994. [Google Scholar]