

Abstract
Cellular processes are mediated by complex networks of molecular interactions. Dissection of their role most commonly is achieved by using genetic mutations that alter, for example, protein–protein interactions. Small molecules that accomplish the same result would provide a powerful complement to the genetic approach, but it generally is believed that such molecules are rare. There are several natural products, however, that illustrate the feasibility of this approach. Split-pool synthesis now provides a simple mechanical means to prepare vast numbers of complex, even natural product-like, molecules individually attached to cell-sized polymer beads. Here, we describe a genetic system compatible with split-pool synthesis that allows the detection of cell-permeable, small molecule inhibitors of protein–protein interactions in 100- to 200-nl cell culture droplets, prepared by a recently described technique that arrays large numbers of such droplets. These “nanodroplets” contain defined media, cells, and one or more beads containing ≈100 pmol of a photoreleasable small molecule and a controlled number of cells. The engineered Saccharomyces cerevisiae cells used in this study express two interacting proteins after induction with galactose whose interaction results in cell death in the presence of 5-fluoroorotic acid (inducible reverse two-hybrid assay). Disruption of the interaction by a small molecule allows growth, and the small molecule can be introduced into the system hours before induction of the toxic interaction. We demonstrate that the interaction between the activin receptor R1 and the immunophilin protein FKBP12 can be disrupted by the small molecule FK506 at nanomolar concentrations in nanodroplets. This system should provide a general method for selecting cell-permeable ligands that can be used to study the relevance of protein–protein interactions in living cells or organisms.
Small molecule ligands have been invaluable in exploring the cellular function of proteins (1–5). Binding of these ligands causes activation or inactivation of their target proteins, analogous to effects produced by gain- or loss-of-function mutations in the corresponding genes. Conditional mutations such as temperature-sensitive alleles (ref. 6 and references therein) or dominant negative mutations (7), have been found to be especially useful. Use of cell-permeable small molecule ligands to alter protein function is attractive because of an inherent conditionality—the cellular activity of the target proteins can be controlled simply and rapidly by adding or removing the compound.
Genome sequencing projects are uncovering a large number of novel proteins with unknown functions. Understanding the functions of every protein encoded by a genome is a primary goal of biology (8). Systematic gene knockout experiments in the yeast Saccharomyces cerevisiae currently are underway to generate ≈6,000 deletion strains (9). One of the limitations of this approach is that an even larger number of strains will need to be constructed with multiple gene deletions to study genetic epistasis. An alternative is to alter the function of the gene products directly through the use of small molecule ligands. Ligands for different proteins can be used in combination to activate or inactivate their target proteins either sequentially or simultaneously. The diversity in small organic compounds that can be generated by combinatorial chemistry (10) promises to match the diversity in natural protein targets. By splitting and then pooling reaction vessels during a multistep synthesis (split-pool) (11), one bead-one compound libraries of enormous size and diversity can be generated whereas encoding techniques (12–14) can reveal the synthesis history and therefore identity of each compound, retrievable by sequencing the encoding tags. The linking of compounds to polymer beads by photocleavable chemistry (15) permits functional assays in solution after photolysis, and decoding allows subsequent identification of active compounds. By these means, it is possible that small molecule ligands might one day be found for any protein encoded by a genome.
Protein–protein interactions are the basis of numerous cellular processes. Inappropriately constitutive interactions can result in neoplastic states (e.g., see ref. 16). The ability to interfere with these interactions will lead to an understanding of the normal processes and possibly to the control of medical disorders. The view that protein–protein interactions are difficult to disrupt by small organic compounds has been reinforced by the general lack of success thus far in screens for such inhibitors. Progress might be achieved with improved methods for the split-pool synthesis of natural product-like compounds and effective methods for their screening. We now report a genetic system compatible with split-pool synthesis to identify cell-permeable, organic compounds that disrupt specific protein–protein interactions.
The yeast two-hybrid system (17) has proven to be a powerful genetic method for identifying novel protein–protein interactions (18–22). Variants such as the one-hybrid (23, 24) and three-hybrid (25–29, 46, 47) systems have also been developed for detecting protein–DNA, protein-RNA, protein–protein, or protein–small molecule interactions. Recently, reverse two-hybrid (30–31) and split-hybrid (32) assays have been reported that select for mutations in the interacting proteins or for other proteins that disrupt a given protein–protein interaction. The possibility of using these systems to discover small molecules that disrupt protein–protein interactions has been suggested by these authors. There are, however, several problems that necessitate the development of a modified genetic selection system.
The first problem is that the interacting proteins should be expressed in an inducible way such that the potential small molecule inhibitor is present before the synthesis of the toxic reporter gene product. In selecting for mutations or other proteins that prevent protein–protein interactions, transformants that produce noninteracting proteins will survive (30, 31, 48). But small molecule screens require the presence of the wild-type interacting forms, which is lethal to the cell. We have solved this problem by constructing an inducible reverse two-hybrid system using Gal and the GAL1 promoter (33) to induce expression of the interacting proteins over a period of hours while using UV irradiation to release potential small molecule inhibitors over a period of seconds.
The second problem is that, although small molecule screens will best be performed in liquid culture, cross-feeding of amino acids can occur between cells harboring selectable markers encoding enzymes that are critical for amino acid biosynthesis. This can lead to viability by stochastic loss of a plasmid encoding one of the interacting proteins rather than by small molecule-based disruption of the protein–protein interaction. Although chromosomal integration of two-hybrid plasmids might in principle solve this problem, it is impractical when a systematic screen with all protein–protein pairs in the proteome is considered (34). We have instead solved this problem by substituting antibiotic resistance markers for the auxotrophic markers used in previous reverse two-hybrid and split-hybrid systems.
Two other problems are caused by the facility with which small molecules rapidly diffuse in liquid culture and by the fact that the quantities of compounds attached to individual beads after split-pool synthesis are miniscule (≈100 pmol). We have solved both of these problems by using recently developed techniques for generating large numbers of small volume (≈100–200 nl) droplets (nanodroplets) that contain beads, cells, and defined media (35, 36). These miniaturization techniques allow large numbers of cell-based assays to be performed in liquid culture without complications arising from diffusion, and the techniques provide high concentrations of ligands through the photochemically controlled release of compounds from individual beads.
In this paper, we report a “small molecule reverse two-hybrid system” (Fig. 1) for selecting small molecule inhibitors of protein interactions. In this system, the interaction between one protein fused to the LexA DNA-binding domain and another fused to the B42 transcriptional activation domain (AD) recruits the AD proximal to a promoter containing LexA binding sites upstream of a URA3 reporter gene. Both expression constructs are repressed by the presence of Glc in the media and activated by shifting to Gal-containing media. The Ura3p-expressing cells are killed in medium containing the pro-toxin 5-fluoroorotic acid (5-FOA) (49). If a small molecule (L) disrupts the intracellular protein–protein interaction, proximity of the AD to the promoter is removed, and transcription of the URA3 gene is diminished, allowing cell survival in 5-FOA. To demonstrate the feasibility of detecting small molecules that disrupt protein–protein interactions by genetic means, the association of FKBP12 with R1 of the transforming growth factor β receptor superfamily type I receptor (37) was disrupted by nanomolar concentrations of FK506 both on an agar plate and in nanodroplets. This system should facilitate the discovery of small molecule ligands both as research tools and therapeutic agents.
Figure 1.
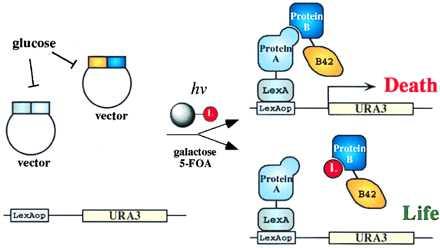
General schematic of the inducible (small molecule) reverse two-hybrid system designed to detect small molecule inhibitors of protein–protein interactions. The expression of the interacting proteins is controlled by the GAL1 promoter, which is repressed in Glc. After Gal induction, the two interacting protein partners are synthesized, and their association in turn induces the synthesis of a toxic gene product, leading to death, unless an inhibitor of the protein–protein interaction is present in the cell. Only cells with this disruption should be selected.
MATERIALS AND METHODS
General.
Standard protocols for molecular biology (38) and yeast genetics (39) were used. DNA sequences for yeast genes were retrieved from the Saccharomyces Genome Database (http://genome-www.stanford.edu/saccharomyces/).
Construction of Reporter Yeast Strains.
The LexAop–URA3 reporter yeast strains are YL(6, 4)LU (MATa, his3, trp1, leu2::LexAop6–LEU2, ura3::LexAop4–SPO13tata–URA3), and YL(2, 4)LU (MATa, his3, trp1, leu2::LexAop2–LEU2, ura3::LexAop4–SPO13tata–URA3).
To generate the LexAop–URA3 reporter construct (Fig. 2A), pRS306 (40) was digested with HindIII and EcoRI, blunted with Klenow (NEB, Beverly, MA), and recircularized to generate pRS306* that lacks HindIII, EcoRV, and EcoRI sites in the polylinker. The 773-bp AatII→EcoRV fragment in pRS306* was replaced by a piece of DNA consisting of −257 (with AatII site added) → −223(HindIII) of the URA3 gene, −213(HindIII) → −170(EcoRI) of the SPO13 gene from EGY48, and −170 (with the EcoRI site of SPO13 added) → +189 (EcoRV of URA3) of the SPO13–URA3 fusion gene from MaV95 (31), all obtained through yeast colony PCR. The resulting plasmid pJH1 contains unique HindIII and EcoRI sites in the promoter region. A 78-bp oligonucleotide (after S. Hanes, SUNY-Albany) containing four copies of the LexA operator from the ColE1 gene was inserted into pJH1 at the HindIII and EcoRI sites, generating pJH3 (Fig. 2A).
Figure 2.
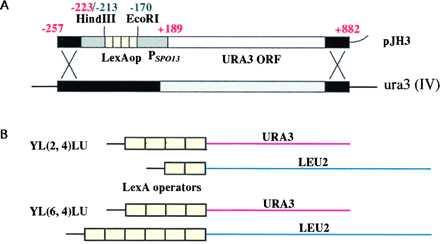
(A) Reporter constructs used for homologous recombination. The LexAop4–SPO13tata–URA3 reporter construct contains four LexA operators upstream of a SPO13 promoter–URA3 fusion reporter gene. Nucleotide numbering is relative to the translation start codon ATG, where A is +1. Red numbering represents nucleotides from the URA3 gene, and green represents those from the SPO13 gene. (B) Schematic representation of reporter yeast strains.
In EGY48 and EGY191 strains (19), although the nature of the ura3 mutation is not known, the similarity in size to the wild-type gene and the presence of nucleotides −257 to −228 and 885 to 851 was revealed by PCR (unpublished result). These sequences were used in the reporter constructs (Fig. 2A). This permits a double-crossing over event necessary for gene replacement. Approximately 10 μg of the reporter plasmid pJH3 was digested with AatII and SmaI and desalted by QIAquick PCR purification kit (Qiagen, Chatsworth, CA), and ≈5 μg was used to transform EGY48 or EGY191 carrying pCALex-AD (TRP1), encoding LexA fused to an AD. Transformants that grew on Sc−Trp −Ura plates were examined for Lex–AD dependency of the Ura3p+ phenotype by cosegregation of Trp1p− and Ura3p− phenotypes, after spontaneous cure of the pCALex-AD plasmid in yeast extract/peptone/dextrose (YPD) liquid culture and subsequent selection on a 5-FOA plate. Clones that showed cosegregation were used to establish the small molecule reverse two-hybrid reporter strains after retransforming with pCALex–AD to verify the reappearance of the Ura3p+ phenotype on Sc–Trp–Ura plates.
Construction of Two-Hybrid Protein Expression Vectors.
One set of these vectors (Table 1) carries amino acid auxotrophic selection markers useful for experiments performed on agar plates. The other set carries antibiotic resistance markers (Fig. 3) suitable for selections done both in liquid media and on agar plates. pCGLex was constructed by ligation of three pieces: the 0.53-kb KpnI → HindIII GAL1 promoter from pJG4-5 (41), the 0.63-kb HindIII → EcoRI LexA sequence from pEG202 (19), and the 5-kb EcoRI → KpnI fragment of pPC86 (42). p2GLex was generated by replacing the 3.9-kb ScaI → EcoRI partial Amp–pBR ori–ADH1 promoter–LexA fragment of pEG202 with the 2.9-kb ScaI → EcoRI partial Amp–ColE1 ori–GAL1 promoter–LexA fragment from pCGLex. To construct pCGB42, pJG4-5 was cut with KpnI, blunted with Klenow, and ligated with an ApaI linker; the 0.53-kb ApaI → EcoRI GAL1 promoter–B42 sequence was then ligated to the 6.5-kb EcoRI → ApaI fragment of pPC16 (42).
Table 1.
Two-hybrid protein expression vectors
| Two-hybrid vectors | Promoter | Replication origin | Selectable marker | |
|---|---|---|---|---|
| LexA fusion | p2GLex | GAL1 | 2 μ | HIS3 |
| pCGLex | GAL1 | CEN6/ARSH4 | TRP1 | |
| pEG202 | ADH1 | 2 μ | HIS3 | |
| pCALex | ADH1 | CEN6/ARSH4 | TRP1 | |
| p2GLex/Zeo | GAL1 | 2 μ | Zeo-r | |
| pCGLex/Zeo | GAL1 | CEN6/ARSH4 | Zeo-r | |
| B42 fusion | pJG4-5 | GAL1 | 2 μ | TRP1 |
| pCGB42 | GAL1 | CEN6/ARSH4 | HIS3 | |
| p2AB42 | ADH1 | 2 μ | TRP1 | |
| pCAB42 | ADH1 | CEN6/ARSH4 | HIS3 | |
| p2GB42/Kan | GAL1 | 2 μ | Kan-r | |
| pCGB42/Kan | GAL1 | CEN6/ARSH4 | Kan-r | |
Figure 3.
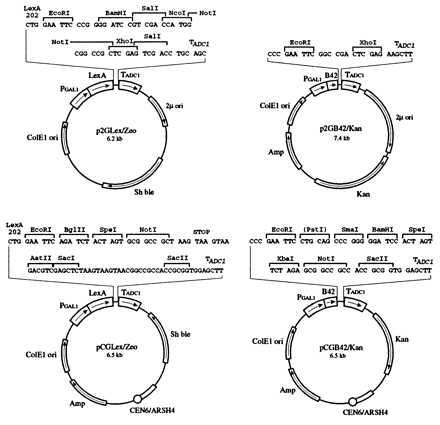
Vectors for inducible two-hybrid protein expression. Expression of the two interacting proteins is under the control of the GAL1 promoter. Expression vectors contain antibiotic markers for selection. Both centromeric and 2-μ vectors were designed.
Four corresponding vectors were generated by replacing TRP1 or HIS3 genes from the above plasmids with antibiotic resistance markers. The LexA fusion vector uses the Streptoalloteichus hindustanus bleomycin gene encoding resistance to zeocin, as a selectable marker in both Escherichia coli and S. cerevisiae. The B42 fusion vector uses the E. coli transposon Tn903-encoded kanamycin resistance (aminoglycoside phosphotransferase) gene (43) as a selectable marker, which confers resistance to geneticin in S. cerevisiae. Further details of this construction will be made available on request.
Beads Linked to FK506.
FK506 was attached to S NH2 beads (Rapp Polymere) through a photolabile linker (15) via an olefin linkage at the C21 side chain of FK506 (Derek S. Tan and S.L.S., unpublished results).
Reverse Two-Hybrid Assays for Small Molecule Inhibition of Protein–Protein Interaction.
Yeast reporter strains were transformed with 0.5 μg of each two-hybrid plasmid DNA. After transforming cells with plasmids containing Zeo-r and Kan-r markers, the cells were resuspended in 1 ml yeast extract/peptone/dextrose and incubated at room temperature for 3 h to allow expression of the antibiotic resistance genes before plating on selective medium–yeast extract/peptone/dextrose containing 200 μg/ml each of zeocin (Invitrogen) and geneticin (Sigma). Double transformants were picked for protein–protein interaction or dissociation assays.
RESULTS
Construction of an Inducible Reverse Two-Hybrid System.
The ideal genetic system would have a reporter that converts a desired small molecule-dependent event into a positive growth phenotype instead of a negative loss of growth phenotype because detection of growth already ensures that the rescuing compound is not cytotoxic. We constructed vectors encoding a DNA-binding domain (LexA) fused to one protein (A) and an AD (B42) fused to another protein (B) (Fig. 1). Expression of the two-hybrid proteins is controlled by the GAL1 promoter. The use of a Gal-inducible system requires that the Gal metabolism pathway be intact in the yeast strain. This rules out the use of the Gal4 DNA-binding domain-based forward or reverse two-hybrid yeast strains (e.g., ref. 31) and one LexA-based system (21, 32) in which the GAL4 and GAL80 genes are inactivated. The other LexA-based system, the EGY series of yeast strains (19), has intact pathways for Gal metabolism. It also contains a superior forward reporter gene, LEU2, and it was therefore used as the starting point for the construction of inducible reverse two-hybrid strains. URA3 (orotidine-5′-phosphate decarboxylase) gene was chosen as the reverse reporter gene because it is the best among several markers capable of double selection (39). Also, the fusion of SPO13 promoter to the URA3 ORF has been reported to generate a reporter construct with very low basal levels of transcription (31).
A LexAop–URA3 reporter yeast strain, YL(6, 4)LU, was constructed by replacing the ura3 locus in EGY48 (19) with the LexAop4–SPO13tata–URA3 reporter construct (Fig. 2A), which contains four ColE1-type LexA operators upstream of a SPO13 promoter–URA3 fusion reporter gene. The resulting strain contains two chromosomal reporters, the original LEU2 and the new URA3, driven by six and four LexA operators, respectively (Fig. 2B). The presence of two distinct reporter genes, having only the LexA operator sequences in common, provides a means to reduce false-positives in two-hybrid assays. Another reporter strain, YL(2, 4)LU, was constructed similarly from EGY191 (19) with two and four LexA operators driving LEU2 and URA3 reporters, respectively (Fig. 2B). The presence of LexA operator sites on a promoter other than the one directing the expression of the reporter gene being assayed allows further modulation of the transcription of the reporter gene, depending on the relative number of LexA binding sites in each promoter (unpublished data). Cells that carry the two-hybrid protein expressing vectors are induced to express the interacting proteins and are selected against in 5-FOA containing medium. Only cells that have acquired a small molecule that prevents the two-hybrid interaction will fail to express Ura3p and be able to grow on 5-FOA-containing media. For this assay to succeed, the cells must not have already accumulated the stable (unpublished data; see also ref. 44) Ura3p protein, e.g., during the establishment of double transformants if the two-hybrid proteins are constitutively expressed.
FK506 Inhibits R1–FKBP12 Interaction on Agar in the Small Molecule Reverse Two-Hybrid System.
We tested this system by using the natural product FK506, which is known to interfere with the interaction between FKBP12 and the transforming growth factor β type I receptor R4 by binding to FKBP12 (37). This protein–protein interaction is one of many discovered through the use of the forward two-hybrid screen. We challenged the system with the stronger protein–protein interaction (37) between FKBP12 and R1, an activin receptor of the transforming growth factor β type I receptor superfamily. The cytoplasmic domain of R1 [R1(C)] was fused to the LexA DNA binding domain, and FKBP12 was fused to the B42 activation domain.
As shown in Fig. 4A, transformants containing both hybrid proteins showed readily detectable growth in Gal medium lacking Leu or Ura, indicating that both reporter genes were expressed. Transformants lacking either one of the proteins did not grow, indicating that reporter gene expression was dependent on both interacting partners. The growth pattern on the 5-FOA/Gal plate is complementary to that on the Ura dropout plate, indicating that negative selection using URA3 was observed as expected. Only cells expressing both interacting proteins failed to grow on these plates. As a control, we used 5-FOA/Glc plates in which the fusion proteins were not expressed, and none of the transformants failed to grow. The lack of growth in 5-FOA/Gal medium for FKBP12–R1 double transformants can be suppressed by the addition of FK506 in a concentration-dependent manner. The experiments presented in Fig. 4A were performed using expression vectors carrying auxotrophic markers. Similar results were obtained with corresponding vectors carrying antibiotic resistance markers (data not shown). In these experiments, zeocin and geneticin were added to the medium instead of dropping out His and Trp.
Figure 4.
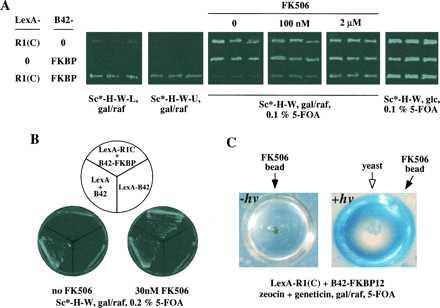
FK506 specifically interferes with the interaction between FKBP12 and the activin type I receptor R1 as detected in the small molecule reverse two-hybrid system. Sc* refers to synthetic complete (Sc) medium without any sugar; gal/raf, 2% galactose plus 1% raffinose; and glc, 2% glucose. (A) Phenotypes of R1C–FKBP12 interaction and its dissociation by FK506. Yeast transformants with both vectors appear within 24–36 h on Sc-H-W plates. Colonies were picked and patched on various selective plates for reported gene assays. Each pairwise transformation was analyzed by six independent transformants, three of which are shown here. (B) FK506-mediated 5-FOA resistance is specific to R1C–FKBP12 transformants. (C) Detection of the FK506 effect in liquid media using arrayed nanodroplets (≈200-nl vol) (36). The droplets containing yeast, medium, and FK506 beads were formed in polydimethylsiloxane plastic molded to contain wells 40 μm in depth, 1 mm in diameter, and 250 μm apart from each other.
To rule out the formal possibility that the effect of FK506 may result from other modes of action, such as direct inhibition of Ura3p activity, prevention of LexA DNA binding, or B42 AD function, we added FK506 to LexA-B42 transformants. Death on 5-FOA plates caused by this activator was not overcome by FK506 (Fig. 4B). Thus, the rescue by FK506 is specific to the FKBP12–R1 complex. Presumably FK506 binding to FKBP12 displaces R1(C) from the protein complex.
Despite the abundance of endogenous FK506-binding proteins, the interaction of R1(C) and FKBP12 in this inducible reverse two-hybrid system is highly sensitive to inhibition by FK506: 30 nM FK506 rescues the lethality of the double transformants in 0.2% 5-FOA. That FK506 can inhibit the interaction between R1 and FKBP12 is consistent with the previous observation, in the forward two-hybrid assay using β-galactosidase as a reporter, that the weaker interaction between R4 and FKBP12 is inhibited by FK506. In these experiments, 1 μM FK506 inhibited β-galactosidase activity by 50% (37). However, using a forward reporter in a related assay, we failed to detect an inhibitory effect on R1 and FKBP12, using up to 5 μM of FK506 (a concentration that partially inhibits yeast growth). A possible explanation for this difference in detection sensitivity may be that, in the forward two-hybrid assay, if even a fraction of R1–FKBP12 protein complexes remains intact, the reporter gene product (and thus the missing amino acid) can continue to be synthesized. This would allow cell growth, albeit at a reduced rate. The reduced sensitivity of the forward vs. reverse two-hybrid assay suggests that the split-hybrid system (32) may be difficult to adapt to small molecule screening because the reduction in the repressor resulting from incomplete disruption by an inhibitor may be insufficient to derepress the auxotrophic reporter gene. In contrast, in the reverse two-hybrid assay, the total amount of 5-FOA in the medium is fixed. A threshold concentration of 5-FOA can be established so that a significant difference in the cellular Ura3p levels will be translated into a life-or-death difference. For example, with R1 and FKBP12, we found that lowering the 5-FOA concentration from 0.1 to 0.05% abolished its differential toxicity to cells expressing both proteins and control cells expressing only one of the two proteins. Also, preculturing cells in Gal for 2 h renders the cells sensitive to 0.1% 5-FOA despite the presence of 0.1 μM FK506. This observation again emphasizes the importance of inducible protein expression for small molecule screening.
FK506 Disruption of R1–FKBP12 Interaction Can Be Selected in Liquid Medium Using Arrayed Nanodroplets.
Two techniques recently have been developed to generate large numbers of tiny droplets of ≈100- to 200-nl vol (called nanodroplets) containing synthesis beads, defined media, and a controlled number of cells (35, 36). In Fig. 4C, we performed the small molecule disruption experiment in arrayed nanodroplets. Growth in 5-FOA for FKBP12–R1 double transformants was observed only in droplets that contained a bead (purple/brown color) covalently linked to FK506 that had been irradiated with long wavelength UV light to release FK506. For this proof-of-principle experiment, we attached a natural product, FK506, to the beads covalently using a photolabile linker, in analogy to previous synthetic libraries of greater than 1 million encoded compounds (45).
DISCUSSION
The split-pool synthesis strategy offers an opportunity to develop new biological reagents that can facilitate an understanding of the cellular function of proteins. One possibility is to use small molecule ligands to disrupt specific protein–protein interactions in cells. We have developed a genetic system to select small organic compounds that have this property.
We have used the Gal-inducible promoter from the GAL1 gene to control the expression of one or both of the interacting proteins. This ensures that expression of the proteins is repressed in Glc medium during the establishment of the double transformants. When cells are switched to Gal medium containing both 5-FOA and beads covalently attached to small molecules, the fusion proteins are synthesized in ≈3 h. By using a recently developed method to convert the medium mixture into a large collection of tiny droplets containing the individual beads and engineered cells, many experiments can be run simultaneously. UV irradiation of a collection of such nanodroplets (≈6,500 in a standard 10-cm dish) results in the release of the small molecules into the nanodroplets. Because the preparation of droplets and release of compounds occurs within minutes, the inhibitors are present long before the synthesis of the toxic combination of interacting proteins. In the proof-of-concept experiment reported herein, the natural product FK506 was released into nanodroplets containing cells that several hours earlier had been induced to express the toxic combination of interacting proteins FKBP12 and the cytoplasmic tail of the activin receptor R1. Cells were able to grow only in nanodroplets containing the released FK506, indicating that the ability of FK506 to disrupt this protein–protein interaction can be detected by the appearance of microcolonies in the nanodroplets (Fig. 4). The assay can be adjusted in terms of the selection of the carbon source, plasmid replication origin (Fig. 3), promoter, and the number of LexA-binding sites. These parameters could be important when, for example, protein–protein interactions of varying affinity are under consideration.
Several three-hybrid systems have been reported that use small molecule dimerizers to recruit an AD to a DNA-binding domain, thereby up-regulating the transcription of a target gene (27–29, 35). In one case, this type of system was used to isolate cDNAs encoding proteins that bind to a known small molecule (29) and, in conjunction with a method to prepare small droplets containing beads and cells (35, 36), it might also be used to expedite isolation of small molecules that can bind to any protein fused to an AD (35). A variation on this theme also was used to detect the intracellular binding of a small molecule to a protein by recruiting a nuclear localization sequence to a target protein fused to green fluorescent protein, thereby causing fluorescence previously observed in the cytoplasm to be restricted to the nucleus of the cell (27). These types of screens have the potential for discovering a wide range of small molecules that bind various regions of a protein target. However, such assays require the use of dumbbell-shaped small molecules comprised of a constant “anchor” end and a variable “presented” end. The requirement for the two linked ends might affect functional groups that could otherwise interact with target proteins and might diminish cell permeability relative to the smaller monomeric entities. In contrast, the reverse two-hybrid format described here results in the selection of unmodified ligands, free of the anchor end. The ligands that are recovered should act specifically on one of the two chosen interacting domains. These two methods should complement each other in efforts to discover small ligands with biological and medical consequences.
Future Applications.
The number of interacting protein pairs to be screened by the small molecule reverse two-hybrid system at the same time need not be limited, nor is the assignment of their identities necessary before carrying out the screen. In principle, a forward two-hybrid screen could be performed using a target protein and LEU2 or the forward URA3 as a reporter gene; the positive colonies would then be gathered, expression would be silenced by incubating in Glc, and a small molecule reverse two-hybrid screen would then be performed. Alternatively, a library containing all interacting components of a proteome (34) might be used as the starting material for a small molecule interference screening. In this scenario, each nanodroplet would contain the full collection of protein pairs and one synthesis bead. The collection of protein–protein interactions can be screened simultaneously because only combinations in which a given compound interferes with a given protein–protein interaction should give rise to a colony. The compound may be identified by decoding tags attached to the bead and the protein pairs by sequencing the plasmid from the colony. It also should be possible to extend the technique to interactions between a protein and other types of ligands (such as nucleic acids, carbohydrates, and second messengers).
The small molecule reverse two-hybrid system should facilitate efforts to combine the power of yeast genetics and combinatorial chemistry. The ultimate goal is to reach the point where ligands are available that alter the function of any target protein, with high specificity and affinity.
Acknowledgments
We thank S. Hanes, E. Golemis, X.-F. Zheng, X.-Y. Zhang, M. Vidal, T. Wang, and J. Chant for providing us with plasmids and strains, D. S. Tan for preparing the TentaGel–FK506 reagent, and G. L. Verdine for sharing equipment. We thank R. Ward, S. Hanes, M. Vidal, and all members of the Schreiber group for helpful discussions and comments. J.H. is a Research Associate and S.L.S. is an Investigator at the Howard Hughes Medical Institute.
ABBREVIATIONS
- AD
activation domain
- FOA
fluoroorotic acid
References
- 1.Yamamoto K R. Annu Rev Genet. 1985;19:209–252. doi: 10.1146/annurev.ge.19.120185.001233. [DOI] [PubMed] [Google Scholar]
- 2.Evans R M. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schreiber S L. Science. 1991;251:283–287. doi: 10.1126/science.1702904. [DOI] [PubMed] [Google Scholar]
- 4.Klausner R D, Donaldson J G, Lippincott-Schwartz J. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schreiber S L. Chem Eng News. 1992;26:22–32. [Google Scholar]
- 6.Dohmen R J, Wu P, Varshavsky A. Science. 1994;263:1273–1276. doi: 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
- 7.Herskowitz I. Nature (London) 1987;329:219–222. doi: 10.1038/329219a0. [DOI] [PubMed] [Google Scholar]
- 8.Lander E S. Science. 1996;274:536–539. doi: 10.1126/science.274.5287.536. [DOI] [PubMed] [Google Scholar]
- 9.Goffeau A, Barrell B G, Bussey H, Davis R W, Dujon B, Feldmann H, Galibert F, Hoheisel J D, Jacq C, Johnston M, Louis E J, Mewes H W, Murakami Y, Philippsen P, Tettelin H, Oliver S G. Science. 1996;274:546. doi: 10.1126/science.274.5287.546. , 563–567. [DOI] [PubMed] [Google Scholar]
- 10.Szostak, J., ed. (1997) Chem. Rev. (Washington, DC) 97, 347–510.
- 11.Furka A, Sebestyen F, Asgedom M, Dibo G. Int J Pept Protein Res. 1991;37:487–493. doi: 10.1111/j.1399-3011.1991.tb00765.x. [DOI] [PubMed] [Google Scholar]
- 12.Brenner S, Lerner R A. Proc Natl Acad Sci USA. 1992;89:5381–5383. doi: 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohlmeyer M H J, Swanson R, Dillard L W, Reader J C, Asouline G, Kobayashi R, Wigler M, Still W C. Proc Natl Acad Sci USA. 1993;90:10922–10926. doi: 10.1073/pnas.90.23.10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nestler H P, Bartlett P A, Still W C. J Org Chem. 1994;59:4723–4724. [Google Scholar]
- 15.Brown B B, Wagner D S, Geysen H M. Mol Divers. 1995;1:4–12. doi: 10.1007/BF01715804. [DOI] [PubMed] [Google Scholar]
- 16.Hall M, Peters G. Adv Cancer Res. 1996;68:67–108. doi: 10.1016/s0065-230x(08)60352-8. [DOI] [PubMed] [Google Scholar]
- 17.Fields S, Song O. Nature (London) 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 18.Fields S, Sternglanz R. Trends Genet. 1994;10:286–292. doi: 10.1016/0168-9525(90)90012-u. [DOI] [PubMed] [Google Scholar]
- 19.Golemis E A, Gyuris J, Brent R. In: Current Protocols in Molecular Biology. Ausubel F M, Brent R, Kingston R, Moore D, Seidman J, Smith J A, Struhl K, editors. New York: Wiley; 1996. p. 20.1. [Google Scholar]
- 20.Allen J B, Walberg M W, Edwards M C, Elledge S J. Trends Biochem Sci. 1995;20:511–516. doi: 10.1016/s0968-0004(00)89119-7. [DOI] [PubMed] [Google Scholar]
- 21.Vojtek A B, Hollenberg S M. Methods Enzymol. 1995;255:331–342. doi: 10.1016/s0076-6879(95)55036-4. [DOI] [PubMed] [Google Scholar]
- 22.Aronheim A, Zandi E, Hennemann H, Elledge S J, Karin M. Mol Cell Biol. 1997;17:3094–3102. doi: 10.1128/mcb.17.6.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M M, Reed R R. Nature (London) 1993;364:121–126. doi: 10.1038/364121a0. [DOI] [PubMed] [Google Scholar]
- 24.Li J J, Herskowitz I. Science. 1993;262:1870–1874. doi: 10.1126/science.8266075. [DOI] [PubMed] [Google Scholar]
- 25.SenGupta D J, Zhang B, Kraemer B, Pochart P, Fields S, Wickens M. Proc Natl Acad Sci USA. 1996;93:8496–8501. doi: 10.1073/pnas.93.16.8496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Aelst L, Barr M, Marcus S, Polverino A, Wigler M. Proc Natl Acad Sci USA. 1993;90:6213–6217. doi: 10.1073/pnas.90.13.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belshaw P J, Ho S N, Crabtree G R, Schreiber S L. Proc Natl Acad Sci USA. 1996;93:4604–4607. doi: 10.1073/pnas.93.10.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rivera V M, Clackson T, Natesan S, Pollock R, Amara J F, Keenan T, Magari S R, Phillips T, Courage N L, Cerasoli F, Jr, Holt D A, Gilman M. Nat Med. 1996;2:1028–1032. doi: 10.1038/nm0996-1028. [DOI] [PubMed] [Google Scholar]
- 29.Licitra E J, Liu J O. Proc Natl Acad Sci USA. 1996;93:12817–12821. doi: 10.1073/pnas.93.23.12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leanna C A, Hannink M. Nucleic Acids Res. 1996;24:3341–3347. doi: 10.1093/nar/24.17.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vidal M, Brachmann R K, Fattaey A, Harlow E, Boeke J D. Proc Natl Acad Sci USA. 1996;93:10315–10320. doi: 10.1073/pnas.93.19.10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shih H M, Goldman P S, DeMaggio A J, Hollenberg S M, Goodman R H, Hoekstra M F. Proc Natl Acad Sci USA. 1996;93:13896–13901. doi: 10.1073/pnas.93.24.13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnston M, Davis R W. Mol Cell Biol. 1984;4:1440–1448. doi: 10.1128/mcb.4.8.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartel P L, Roecklein J A, SenGupta D, Fields S. Nat Genet. 1996;12:72–77. doi: 10.1038/ng0196-72. [DOI] [PubMed] [Google Scholar]
- 35.Borchardt, A. B., Liberles, S. D., Biggar, S., Crabtree, G. R. & Schreiber, S. L. (1997) Chem. Biol., in press. [DOI] [PubMed]
- 36.You, A., Jackman, R. J., Whitesides, G. M. & Schreiber, S. L. (1997) Chem. Biol., in press. [DOI] [PubMed]
- 37.Wang T, Donahoe P K, Zervos A S. Science. 1994;265:674–676. doi: 10.1126/science.7518616. [DOI] [PubMed] [Google Scholar]
- 38.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 39.Guthrie C, Fink G R, editors. Guide to Yeast Genetics and Molecular Biology. San Diego: Academic; 1991. [Google Scholar]
- 40.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gyuris J, Golemis E, Chertkov H, Brent R. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- 42.Chevray P M, Nathans D. Proc Natl Acad Sci USA. 1992;89:5789–5793. doi: 10.1073/pnas.89.13.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guldener U, Heck S, Fielder T, Beinhauer J, Hegemann J H. Nucleic Acids Res. 1996;24:2519–2524. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ronne H, Rothstein R. Proc Natl Acad Sci USA. 1988;85:2696–2700. doi: 10.1073/pnas.85.8.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Combs A P, Kapoor T M, Feng S, Chen J K, Daude-Snow L, Schreiber S L. J Am Chem Soc. 1996;118:287–288. [Google Scholar]
- 46.Chiu M I, Katz H, Berlin V. Proc Natl Acad Sci USA. 1994;91:12574–12578. doi: 10.1073/pnas.91.26.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stan R, McLaughlin M M, Cafferkey R, Johnson R K, Rosenberg M, Livi G P. J Biol Chem. 1994;269:32027–32030. [PubMed] [Google Scholar]
- 48.Vidal M, Braun P, Chen E, Boeke J D, Harlow E. Proc Natl Acad Sci USA. 1996;93:10321–10326. doi: 10.1073/pnas.93.19.10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boeke J D, LaCroute F, Fink G R. Mol Gen Genet. 1984;197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]