

Abstract
A prothrombotic state in obesity may be partially responsible for the higher incidence of atherosclerotic complications. However the factors responsible for this prothrombotic state, linked with high levels of plasminogen activator inhibitor-1 (PAI-1), are not fully known. Leptin is elevated in obesity and studies have shown a positive correlation between leptin and PAI-1 levels in human subjects, along with a negative correlation with tissue type plasminogen activator (tPA). We tested the hypothesis that leptin induces PAI-1 and inhibits tPA expression using human coronary artery endothelial cells (HCAEC) in culture as these cells play an important role in atherosclerosis. We demonstrate that leptin induces the transcription and translation of PAI-1 in HCAEC. The leptin dependent upregulation of PAI-1 mRNA and protein was comparable to insulin-induced PAI-1 expression. We show leptin concentration (0-150 ng/ml) dependent increases in PAI-1 mRNA and protein after six and twelve hours of leptin administration respectively. Increased intracellular PAI-1 expression correlates with increased PAI-1 activity in conditioned media and inhibition of specific ERK1/2 pathway by treatment with PD98059 (20 to 40 μM) inhibits leptin dependent PAI-1 expression. However no changes in tPA expression were seen with time or increasing concentrations of leptin. Also leptin treatment did not alter total tPA concentration or tPA activity in conditioned media. In conclusion, our study shows that leptin up-regulates the expression of PAI-1 in vascular endothelial cells via activation of ERK1/2 but does not regulate tPA expression. These studies demonstrate a novel mechanism for the prothrombotic role of leptin in development of atherosclerosis.
Keywords: Leptin, Plasminogen activator inhibitor, atherosclerosis, obesity, fibrinolysis
Introduction
Obesity is associated with increased risk for cardiovascular disease and is characterized by systemic inflammation and a prothrombotic state [1; 2]. The link between obesity, inhibition of fibrinolysis, and elevation of plasminogen activator inhibitor-1 (PAI-1) is regarded as a central component for this prothrombotic state [3; 4; 5]. Several clinical studies have shown an association between high plasma PAI-1 and coronary artery disease, myocardial infarction, and recurrence of myocardial infarction [6; 7; 8; 9].
PAI-1 belongs to the superfamily of serine protease inhibitors and plays an important role in regulation of fibrinolysis and proteolysis by inhibiting tissue-type plasminogen activator (tPA), and urokinase-type plasminogen activator [9; 10]. Both these processes are important in development and progression of atherosclerosis. Studies in animal models have indicated that increased PAI-1 activity enhances thrombosis, and antibodies against PAI-1 prevent the progression of thrombosis [11; 12; 13]. PAI-1 deficiency is known to protect against atherosclerotic progression,[14] and transgenic mice that express a stable form of human PAI-1 develop coronary artery thrombosis [15]. These studies suggest an important role for PAI-1 in development of cardiovascular disease.
Obesity is also associated with high levels of leptin, an adipokine independently associated with increased risk for cardiovascular disease [16; 17; 18; 19]. Leptin has been implicated in platelet activation and aggregation as well as intravascular thrombosis [16]. A positive correlation between PAI-1 and plasma leptin has been demonstrated in men with ischemic heart disease [20], and in premenopausal women [21]. In a Swedish population based study, leptin positively correlated with PAI-1 levels and inversely with tPA levels [22]. Similar correlations between leptin, PAI-1 and tPA were also shown in a study of hypertensive overweight subjects [23]. These correlations appear independent of BMI, body fat content, age and gender, and point to a probable role of leptin in regulation of PAI-1 expression and activity. Even though several studies have demonstrated these correlations between leptin, PAI-1, and tPA levels, the cause and effect relationship has not been investigated at a molecular level. Therefore we tested the hypothesis that leptin increases PAI-1 expression and decreases tPA expression using cultured human vascular endothelial cells.
Methods
Experiments were performed using primary human coronary artery endothelial cells (HCAEC, from Cambrex, Walkersville, MD). Cells were grown in endothelial growth media-2 (EGM-2, Cambrex) supplemented with growth factors and 2 % fetal bovine serum. All experiments were performed at 3-5 passages with 70-80 % confluency after overnight incubation in serum and growth factor free media [24].
To determine the role of leptin in the regulation of PAI-1 the cells were incubated with either zero leptin and zero insulin (control), 100 ng/ml leptin (Sigma, St Louis, MI), or 100 mM insulin (positive control; Sigma, St Louis, MI) for 6 hours for mRNA and 12 hours for protein analysis [25]. For the dose dependent studies, the cells were incubated with increasing concentration of leptin (0-150 ng/ml) either for 6 hours (real time mRNA analysis), or 12 hours (intracellular protein expression analysis) or 24 hours (activity analysis in conditioned media). Buffer [15 mM HCl (500 μl), 7.5 mM NaOH (300 μl)] used to make leptin stock was used as a zero leptin vehicle control. To determine the role of mitogen activated protein kinases (MAPK) pathways on leptin dependent PAI-1 expression, specific ERK1/2 inhibitor PD98059 (20-40 μM) (Sigma, St Louis, MI) and P38 inhibitor SB203580 (5-20 μM) (Sigma, St Louis, MI) were used. The cells were treated with increasing concentrations of the inhibitors for 30 minutes prior to leptin treatment.
Real time RNA analyses were performed to determine the effect of leptin treatment on PAI-1 transcription. Total RNA was extracted from the treated cells using a RNA isolation kit (Invitrogen, Carlsbad, CA) according to manufacturer's instructions. cDNA was synthesized using 10 ng RNA per sample with high capacity cDNA Archive kit (Applied Biosystems, Foster City, CA). Commercially available TaqMan probe for PAI-1 and GAPDH (endogenous control; Applied Biosystems, Foster City, CA) were used in standard conditions as recommended by the manufacturer to determine the level of PAI-1 transcription in the treated cells. Absolute values of PAI-1 and GAPDH were calculated using standards. The PAI-1 value obtained was divided by GAPDH value (endogenous control) to obtain a normalized target value ratio. Results are expressed as fold increases as compared to vehicle (zero leptin) control.
Western blot analysis was done to quantify PAI-1 and tPA protein in experimental cell lysates. The cells were lysed immediately after each experiment using lysis buffer containing 50 mM NaCl, 50 mM NaF, 50 mM Na4O7P2, 5 mM EDTA, 5 mM EGTA, 0.1 mM Na3VO4, 1 % Triton X-100, 10 mM HEPES, pH 7.4. Equal amounts of protein from each sample were loaded and transferred to PVDF membranes. The membranes were blocked with 5 % non fat milk and incubated with specific primary antibody against PAI-1 (Molecular innovations, Southfield, MI), tPA (Molecular innovations, Southfield, MI) and GAPDH (Abcam Inc, Cambridge, MA). Membranes were then incubated with HRP-conjugated secondary antibodies and developed with enhanced chemiluminescence (Amersham biosciences, U.K.). The optical density of the band was measured using Scion Image software (Scion Corp.). The protein expression levels were normalized to GAPDH and expressed as relative densitometric units. The results are expressed as fold increases as compared to vehicle (zero leptin) control.
To determine the effect of leptin treatment on secreted PAI-1 and tPA activity, the conditioned media from each experiment was collected, centrifuged to remove any cellular debris and stored at -20 °C until further analysis. Total tPA, tPA activity and PAI-1 activity were determined in culture supernatants using commercially available assay kits (Molecular innovations, Southfield, MI). The quantitation was carried out according to the manufacturer's instructions.
All experiments were performed at least three times. Data are presented as mean ± SEM. Pairwise analysis and statistical significance was determined using Wilcoxon rank sum test, with level of significance set at p < 0.05.
Results
Leptin induces PAI-1 expression
Incubation of HCAEC in the presence of leptin (100 ng/ml) resulted in a significant increase in PAI-1 mRNA (figure 1A) and protein expression (figure 1B). Insulin was used as a positive control in our experiments [26]. We observed a comparable increase in PAI-1 transcription and translation with leptin and insulin treatment. To further evaluate the role of leptin in PAI-1 regulation, we demonstrate leptin dose-dependent effects on PAI-1 mRNA (figure 2A) (p=0.02), protein expression (figure 2B) (p= 0.01) and activity in conditioned media (figure 2C) (p=0.0001). Significant increases in PAI-1 protein are seen only at high leptin concentrations (≥50 ng/ml) and not at low leptin concentrations (≤ 20 ng/ml). These higher concentrations of leptin (≥50 ng/ml) are consistent with those observed in the obese population.
Figure 1.
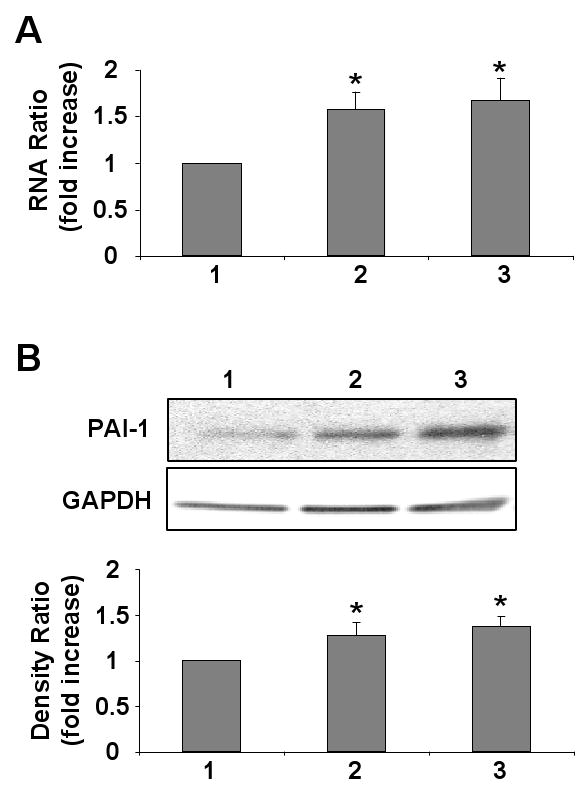
Leptin up-regulates PAI-1 transcription and translation in HCAEC. A) Quantitative mRNA analysis from four independent experiments showing increased PAI-1 mRNA transcription following leptin treatment. B) Representative Western blot and densitometry graph from four independent experiments showing increased PAI-1 expression following leptin treatment. The cells were treated with 1: Vehicle alone (control); 2: Leptin (100 ng/ml); and 3: Insulin (100 mM). Data are presented as mean ± SEM. Asterisk denotes statistical significance as determined by Wilcoxon rank sum test compared to control experiment.
Figure 2.
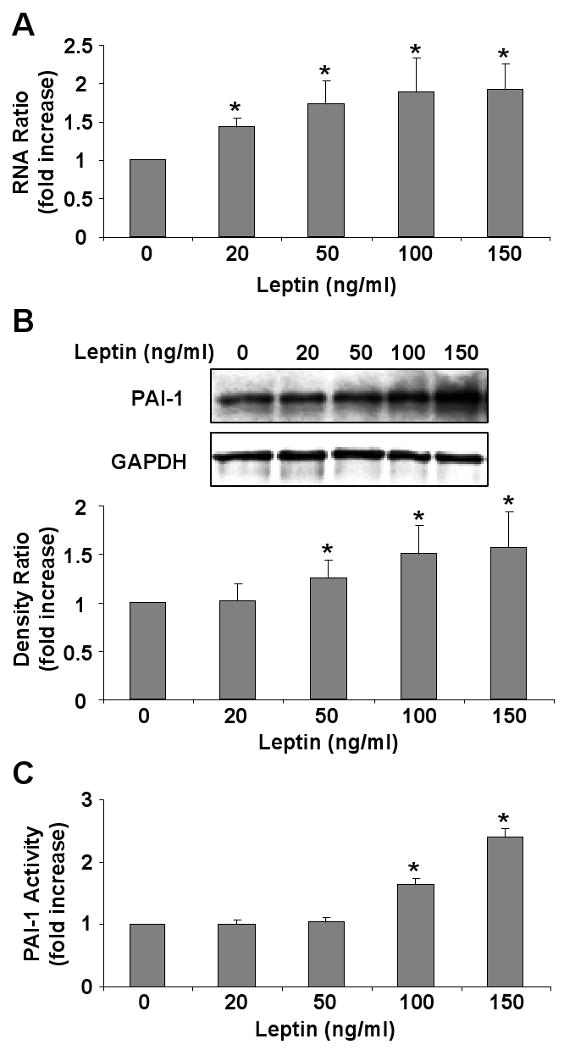
Leptin induces PAI-1 in a dose dependent manner. A) Quantitative mRNA analysis from four independent experiments showing leptin concentration dependent increases in PAI-1 mRNA. B) Representative Western blot and densitometry graph from four independent experiments showing leptin dose dependent increases in PAI-1 expression following leptin treatment. C) Graph showing increased leptin-dose dependent PAI-1 activity in conditioned media from four independent experiments. Data are presented as mean ± SEM. Asterisk denotes statistical significance as determined by Wilcoxon rank sum test compared to vehicle (zero leptin) control (p<0.05).
Leptin dependent PAI-1 expression is mediated by ERK1/2 activation
Pretreatment of HCAEC with ERK1/2 specific inhibitor PD98059 attenuated the leptin dependent increased expression of PAI-1 (figure 3A). Increasing doses of PD98059 (20-40 μM) caused a dose dependent decreases in PAI-1 protein expression (p=0.01). Maximum inhibition was seen at 40 μM concentration of PD98059 when PAI-1 expression was comparable to that of control (zero leptin) treated cells. Pretreatment with increasing concentrations of p38 inhibitor SB203580 (5-20 μM) did not change PAI-1 expression (figure 3B) (p=0.18). Our data thus show that leptin upregulates PAI-1 expression through activation of the ERK1/2 pathway and not the p38 pathway.
Figure 3.
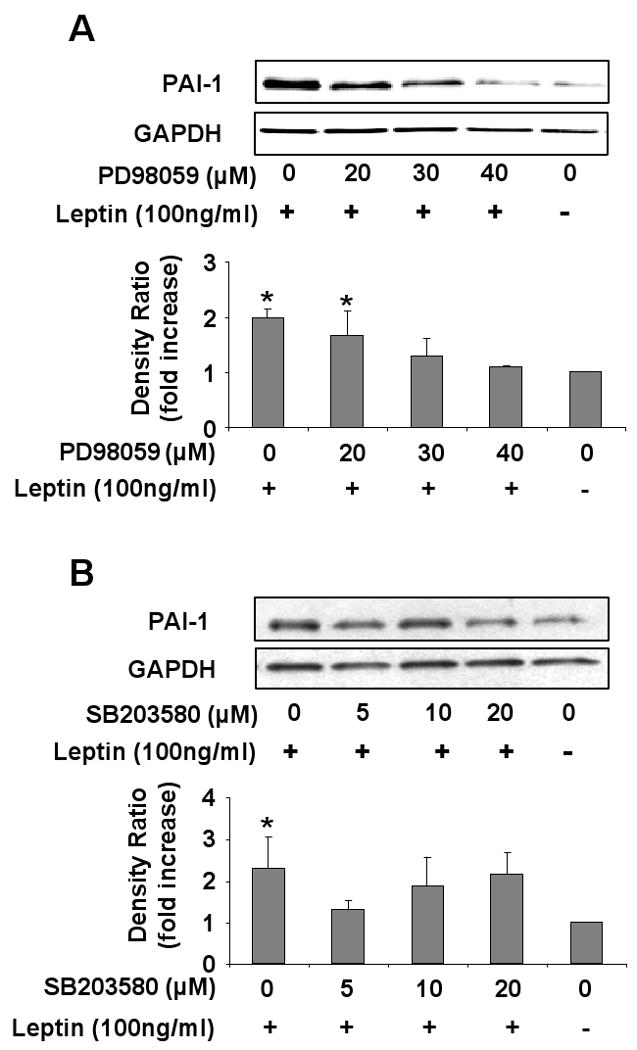
Leptin dependent PAI-1 expression is mediated by ERK1/2 pathway. A) Representative Western blot and densitometry graph from four independent experiments showing inhibition of leptin-induced PAI-1 expression in the presence of increasing concentration of ERK1/2 inhibitor (PD98059) B) Representative Western blot and densitometry graph from four experiments showing absence of any effect of increasing concentration of p38 inhibitor (SB203580) Data are presented as mean ±SEM. Asterisk denotes statistical significance as determined by Wilcoxon rank sum test compared to vehicle (zero leptin) control (p<0.05).
Leptin does not decrease tPA expression
Incubation of HCAEC with leptin did not alter tPA protein expression level at any time point up to 24 hours of treatment (figure 4A) (p=0.93) or with treatment with increasing concentrations of leptin (0-150 ng/ml) (figure 4B) (p=0.17). Also no significant changes in either total tPA (figure 4C) (p=0.31) or tPA activity (figure 4D) (p=0.10) were observed in conditioned media after 24 hours of treatment with increasing concentrations of leptin (0-150 ng/ml).
Figure 4.
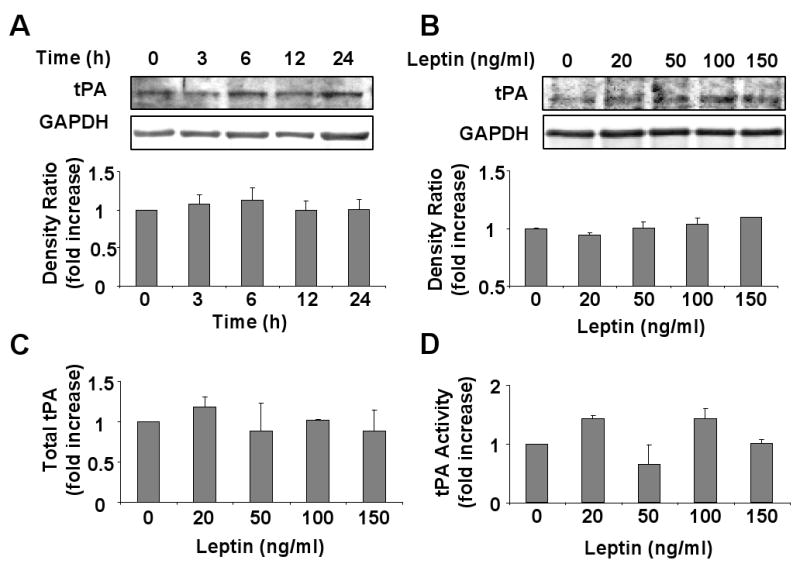
Leptin does not regulate tPA protein expression or activity. A) Representative Western blot and densitometry graph from four independent experiments showing absence of any changes in tPA expression with time after leptin (100 ng/ml) treatment. B) Representative Western blot and densitometry graph from four independent experiments showing no changes in tPA expression after treatment with increasing concentration of leptin. C) Graph from four independent experiments showing no changes in total tPA concentration in the conditioned media after treatment with increasing leptin concentrations. D) Graph from four independent experiments showing no changes in tPA activity in conditioned media from cells treated with increasing leptin concentrations. Data are presented as mean ±SEM.
Discussion
The major findings of this study include, first, that leptin up-regulates the expression of PAI-1 in vascular endothelial cells via ERK1/2 activation; and second, tPA levels do not appear to be regulated by leptin. Our study thus partially supports our hypothesis in that leptin upregulates PAI-1 expression, but leptin does not inhibit tPA expression. This indicates that leptin targeted therapeutic interventions against obesity would potentially improve the associated prothrombotic state as well.
We demonstrate that leptin induces PAI-1 mRNA transcription, and protein expression in vascular endothelial cells, through ERK1/2 activation. The induction levels seen with leptin is comparable to that reported for other ligands including CRP and insulin in vascular endothelial cells [25]. Even the moderate leptin-dependent PAI-1 induction in vascular endothelial cells is important as it could be potentially damaging to the atherosclerotic lesion. The increased localized PAI-1 expression in the endothelium could promote formation of plaque with lipid laden cores and thin fibrous caps, which are more prone to rupture [27; 28]. Indeed the presence of PAI-1 has been shown in human atherosclerotic arteries and the increased expression of PAI-1 has been shown to be directly proportional to the degree of atherosclerosis [29]. Therefore, the increased expression and activity of PAI-1 in obese individuals may be the link to increased cardiovascular risk in this population [11; 30]. Our data is consistent with several in-vivo and in-vitro studies which have suggested a proatherogenic role of leptin. While leptin at physiological levels may be protective, high leptin levels are proatherogenic [16]. In our studies we demonstrate that leptin at normal physiological levels does not increase PAI-1 protein expression but does so only at higher physiological levels of leptin (≥ 50 ng/ml). These data are consistent with animal studies of leptin concentrations affecting atherosclerotic progression [16; 31].
In obesity, leptin levels are increased along with cardiovascular risk. So even though leptin activity is impaired at the central level, leptin may still signal in several peripheral cell types which may be responsible for its independent association with higher cardiovascular risk [16; 19; 32; 33]. Our study thus provides an additional molecular mechanism for the proatherogenic role of leptin and its association with increased cardiovascular risk. Further, our data indicates a molecular mechanistic explanation for several studies which report that leptin and PAI-1 levels correlate independent of BMI, age and body fat content [21; 22; 23; 34; 35]. Additional studies showing parallel changes in leptin and PAI-1 levels during weight loss, exercise, fasting, gastric restriction surgery, and aging are also consistent with our findings [35; 36; 37]. However, leptin deficient ob/ob mice are characterized by high levels of PAI-1 expression [38]. This lack of consistency with our data could be explained by the presence of other factors, such as insulin and clock protein which may also influence PAI-1 expression in ob/ob mice [39]. A limitation of our study is that it was performed in vitro. In an in-vivo environment several other factors could together play an important role in regulation of both leptin and PAI-1. A prior study investigating the effect of leptin on PAI-1 regulation in human adipocytes failed to demonstrate any relationship [40]. However in this study, supra-physiological concentrations of leptin were used and PAI-1 was determined only after 4 days of culture. This could also be indicative of a cell type-specific effect of leptin. Previous studies with insulin have also suggested tissue specificity with upregulation of PAI-1 expression occurring in hepatocytes and not in adipocytes [5]. However, considering that the main emphasis of the study was to determine the cardiovascular consequences of increased leptin in obesity, we limited our study to vascular endothelial cells as they are known to play an important role in the development and progression of atherosclerosis [41]. However the role of leptin in determining the systemic levels of PAI-1 needs further investigation by considering its ability to regulate PAI-1 expression in adipocytes and hepatocytes.
Previous studies have shown that ERK 1/2 pathway is involved in PAI-1 induction; we therefore sought to determine the role of ERK1/2 in leptin-dependent PAI-1 activation [42]. We have previously demonstrated that leptin can activate ERK1/2 via specific leptin receptor in HCAEC [24]. In our present study we show that leptin induces PAI-1 expression via the activation of ERK1/2 pathway. With increasing concentration of ERK1/2 inhibitor (PD98059) the PAI-1 expression reduced to the basal levels as seen without leptin treatment. This indicates that leptin signals PAI-1 activation mainly via this pathway, therefore we did not examine the role of other leptin signaling pathways such as JAK/STAT3, AKT and AMPK in our study.
tPA activity is an important determinant of the coagulation-fibrinolysis balance. High PAI-1 increasingly inhibits tPA activity which disturbs this balance leading to a prothrombotic state. Even though increased PAI-1 is mainly responsible for the decreased fibrinolysis it would be important to investigate if leptin could down regulate tPA expression as well. We show that leptin does not regulate tPA expression by western blot analysis as well as total protein quantification in the conditioned media. The tPA activity did not change with increasing concentrations of leptin in a dose dependent manner in the conditioned media. Also no significant correlation was observed between PAI-1 activity and tPA activity in the leptin dose dependent studies. However, we could still speculate that the negative correlation seen between tPA activity and leptin in human subjects results solely from the leptin-induced increases in PAI-1 [22].
From the clinical perspective, treatment of acute thrombotic events such as myocardial infarction and stroke with thrombolytic agents is proven to be efficacious. However they are not a viable option for long term administration and a need exists for development of new therapeutics for long term use. Antibodies or specific inhibitors of PAI-1 activity have shown promising results in the prevention of atherosclerosis [11], and thrombosis [43]. Therefore development of novel therapeutics aimed at down regulating the expression of PAI-1 could be an important strategy for long term use. Our study provides initial evidence for targeting leptin as a therapeutic strategy for modulating PAI-1 expression and activity.
In summary, leptin induces PAI-1 expression in vascular endothelial cells which may play an important role in progression of atherosclerosis. Therefore, targeting leptin for treatment of atherosclerosis in obesity may be beneficial in lowering the PAI-1 dependent prothrombotic state as well.
Acknowledgments
This work was supported by an American Heart Association Postdoctoral Grant (0725787Z) to PS; and National Institutes of Health grant (R01 HL73211 and R01 HL65176) to VKS.
Abbreviations
- PAI-1
plasminogen activator inhibitor-1
- tPA
tissue type plasminogen activator
- HCAEC
human coronary artery endothelial cells
- MAPK
mitogen activated protein kinases
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. Jama. 1999;282:1523–9. doi: 10.1001/jama.282.16.1523. [DOI] [PubMed] [Google Scholar]
- 2.Libby P, Ridker PM. Inflammation and atherosclerosis: role of C-reactive protein in risk assessment. Am J Med. 2004;116 6A:9S–16S. doi: 10.1016/j.amjmed.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Faber DR, de Groot PG, Visseren FL. Role of adipose tissue in haemostasis, coagulation and fibrinolysis. Obes Rev. 2009;10:554–63. doi: 10.1111/j.1467-789X.2009.00593.x. [DOI] [PubMed] [Google Scholar]
- 4.Rosito GA, D'Agostino RB, Massaro J, Lipinska I, Mittleman MA, Sutherland P, Wilson PW, Levy D, Muller JE, Tofler GH. Association between obesity and a prothrombotic state: the Framingham Offspring Study. Thromb Haemost. 2004;91:683–9. doi: 10.1160/th03-01-0014. [DOI] [PubMed] [Google Scholar]
- 5.Mertens I, Van Gaal LF. Obesity, haemostasis and the fibrinolytic system. Obes Rev. 2002;3:85–101. doi: 10.1046/j.1467-789x.2002.00056.x. [DOI] [PubMed] [Google Scholar]
- 6.Hamsten A, de Faire U, Walldius G, Dahlen G, Szamosi A, Landou C, Blomback M, Wiman B. Plasminogen activator inhibitor in plasma: risk factor for recurrent myocardial infarction. Lancet. 1987;2:3–9. doi: 10.1016/s0140-6736(87)93050-9. [DOI] [PubMed] [Google Scholar]
- 7.Wieczorek I, Ludlam CA, Fox KA. Tissue-type plasminogen activator and plasminogen activator inhibitor activities as predictors of adverse events in unstable angina. Am J Cardiol. 1994;74:424–9. doi: 10.1016/0002-9149(94)90896-6. [DOI] [PubMed] [Google Scholar]
- 8.Juhan-Vague I, Alessi MC. Plasminogen activator inhibitor 1 and atherothrombosis. Thromb Haemost. 1993;70:138–43. [PubMed] [Google Scholar]
- 9.Collen D, Lijnen HR. Molecular basis of fibrinolysis, as relevant for thrombolytic therapy. Thromb Haemost. 1995;74:167–71. [PubMed] [Google Scholar]
- 10.Lijnen HR. Pleiotropic functions of plasminogen activator inhibitor-1. J Thromb Haemost. 2005;3:35–45. doi: 10.1111/j.1538-7836.2004.00827.x. [DOI] [PubMed] [Google Scholar]
- 11.Levi M, Biemond BJ, van Zonneveld AJ, ten Cate JW, Pannekoek H. Inhibition of plasminogen activator inhibitor-1 activity results in promotion of endogenous thrombolysis and inhibition of thrombus extension in models of experimental thrombosis. Circulation. 1992;85:305–12. doi: 10.1161/01.cir.85.1.305. [DOI] [PubMed] [Google Scholar]
- 12.Friederich PW, Levi M, Biemond BJ, Charlton P, Templeton D, van Zonneveld AJ, Bevan P, Pannekoek H, ten Cate JW. Novel low-molecular-weight inhibitor of PAI-1 (XR5118) promotes endogenous fibrinolysis and reduces postthrombolysis thrombus growth in rabbits. Circulation. 1997;96:916–21. [PubMed] [Google Scholar]
- 13.Vaughan DE, Declerck PJ, Van Houtte E, De Mol M, Collen D. Reactivated recombinant plasminogen activator inhibitor-1 (rPAI-1) effectively prevents thrombolysis in vivo. Thromb Haemost. 1992;68:60–3. [PubMed] [Google Scholar]
- 14.Eitzman DT, Westrick RJ, Xu Z, Tyson J, Ginsburg D. Plasminogen activator inhibitor-1 deficiency protects against atherosclerosis progression in the mouse carotid artery. Blood. 2000;96:4212–5. [PubMed] [Google Scholar]
- 15.Eren M, Painter CA, Atkinson JB, Declerck PJ, Vaughan DE. Age-dependent spontaneous coronary arterial thrombosis in transgenic mice that express a stable form of human plasminogen activator inhibitor-1. Circulation. 2002;106:491–6. doi: 10.1161/01.cir.0000023186.60090.fb. [DOI] [PubMed] [Google Scholar]
- 16.Beltowski J. Leptin and atherosclerosis. Atherosclerosis. 2006;189:47–60. doi: 10.1016/j.atherosclerosis.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Wolk R, Berger P, Lennon RJ, Brilakis ES, Johnson BD, Somers VK. Plasma leptin and prognosis in patients with established coronary atherosclerosis. J Am Coll Cardiol. 2004;44:1819–24. doi: 10.1016/j.jacc.2004.07.050. [DOI] [PubMed] [Google Scholar]
- 18.Wallace AM, McMahon AD, Packard CJ, Kelly A, Shepherd J, Gaw A, Sattar N. Plasma leptin and the risk of cardiovascular disease in the west of Scotland coronary prevention study (WOSCOPS) Circulation. 2001;104:3052–6. doi: 10.1161/hc5001.101061. [DOI] [PubMed] [Google Scholar]
- 19.Mark AL, Correia ML, Rahmouni K, Haynes WG. Selective leptin resistance: a new concept in leptin physiology with cardiovascular implications. J Hypertens. 2002;20:1245–50. doi: 10.1097/00004872-200207000-00001. [DOI] [PubMed] [Google Scholar]
- 20.Thogersen AM, Soderberg S, Jansson JH, Dahlen G, Boman K, Nilsson TK, Lindahl B, Weinehall L, Stenlund H, Lundberg V, Johnson O, Ahren B, Hallmans G. Interactions between fibrinolysis, lipoproteins and leptin related to a first myocardial infarction. Eur J Cardiovasc Prev Rehabil. 2004;11:33–40. doi: 10.1097/01.hjr.0000116824.84388.a2. [DOI] [PubMed] [Google Scholar]
- 21.De Mitrio V, De Pergola G, Vettor R, Marino R, Sciaraffia M, Pagano C, Scaraggi FA, Di Lorenzo L, Giorgino R. Plasma plasminogen activator inhibitor-I is associated with plasma leptin irrespective of body mass index, body fat mass, and plasma insulin and metabolic parameters in premenopausal women. Metabolism. 1999;48:960–4. doi: 10.1016/s0026-0495(99)90190-7. [DOI] [PubMed] [Google Scholar]
- 22.Soderberg S, Olsson T, Eliasson M, Johnson O, Ahren B. Plasma leptin levels are associated with abnormal fibrinolysis in men and postmenopausal women. J Intern Med. 1999;245:533–43. doi: 10.1046/j.1365-2796.1999.00472.x. [DOI] [PubMed] [Google Scholar]
- 23.Skurk T, van Harmelen V, Lee YM, Wirth A, Hauner H. Relationship between IL-6, leptin and adiponectin and variables of fibrinolysis in overweight and obese hypertensive patients. Horm Metab Res. 2002;34:659–63. doi: 10.1055/s-2002-38253. [DOI] [PubMed] [Google Scholar]
- 24.Singh P, Hoffmann M, Wolk R, Shamsuzzaman AS, Somers VK. Leptin induces C-reactive protein expression in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:e302–7. doi: 10.1161/ATVBAHA.107.148353. [DOI] [PubMed] [Google Scholar]
- 25.Devaraj S, Xu DY, Jialal I. C-reactive protein increases plasminogen activator inhibitor-1 expression and activity in human aortic endothelial cells: implications for the metabolic syndrome and atherothrombosis. Circulation. 2003;107:398–404. doi: 10.1161/01.cir.0000052617.91920.fd. [DOI] [PubMed] [Google Scholar]
- 26.Grenett HE, Benza RL, Li XN, Aikens ML, Grammer JR, Brown SL, Booyse FM. Expression of plasminogen activator inhibitor type I in genotyped human endothelial cell cultures: genotype-specific regulation by insulin. Thromb Haemost. 1999;82:1504–9. [PubMed] [Google Scholar]
- 27.Sobel BE. Increased plasminogen activator inhibitor-1 and vasculopathy. A reconcilable paradox. Circulation. 1999;99:2496–8. doi: 10.1161/01.cir.99.19.2496. [DOI] [PubMed] [Google Scholar]
- 28.Sobel BE, Taatjes DJ, Schneider DJ. Intramural plasminogen activator inhibitor type-1 and coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:1979–89. doi: 10.1161/01.ATV.0000091250.53231.4D. [DOI] [PubMed] [Google Scholar]
- 29.Schneiderman J, Sawdey MS, Keeton MR, Bordin GM, Bernstein EF, Dilley RB, Loskutoff DJ. Increased type 1 plasminogen activator inhibitor gene expression in atherosclerotic human arteries. Proc Natl Acad Sci U S A. 1992;89:6998–7002. doi: 10.1073/pnas.89.15.6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mutch NJ, Wilson HM, Booth NA. Plasminogen activator inhibitor-1 and haemostasis in obesity. Proc Nutr Soc. 2001;60:341–7. doi: 10.1079/pns200199. [DOI] [PubMed] [Google Scholar]
- 31.Schafer K, Halle M, Goeschen C, Dellas C, Pynn M, Loskutoff DJ, Konstantinides S. Leptin promotes vascular remodeling and neointimal growth in mice. Arterioscler Thromb Vasc Biol. 2004;24:112–7. doi: 10.1161/01.ATV.0000105904.02142.e7. [DOI] [PubMed] [Google Scholar]
- 32.Romero-Corral A, Sierra-Johnson J, Lopez-Jimenez F, Thomas RJ, Singh P, Hoffmann M, Okcay A, Korinek J, Wolk R, Somers VK. Relationships between leptin and C-reactive protein with cardiovascular disease in the adult general population. Nat Clin Pract Cardiovasc Med. 2008;5:418–25. doi: 10.1038/ncpcardio1218. [DOI] [PubMed] [Google Scholar]
- 33.Martin SS, Qasim A, Reilly MP. Leptin resistance: a possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J Am Coll Cardiol. 2008;52:1201–10. doi: 10.1016/j.jacc.2008.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mertens I, Considine RV, Van der Planken M, Van Gaal LF. Hemostasis and fibrinolysis in non-diabetic overweight and obese men and women. Is there still a role for leptin? Eur J Endocrinol. 2006;155:477–84. doi: 10.1530/eje.1.02239. [DOI] [PubMed] [Google Scholar]
- 35.Eriksson M, Johnson O, Boman K, Hallmans G, Hellsten G, Nilsson TK, Soderberg S. Improved fibrinolytic activity during exercise may be an effect of the adipocyte-derived hormones leptin and adiponectin. Thromb Res. 2008;122:701–8. doi: 10.1016/j.thromres.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 36.Rangemark C, Hedner JA, Carlson JT, Gleerup G, Winther K. Platelet function and fibrinolytic activity in hypertensive and normotensive sleep apnea patients. Sleep. 1995;18:188–94. doi: 10.1093/sleep/18.3.188. [DOI] [PubMed] [Google Scholar]
- 37.van Dielen FM, Buurman WA, Hadfoune M, Nijhuis J, Greve JW. Macrophage inhibitory factor, plasminogen activator inhibitor-1, other acute phase proteins, and inflammatory mediators normalize as a result of weight loss in morbidly obese subjects treated with gastric restrictive surgery. J Clin Endocrinol Metab. 2004;89:4062–8. doi: 10.1210/jc.2003-032125. [DOI] [PubMed] [Google Scholar]
- 38.Samad F, Loskutoff DJ. Tissue distribution and regulation of plasminogen activator inhibitor-1 in obese mice. Mol Med. 1996;2:568–82. [PMC free article] [PubMed] [Google Scholar]
- 39.Oishi K, Ohkura N, Wakabayashi M, Shirai H, Sato K, Matsuda J, Atsumi G, Ishida N. CLOCK is involved in obesity-induced disordered fibrinolysis in ob/ob mice by regulating PAI-1 gene expression. J Thromb Haemost. 2006;4:1774–80. doi: 10.1111/j.1538-7836.2006.02032.x. [DOI] [PubMed] [Google Scholar]
- 40.Aprath-Husmann I, Rohrig K, Gottschling-Zeller H, Skurk T, Scriba D, Birgel M, Hauner H. Effects of leptin on the differentiation and metabolism of human adipocytes. Int J Obes Relat Metab Disord. 2001;25:1465–70. doi: 10.1038/sj.ijo.0801737. [DOI] [PubMed] [Google Scholar]
- 41.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 42.Demyanets S, Kaun C, Rychli K, Rega G, Pfaffenberger S, Afonyushkin T, Bochkov VN, Maurer G, Huber K, Wojta J. The inflammatory cytokine oncostatin M induces PAI-1 in human vascular smooth muscle cells in vitro via PI 3-kinase and ERK1/2-dependent pathways. Am J Physiol Heart Circ Physiol. 2007;293:H1962–8. doi: 10.1152/ajpheart.01366.2006. [DOI] [PubMed] [Google Scholar]
- 43.Elokdah H, Abou-Gharbia M, Hennan JK, McFarlane G, Mugford CP, Krishnamurthy G, Crandall DL. Tiplaxtinin, a novel, orally efficacious inhibitor of plasminogen activator inhibitor-1: design, synthesis, and preclinical characterization. J Med Chem. 2004;47:3491–4. doi: 10.1021/jm049766q. [DOI] [PubMed] [Google Scholar]