

i. Summary
Experiments aimed at analyzing the response of blood vessels to mechanical injury and ensuing remodeling responses often employ the highly characterized carotid artery balloon injury model in laboratory rats. This approach utilizes luminal insertion of a balloon embolectomy catheter into the common carotid artery with inflation and withdrawal resulting in an injury characterized by vascular endothelial cell (EC) denudation and medial wall distension. The adaptive response to this injury is typified by robust vascular smooth muscle cell (SMC) replication and migration, SMC apoptosis and necrosis, enhanced synthesis and deposition of extracellular matrix (ECM) components, partial vascular EC regeneration from the border zones, luminal narrowing and establishment of a neointima in time-dependent fashion. Evaluation of these adaptive responses to blood vessel injury can include acute and longer-term qualitative and quantitative measures including expression analyses, activity assays, immunostaining for a plethora of factors and signals, and morphometry of neointima formation and gross mural remodeling. This chapter presents a logical continuation of Chapter in this series that offers details for performing the rat carotid artery balloon injury model in a standard laboratory setting by providing commonly used protocols for performing histological and morphometric analyses in such studies. Moreover, procedures, caveats, and considerations included in this chapter are highly relevant for alternative animal vascular physiology/pathophysiology studies and in particular those related to mechanisms of vascular injury and repair. Included in this chapter are specifics for in situ perfusion-fixation, tissue harvesting and processing for both snap-frozen and paraffin-embedded protocols, specimen embedding and sectioning, slide preparation, several standard histological staining steps, and routine morphological assessment. Included in Notes are important caveats and considerations for practical use of these methods.
Keywords: balloon injury, embedding, harvesting, histology, medial wall, microscopy, morphometry, neointima, perfusion-fixation, processing, rat carotid artery, remodeling, sectioning, staining
1. Introduction
Use of histological and morphometric techniques is essential for examination of disease processes as well as for the investigation of many of the mechanisms that contribute to a wide variety of tissue pathologies. As a logical continuation of varied animal survival surgeries, these techniques involve tissue fixation and harvesting from the animal post-mortem, tissue processing, embedding, and sectioning, microscope slide preparation, tissue staining for a multitude of cellular and/or molecular components in the specimen, and analysis via microscopy by the investigator(s). Germane to animal vascular studies and specifically the rat carotid artery balloon injury model (described in Chapter of this series), this chapter details commonly used “histo-techniques” for routine performance of several of these key steps. Particular emphases are placed on protocols for performing in situ perfusion-fixation, tissue harvesting for both fixed and fresh samples, several commonly-employed staining procedures, and standardized morphometric analyses. Based on the breadth of histological and morphometric protocols currently available, many of which are suitable for vascular tissues, this chapter is not intended to serve as a comprehensive summation of all techniques and strategies but rather as a general guide for continuance of studies employing the rat carotid artery balloon injury model. Thus, only brief outlines of protocols are provided for several of these steps. For interested readers, excellent comprehensive resources are available for consultation on a wide variety of histological and morphometric techniques (1, 2), including a summary of procedures specific to animal vascular studies (3). In conclusion of this chapter, Notes provides important considerations for practical utility of these protocols.
2. Materials
Materials needed to perform these procedures are listed below in sub-headings for the various steps. Where appropriate, brief mention of their use is included along with item numbers, name-brands, and/or manufacturer preferred by the author.
2.1. In situ perfusion-fixation
-
Solutions:
-
Supplies:
ample lighting
rack and clamps (for perfusion-fixation apparatus)
3-way stopcocks with luer-lock end (used for attachment of tubing sections)
2-way stopcocks with luer-lock end (used for attachment of short section of tubing to the over-the-needle catheter)
tubing (3/16″ inside diameter, 5/16″ outside diameter vinyl, Fisher Scientific)
needles (26 g. for anesthetic)
syringes (1 ml for anesthetic and for washing lumen ex vivo; 50–60 ml capacity for perfusion-fixation apparatus)
intravascular over-the-needle catheter (18–20 g., 1½–2.0″; see Note 5) or similar guiding needle collecting pan (for perfusion-fixation and animal waste)
gauze
cotton tipped swabs (have plenty of these available)
face mask or shield, protective goggles, and/or gloves (per institutional guidelines)
tape (cut into sections)
-
surgical instruments:
scissors, large (#RS-6942, Roboz)
scissors, medium (#RS-5910, #RS-5912SC, Roboz)
scissors, fine (#RS-5600, Roboz)
forceps, heart-holding (#RS-5255, #RS-8270, Roboz) or other large serrated-edged
forceps, medium
forceps, small curved (#RS-4935, Roboz)
micro-caliper (#RS-6460, Roboz) or small metric ruler (to measure vessel lengths)
waste container
necropsy bag
2.2. Tissue harvesting
-
For perfusion-fixed tissues:
-
For fresh tissues:
ice and ice cooler (for cleaning vessel ex vivo)
liquid nitrogen (for snap-freezing)
dry ice (for snap-freezing)
methanol (for snap-freezing)
freezing medium (Super Friendly Freeze-It, Fisherbrand, or other suitable cytological fixative)
Petrie dish (for cleaning vessel ex vivo)
gauze
cotton tipped swabs
labeled tubes
deep freezer (−80°C)
alcohol-proof marking pen
2.3. Tissue processing
-
Solutions:
-
Supplies:
processing cassettes with lids (Unisette, Omnisette, or Histosette II, Fisherbrand)
Kimwipes or other fine grade porous tissues (mainly used for small and/or highly valuable tissues)
automated tissue processor (Tissue Tek)
coplin jars or glass staining dishes with covers (if manual processing is performed)
pencil (for labeling embedding blocks/cassettes)
2.4. Embedding
-
For paraffin-embedded tissues:
paraffin (TissuePrep or Tissue Path Paraplast, Fisher, or Paramat)
paraffin repellent (PARA/GARD, TBS)
embedding blocks or rings (Tissue Path, Fisher)
stainless steel base molds (HistoPrep, Fisher)
paraffin embedder with cold plate (an automated machine is preferred)
forceps heater (highly recommended)
-
For frozen tissues:
tissue freezing medium (TFM or O.C.T., TBS)
embedding blocks or rings (Tissue Path, Fisher)
stainless steel base molds (HistoPrep, Fisher)
2.5. Sectioning
-
For paraffin-embedded tissues:
rotary microtome (or other micro-sectioning tool)
microtome stainless-steel blades (disposable)
flotation bath (TissuePrep circular flotation bath, Fisher)
granular gelation for flotation bath (to increase surface tension of water, but is not essential)
fine forceps
small brush
water (diethyl pyrocarbonate (DEPC)-treated; see Section 3.5., Note 8)
-
For frozen tissues:
Cryostat (or other micro-sectioning tool used for frozen specimens)
cryostat blades
2.6. Slide preparation
incubator or slide warmer
microscope slides (Superfrost, Fisher)
2.7. Routine staining procedures for histomorphometry
cover slips/cover glasses
mounting medium (Permount, Fisher)
light microscope (for visual inspection of tissue samples, staining efficacy)
glass jars with lids (to hold staining solutions)
staining solutions (see individual recipes, Section 3.7.)
2.8. Microscopic and morphological evaluation
ultra-fine point permanent marker (Sharpie)
light microscope with 10x, 40x objectives (and other objectives based on personal preference)
complete image analysis system with camera, interface, software of choice, and computer graphical and statistical software of choice
2.9. Endothelial cell regeneration
Evans blue dye (see Note 9)
large beaker
hot water
syringe (1 ml) with needle (26–28 g.)
small micro-scissors (McPherson-Vannas, #RS-5600, Roboz)
small curved forceps (Micro-dissecting tweezers, Pattern 7S, #RS-4935, Roboz)
dissecting dish with silicone pad and pins (# 70540, Electron Microscopy Services)
small ruler or other measuring device (caliper)
3. Methods
Histological preparation of animal tissues following an experimental procedure or intervention generally encompasses the following steps performed in succession: tissue fixation, processing, embedding, sectioning and slide preparation, staining, and microscopic analysis.
3.1. In situ perfusion-fixation
The primary purpose for perfusing the animal carcass immediately following euthanasia is to rid the tissues of interest of resident blood components which can cause interference in ensuing microscopic examination. Once tissues are removed from a body however, they can rapidly undergo autolysis, putrefaction, and environmental degradation. Fixation is a complex series of chemical events that “fixes” or retains tissues as close to their living state as possible and preserves tissue integrity, from correct anatomical orientation to ultrastructure, without rearrangement or loss of vital cellular and/or molecular components. These processes are performed in situ at mean arterial pressure in an effort to maintain the vasculature at normal distending pressures.
Setup
Preparation of all reagents and solutions, catheters, supplies, etc. should be accomplished ahead of the time when performing the perfusion-fixation protocol. This is especially important if acute studies are being conducted when timing is critical. As is the case when performing animal survival surgery, the initial steps of this protocol involve manipulations on a living animal and therefore preparedness and expediency are essential. Prepare PBS or saline (or vasodilator) solutions ahead of time and set in a 37°C incubator until temperature is reached. Prepare fixative at desired concentration or percentage. It is useful to have individual aliquots (60–100 ml, depending on protocol) of each solution readily available to simplify pouring the solutions into the syringes of the perfusion-fixation apparatus.
The perfusion-fixation apparatus (see Figs. 1, 2) must be constructed ahead of time if it is to be used (instead of a perfusion pump, which will not be discussed) for perfusion-fixation. Using a tall rack stand situated on a laboratory bench, attach two clamps to the stand at a level that results in a column of fluid approximately equal to 100 mmHg (see Note 10). This is calculated using the fact that 1.36 cm of water (or fluid of similar viscosity) in a column equals 133 Pascals (1333 dynes/cm2) which also equals 1 mm Hg. A mean pressure of 100 mmHg therefore equates to a column of fluid 136 cm, or approximately 53.5 inches, high (similarly, a mean pressure of 120 mmHg equates to a column of fluid with height 64.25 inches). This is the height for the column of PBS (or vasodilator solution) and for fixative, respectively, each in individual syringes with individual tubing (up to a point). Remove the plungers and protective caps from two 50–60 ml syringes and attach the ends of the tubing cut to appropriate lengths (to make a column 53.5 inches high for 100 mmHg pressure) to the “needle ends” of the syringes (syringes are held upside down). Next, attach the other two ends of the tubing each to one of the available ends of the 3-way stopcock (not the luer-lock end) (see Figs. 1, 2). Now, using an additional short piece of tubing (8–12 inches) attach it to the luer-lock end of the 3-way stopcock and then attach a 2-way stopcock to the other end of this short piece of tubing. To this 2-way stopcock the luer-lock hub of an over-the-needle catheter (used for cardiac puncture and perfusion) will be fastened. Attach the two syringes in an inverted fashion on the clamps already fixed to the rack and let the tubing drape to the floor (see Figs. 1, 2, Note 11). Clearly label one syringe “PBS” or “vasodilator” and the other one “FIXATIVE”.
Figure 1.

A diagram of the apparatus used for performing in situ perfusion-fixation on rat carotid arteries. With the use of a laboratory bench rack, two clamps and syringes, tubing, stopcocks, and an over-the-needle catheter this apparatus can be easily constructed for assistance in performing histomorphometric studies in a variety of animal tissues. This apparatus utilizes two independent columns of fluid that approximate pressures of 100 mmHg (see text for detailed description), and is used for trans-cardial perfusion and fixation of rat carotid arteries with PBS (or vasodilator solution). Tissues are then harvested for processing, embedding, sectioning, and staining with subsequent analytical evaluation.
Figure 2.

Photographs of an in situ perfusion-fixation apparatus. A: complete apparatus; B: close-up photo of over-the-needle catheter (with catheter removed) attached to stopcock and tubing.
When choosing tubing, pick a size that can fit relatively easily but snugly onto the ends of the syringes, the stopcocks, and the luer-lock hub of the over-the-needle catheter. Since pressure equals force (acceleration of gravity) per unit area, then the cross-sectional area of the column (or tubing) will not affect the pressure calculation (see Note 10). The author uses 3/16″ (inside diameter) tubing in his laboratory setup with much success; however, slightly smaller or larger tubing can also be used as long as it fits tightly onto both of the stopcocks and the over-the-needle catheter. Become familiar with opening and closing the stopcocks so that this maneuver can be performed with one hand. Practice using water in both syringes and opening and closing the two in-series stopcocks to see the magnitude and speed of flow from the 2-way stopcock or catheter end (see Note 12). Make sure the apparatus is in proper working condition and that the stopcocks are firmly attached to the tubing and no leaks are apparent prior to performing the protocol. Ahead of the time (~15–20 min) for performing the perfusion-fixation and with both stopcocks in closed position fill the appropriate (labeled) syringe with ~60 ml warmed PBS (or vasodilator) and the other one with ~60 ml fixative (see Note 13). Now, importantly open the stopcocks so that only fixative will flow through the tubing, through the first stopcock and short segment of “common” tubing, and through the distal stopcock and out. Close both stopcocks and remove any trapped air bubbles in the stopcocks and tubing. Next, open the stopcocks to allow only PBS (or dilator solution) to run through the tubing and stopcocks and out. Close the stopcocks to stop flow and purge all air bubbles. The order of performing this preparatory step is very important and critical for adequate perfusion and fixation of tissues (see Note 14). Refill the syringes with PBS and fixative solutions to account for fluid loss in this preparatory step.
Several other steps need to be completed prior to the perfusion protocol. Prepare all surgical instruments with thorough cleaning and proper convenient placement in the surgical area. Make sure adequate numbers of cotton tipped swabs and pieces of gauze are available and located nearby. Cut sections of black 4-0 suture and have these nearby as well. Prepare labeled tubes (using alcohol-proof markers) filled with either fixative (for post-fixation of ~4 hours) or 70% ethanol (to hold tissues following post-fixation and until ready for processing). For each tissue 2 tubes will be made available, one with fixative and the other with 70% ethanol.
Protocol
Once again, before anesthetizing the animal make sure that everything is ready for the protocol so that delays do not occur after the animal has been sedated. If recommended by the institutional animal care and use committee and based on specifics of the research study, the surgeon should choose to wear a face mask or shield, protective goggles, and/or gloves as appropriate. Also, it is recommended to perform this protocol with the perfusion-fixation apparatus situated in a fume hood so that fumes from the fixative do not affect the surgeon or other personnel in the immediate area. Weigh the rat and provide anesthetic (as routine) to achieve a surgical plane of anesthesia. Wait until the animal is fully sedated, and then follow with an overdose amount of anesthetic (recommended 2.5x original dose or according to institutional guidelines). Monitor breathing and heart rate patterns. The animal should be extremely sedated so that overdose is imminent. Needless to say, make sure that the anesthetic does not cause unwanted bias or interfere with the scientific aims of the study.
Place the animal on their back on a surgical tray with the head towards you and tape down the arms and legs. The surgeon may also want to loosely wrap a suture around the teeth of the rat and tape down the ends (to hold the head still). Working quickly, using large serrated-edge scissors make a midline incision starting at the sub-sternal notch through the sternum (and ribcage), moving in a caudal direction. Keep the tips of the scissors up in order to avoid puncture of heart or underlying tissues. Keep cutting through the sternum and ribcage all the way down to the diaphragm. It is important to work rapidly here once the initial cut is made, considering that euthanasia is being induced via pneumothorax (followed by overdose) and the fact that expediency will usually provide more accurate data related to the living condition. Once the incision has been made through the entire ribcage, using large forceps or hemostats gently retract the ribs apart from each other to expose underlying tissues. Do not pull too tightly at this stage, as that could crush the tissues of interest (which lie directly below this opening).
Now the thoracic aorta will be clamped in order to perfuse only the upper thorax, neck, and head of the animal (see Note 13); however, if for scientific or other reasons the investigator chooses to perfuse the entire animal, then this next step can be avoided. Rotate the surgical tray 90° to the right so that now the animal is lying horizontally on the surgical table with head facing the right. Still using the large scissors, at the bottom of the ribcage where the initial cut ended make a longitudinal cut along the diaphragm towards the back on the left side of the animal. The left side of the animal is used in this step because access to the thoracic aorta is straight-forward. Separate the ribs a bit to expose the lungs and gently move those aside to expose the underlying thoracic aorta. This appears as a whitish band running caudally on top of the vertebral column. Clamp the thoracic aorta with large hemostat and lay the hemostat to the side of the animal carcass and out of the way.
Next, rotate the surgical tray another 90° to the right (now the head of the animal is directed away from the surgeon) and using large forceps carefully retract the ribs to further expose the underlying carotid vasculature and the heart and aorta. At this point the surgeon may need to make an additional cut through the ribs to gain visual access to the heart and aorta. If needed to adequately visualize the aortic root, carefully remove any fatty tissues lying on top of the heart. Using heart-holding or blunt forceps gently grasp the heart and insert the over-the-needle catheter tip into the apex and advance it fully into the left ventricle (see Note 5). Retract only the needle leaving the catheter sheath indwelling and advance it straight inwards until it appears in the aortic root immediately as it leaves the heart. If resistance is met, pull back on the catheter and attempt advancing it to the aorta again. Once in place gently attach the end of the catheter onto the luer-lock end of the 2-way stopcock (at end of tubing) and carefully tape down the catheter end and/or tubing to keep it secure.
At this point the surgeon needs to make sure that the animal carcass (along with the catheter in place and tubing attached) is at a point where perfusion will occur at mean arterial pressure (see Figs. 1, 2). If following the steps used by the author this will involve carefully moving the surgical tray containing the animal to the floor just below the perfusion-fixation apparatus. When the animal carcass is in place, using small scissors carefully cut the right atrium to make an outflow and carefully open the stopcocks to allow flow-through of only the warm PBS (or vasodilator solution) through the system. If adequate perfusion takes place the surgeon will be able to immediately see blood leaving the incised right atrium and exiting the body (see Note 15). The color of the outflow will gradually become clearer as blood is removed from the upper thorax, neck, and head regions of the animal. Monitor this outflow to ensure adequate perfusion. Also importantly watch the level of PBS (or dilator) in both the syringe and the tubing to make sure that air does not enter the animal (see Note 16).
When the fluid leaving the right atrium is clear and after an adequate volume of PBS (or dilator solution) has perfused the animal, carefully switch the 3-way stopcock to now allow fixative to flow through the system and to enter the animal. Again, monitor the level of the fixative and make sure that air does not enter the animal. As the fixative flows through the body, sometimes (but not always) the muscles will twitch in tetany as indication that adequate tissue fixation is occurring. Once the tissues are completely fixed “rigor” will set in the upper body and tissues will be hard. At this point remove the catheter from the heart and place the animal carcass on an absorbant pad on the operating table for tissue harvesting (see Note 17). Quickly run liberal volumes of clean water along with disinfectant through both syringes and the entire perfusion-fixation apparatus including the catheter and needle and collect in a pan. The surgeon may want to use heparin on both the needle and over-the-needle catheter to dissolve any existing blood clots that have occurred. The surgeon will now want to proceed with tissue harvesting, but at a convenient later time clean all instruments and clean and sterilize the surgical area with alcohol.
3.2. Tissue harvesting
For perfusion-fixed tissues
The animal carcass should now be on the surgical table with the upper thorax, neck, and head completely fixed and with the carcass situated so that the head of the animal is directed towards the surgeon. The author suggests wearing a face mask at this point. Using forceps and small scissors remove all tissue and muscular fascia surrounding the left carotid artery and expose the entire left carotid vasculature from the bifurcation to the aorta. Carefully free the left carotid artery from all adjacent tissues and especially the underlying connective fascia. Using fine scissors cut the distal end of the carotid artery at the bifurcation, gently lift it up, and separate the entire length of the artery from extraneous tissues in a caudal direction to the aorta. Cut the carotid artery from the aorta and place it in a Petrie dish filled with fixative, making sure to maintain proper orientation of the vessel as it was inside the animal! It helps to have a small piece of gauze soaked in fixative inside the Petrie dish. Gently clean extraneous tissue still attached to the vessel and make sure that the lumen is free of blood clots. If blood components are still resident inside the lumen, gently flush the lumen with fixative (via syringe and needle) to remove these. To ensure proper vessel orientation throughout the processing steps, at this point attach a small loop of suture around the most distal end of the artery and make a note of this. Place the left carotid artery in an appropriately labeled vial containing the same fixative, incubate (post-fix) for an additional 4 hours (see Note 18), and then transfer the tissues to vials containing 70% alcohol until ready for processing. Next perform the exact same protocol for the right carotid artery. Once vessels are in fixative, make all relevant notes about the time of incubation of the samples in fixative, the efficiency/efficacy of the perfusion-fixation protocol, or any other unique observations that might impact the results and/or data interpretation. Place the animal carcass in a necropsy bag, close securely, and place in an appropriate freezer or other container according to institutional guidelines.
The author will now discuss a method that can be used for tissue harvesting if the investigator has incorporated a protocol whereby a certain treatment has been applied to the exposed distal portion of the left carotid artery vasculature immediately following balloon injury (of course, this is based on specifics of the research design but is a commonly used method). In this case, the entire length of the left carotid artery will have been injured, yet only the distal portion (approximately half) will have been treated with a drug, blocking antibody, or other agent (see Fig. 3) (4). During harvesting proceed as usual until the entire length of the common carotid is exposed and freed from underlying and adjacent tissues. Remove the entire length of the left carotid artery (as mentioned before) and place in a Petrie dish filled with fixative making sure to maintain proper orientation of the vessel! Once the vessel is cleaned cut the vessel approximately in half and tie a suture around the distal end of each vessel section. Place each vessel section into individually-labeled tubes (labeled “distal LCA” or “proximal LCA” or similar) containing fixative, post-fix for 4 hours (see Note 18), and then place tissues in 70% alcohol until ready for processing. Harvest the right carotid artery as routine.
Figure 3.

A scheme for treating the balloon-injured distal section of a carotid artery for comparison to the injured untreated proximal section from the same animal (re-drawn from Ref. 4). The entire length of the left carotid artery (LCA) is balloon injured, yet only the distal portion is subjected to a treatment of choice (as determined by the investigator). The right carotid artery (RCA) serves as an uninjured inter-animal control. This scheme can be used for designing appropriate research experiments as well as for histological procedures utilizing inter-animal comparisons. ECA: external carotid artery; ICA: internal carotid artery.
For fresh tissues
When using specific antibodies for immunostaining that work optimally on fresh tissues or when performing certain protocols such as Western blotting, fresh unfixed tissues must be removed from the animal expediently and snap-frozen. Two protocols are pertinent here, one using fresh tissues for cryostat sectioning and for making slides, and one utilizing fresh tissues for Western blotting or similar experiments that incorporate whole un-sectioned tissues.
Prior to performing this method, collect ice in a small cooler and pack it to the top. Lay a Petrie dish on the ice, place a small piece of gauze inside the dish, and fill with PBS. Fill a container with liquid nitrogen and cover. If liquid nitrogen is not available, then partially fill a beaker with methanol, carefully add chunks of dry ice, and allow time for liquid to cool. Label vials accordingly using an alcohol-proof marker (or label the inside of the vial top and/or mark accordingly with tape). Once the animal is completely sedated and placed supine on a surgical tray with arms and legs retracted and with the head towards the surgeon, working quickly cut from just above the sternum through the ribs to the diaphragm (with scissor tips up) and expose the carotid and aortic vasculature and the heart (the same as performed for perfusion-fixation). Carefully separate the ribs to expose the underlying tissues. Control bleeding with ample use of gauze and/or cotton swabs. Using blunt dissection carefully remove the overlying muscles and all adjacent tissues to expose and isolate the left carotid artery. Cut the most distal end of the carotid artery at the bifurcation and hold the cut end gently with fine forceps. Delicately holding the end of the vessel with the forceps cut the remainder of the carotid artery free from associated tissues caudally all the way to the aortic arch and remove the vessel from the animal. Immediately place the artery in ice cold PBS (in the Petrie dish on ice) and promptly clean the artery of blood and extraneous tissue. If tissue is to be used for cryostat sectioning and to make slides, then tie a suture around the distal end of the artery, hold the artery upright inside an embedding block (or ring) situated in a stainless steel base mold on sitting on dry ice in a cooler, and fill the embedding block with tissue freezing medium until the vessel is covered. Keep the block on dry ice until completely frozen and then transport to a deep freezer (−80°C). As mentioned above, if the research plan calls for separate analyses of the distal and proximal sections of the carotid artery (if the distal end has been treated), then similar steps must be taken to appropriately define the individual sections and to maintain their proper orientation. If the tissue is to be used for Western blotting or another protocol that requires fresh whole tissue, once the vessel is clean simply place it in a pre-labeled empty vial and drop it into liquid nitrogen (or ethanol solution with dry ice) to ensure instant freezing. These procedures do not require placing suture around the vessel as the entire vessel section will be used in the ensuing method. Transfer the vials containing vessels to deep freeze until ready for use in a specific protocol. These steps are repeated for the right carotid artery. Follow-up procedures include collecting and thoroughly cleaning all instruments, cleaning and sterilizing the surgery area with 70% alcohol, making relevant notes on the technique and any other important observations, and proper disposal of the animal carcass according to institutional guidelines.
3.3. Tissue processing
For cryostat sectioning since the frozen tissues will go directly from harvesting into embedding in an appropriate freezing medium, no steps are included here specific for processing; therefore, the following descriptions are relevant to perfusion-fixed samples only. After the tissues have been fixed they must be dehydrated and cleared in order for adequate paraffin infiltration, embedding, and sectioning. This step is termed “tissue processing” and involves tissues incubated in a series of graded alcohols, usually 70% through 100%, to remove water. A first step in 30% alcohol can be used for delicate, highly valuable, or susceptible animal tissues. The next step is clearing which removes traces of dehydrant alcohol and uses an agent miscible with the embedding medium. Xylene is the most common clearing agent used for tissue processing for these tissues (see Note 6). The final step of processing is infiltration of the tissues in liquid paraffin or other embedding medium (see Note 7). For protocols involving rat vascular tissues, it is recommended that automated (mechanical) processing be used to ensure reproducibility and consistency from sample to sample. Automated processing involves movement of tissues (in tissue cassettes) through graded alcohols, xylenes, and liquid paraffin on a pre-set time scale. Newer processors have the ability to treat the samples with vacuum and/or heat during processing. These machines also allow individual protocols to be entered based on the desired incubation time in each of the solutions.
Processing of rat carotid arterial tissues starts following in situ fixation, post-fixation (4 hours), and storage of the samples in 70% alcohol at room temperature. At this point the orientation or the vessel will have been ensured with suture placement around the distal end. Using fine forceps carefully remove the vessel from 70% alcohol and place it directly in a pre-labeled (with pencil) embedding cassette and snap the lid closed (see Note 19). A 12-step processing procedure is employed for these tissues suitable for either manual or automated processing. If manual processing is used, then the investigator must first prepare all solutions in glass staining dishes (with lids). Volatile solutions should be placed in an appropriate fume hood. For automated processing, the timing clock must on the apparatus must first be set according to the time schedule for incubations. The following incubation schedule has been used successfully by the author for a variety of animal vascular tissues including rat carotid arteries; however, individual variations (within reason) can be used with anticipated success. Of note, all incubation steps are performed under vacuum and pressure and at room temperature except the final 2 steps (liquid paraffin), which are performed under elevated temperature. The author routinely performs tissue processing during the overnight hours with the sample incubating in liquid paraffin (step 12) and ready for embedding the next morning.
| Processing step: | Bath: | Time (min): |
|---|---|---|
| 1 | 50% ethanol | 120–180 |
| 2 | 80% ethanol | 25 |
| 3 | 80% ethanol | 25 |
| 4 | 95% ethanol | 40 |
| 5 | 95% ethanol | 40 |
| 6 | 100% ethanol | 40 |
| 7 | 100% ethanol | 40 |
| 8 | 100% ethanol | 55 |
| 9 | xylene | 60 |
| 10 | xylene | 90 |
| 11 | paraffin, liquid (at 58–60°C) | 90 |
| 12 | paraffin, liquid (at 58–60°C) | ≥ 90 |
3.4. Embedding
Frozen tissues will have been harvested fresh from the animal, cleaned while immersed in ice cold PBS, suture tied around the distal end to ensure orientation, and quickly embedded in an appropriate medium, frozen, and ready for cryostat sectioning. Generally frozen tissues are embedded in an unfixed state; however, certain procedures require use of a post-fixation step (generally formol-calcium) to reduce chance of diffusion of labile substances (1). Paraffin-embedded tissues in holding cassettes will be immersed in liquid paraffin in the final stage of processing. The use of a paraffin-embedder with a cold plate is highly suggested for this step, although manual embedding can be performed using melted paraffin in a suitable container over a hot plate and an ice bucket or other cold container for wax hardening.
When using a paraffin-embedder, make sure the embedder is pre-heated (at least 3 degrees above the melting point of the wax), that all the paraffin pellets are in liquified form, and that the cold plate is pre-chilled. Run a test embedding block to make sure the machine is fully operational and that the paraffin hardens after a short time on the cold plate. Use of a forceps heater during the embedding procedure is highly recommended and will ease manipulation of the tissue sections. Remove the cassette from the processor (in liquid paraffin) and place it on the heating block of the embedder and gently open. Carefully remove the vessel using fine forceps and place the vessel alone on the heating block (to keep the wax melted). Place a pre-labeled embedding block or ring in a stainless steel base mold and also place this on the heating block. Now, with one hand hold the embedding block and base mold under the paraffin dispenser and with forceps in the other hand carefully pick up the vessel so that it can be embedded in proper orientation (according to the research design) inside the embedding block. The author normally embeds vascular tissues with the suture-end down first, thereby during sectioning the samples are taken first from the most distal portion of the vessel continuing in a proximal direction. Holding the vessel absolutely still in correct orientation and perfectly vertical (only if vessel cross-sections are desired; see Note 20) inside the embedding block, fill the block with liquid paraffin around the vessel, release the vessel from the forceps, and carefully move the filled block to the cold plate. Be careful when releasing the vessel from the forceps as the wax can cause them to stick together and move the vessel out of its vertical position. In short time the block will start to harden. Once the paraffin is completely solidified, move the block from the cold plate, release the stainless steel base mold, and store the block at room temperature until ready for sectioning.
3.5. Sectioning
For paraffin-embedded tissues
After tissues have been embedded in paraffin and hardened, sections must be made and placed on a specimen slide for microscopic inspection and analysis. Prior to this step make sure to pre-heat (~10°C below the melting point of the wax) a flotation bath containing diethyl pyrocarbonate (DEPC)-treated water or other suitable agent (see Note 8). Diethyl pyrocarbonate derivitizes histidine residues and is therefore an effective method to inactivate nucleases including RNAse. Have the flotation bath located adjacent to the microtome and next to a supply of microscope slides. Sectioning of paraffin-embedded tissues is performed with a microtome that allows precisely thin and standardized tissue cross-sections to be cut in repetitive manner. Sections in paraffin can be cut between 3 and 10 microns in thickness, but usually range between 5 and 8 microns. The author uses 5 micron sections for rat carotid arteries to be used in standard histology procedures. Keeping a very sharp microtome knife blade is essential for proper cutting (see Note 21). First, place the embedding block on the specimen holder of the microtome with the vessel to be cut directed towards the investigator. Set the desired thickness of the section to be cut on the microtome control. Slowly rotate the microtome handle and the tissue block will advance the desired distance (5 microns) with each rotation. Prior to the first cut, make sure that the arm holding the embedding block has not advanced a distance that will cut the entire embedding block in half! To be sure to avoid this, make sure the arm is retracted a sufficient distance prior to placing the embedding block in it and prior to rotating the microtome handle. With each microtome cut, the block will move down and over the immovable microtome blade and a thin section of wax (containing the tissue) will be placed on the blade itself. With adequate training and experience, “ribbons” or “sheets” of adjacent serial paraffin sections can be cut from a single block. Next, using forceps or a small brush carefully move the cut paraffin sections to the heated flotation bath nearby that will help to expand the wax and remove wrinkles or folds from the tissues. During this step it is recommended that the trailing end of the wax ribbon or sheet make contact with the water first, thus producing a slight drag that will help remove these wrinkles and folds. Keep the wax sections floating on the water only a short time (≤ 30 sec) before picking them up on the slides (discussed below). Longer times spent floating on the heated water could drastically expand the wax and distort the tissue it contains. Several common problems associated with microtomy and tissue sectioning are described in Note 22 as well as in available resources (1, 2).
Frozen tissues are cut in a similar fashion and employ use of a cryostat, basically a microtome inside a refrigerated container. Temperatures within the cryostat normally range between −15 and −30°C, thus keeping the sections frozen throughout the cutting procedure. In particular, cryostat sectioning of non-fatty unfixed tissues including rat carotid arteries works optimally at around −25°C. Vascular tissues in the freezing embedding medium in the embedding blocks (prepared immediately following harvesting from the animal post-mortem with subsequent cleaning) are placed on the specimen holder of the cryostat and the proper thickness is set on the control. Like microtomy, rotation of the cryostat handle will cut tissue sections at desired thickness. Tissues are not placed in a flotation bath while using the cryostat but instead the cut tissue sections can be directly picked up on microscope slides, dried, and made ready for additional histological approaches.
3.6. Slide preparation
At this step paraffin-embedded sections are floating on DEPC-treated water in the flotation bath and are ready to be picked up on microscope slides. This procedure is straight-forward yet the investigator should attempt to capture the wax sections completely flat on the slide with the wax spread out evenly in all directions. Once the wax sections containing tissue have been picked up on the pre-labeled slides (see Note 23), blot excess water and rest the slide on an angle on absorbant towels until dry. Place the slides flat in a pre-warmed incubator or drying oven (with temperature set at melting point of the wax) or slide warmer for at least 30 minutes to help desiccate the section and to enhance adherence of the section to the slide. If heat might harm certain antigens essential for immunostaining, then avoid this step or simply lower the temperature of the incubator or slide warmer. For delicate or highly valuable tissues it is recommended to incubate the slides at 37°C overnight. Once dried, store the slides in an appropriate dry location until ready for further histological assays. Tissue sections cut on the cryostat are ready to be picked up by a microscope slide following sectioning. These should be rested on an angle on absorbant towels, and when completely dried stored for further analyses.
3.7. Routine staining procedures for histomorphometry
The slides are now ready to undergo one of a variety of histological staining procedures according to the research design and experimental endpoints. A listing of various histological stains useful for animal vascular studies is available (3). In general, the initial steps of all staining procedures involve a reversal of the procedures taken through tissue processing and embedding. In other words, the paraffin wax that impregnated the tissue must now be extracted and the tissue must be rehydrated to allow water-soluble dyes to penetrate the sections. So, before staining can be performed the slides are de-paraffinized in xylenes and rehydrated through serial dilutions of ethanols (100%, 95%, 80%, 50%) to water. Once the wax is removed and the tissues hydrated, then protocols are followed pertinent to the stain desired. Once adequate staining is achieved, then the stained section on the microscope slide is cover-slipped with a thin glass or plastic sheet and with the use of an appropriate adhesive mounting medium. This protects and preserves the tissues and also provides enhanced optical resolution for microscopic viewing. Once the mounting media has dried, slides can be stored at room temperature for an extended period of time (see Note 24). Several commonly-used staining procedures for rodent vascular tissues are briefly described here. These are succinct and generalized guidelines for the reader to follow and the author recommends consulting more thorough references for details of these protocols prior to use (consult Refs. 1, 2).
Of all the dyes used for histomorphometry, a combination of hematoxylin and eosin (H&E) is the most common. Hematoxylin behaves like a basic dye and stains nucleic acids of the cell nucleus blue/black. However, hematoxylin itself is not a stain and the major oxidization product is hematein, an anionic natural dye with color properties. Hematein, though, needs an enhancing agent that strengthens the tissue-dye bond in order for it to stain the tissue. This agent, termed a mordant, is a metal cation such as aluminum, iron, or tungsten. The type of mordant used strongly dictates the types of tissues stained and the final colored product. Eosin, an acidic xanthene dye, stains collagen pink and cellular cytoplasm red. A brief method for H&E staining of paraffin-embedded tissue sections is listed below based on a standardized protocol (2).
-
H&E staining method:
de-paraffinize and rehydrate tissues to distilled water
remove fixative pigments (see Note 25)
stain with a hematoxylin of choice for an appropriate length of time (see Note 26)
wash in slow running tap water until sections become blue (≤ 5 min) (see Note 27)
differentiate in 1% acid alcohol (1% HCl in 70% ethanol) for 5–10 sec
wash in slow running tap water until sections again become blue (10–15 min) or dip slides into 0.05% ammonia water and then wash in running tap water for 5 min (see Note 27)
stain in 1% eosin for 10 min
wash in running tap water for 1–5 min (see Note 27)
dehydrate through serial solutions of graded alcohols, clear with xylene, dry, and coverslip.
Results from H&E staining are blue/black nuclei, red/pink cytoplasm, deep pink muscle fibers and fibrin, and red/orange red blood cells. An example of an H&E-stained rat balloon-injured carotid artery cross-section along with a close-up view in the inset is shown in Figure 4.
Figure 4.
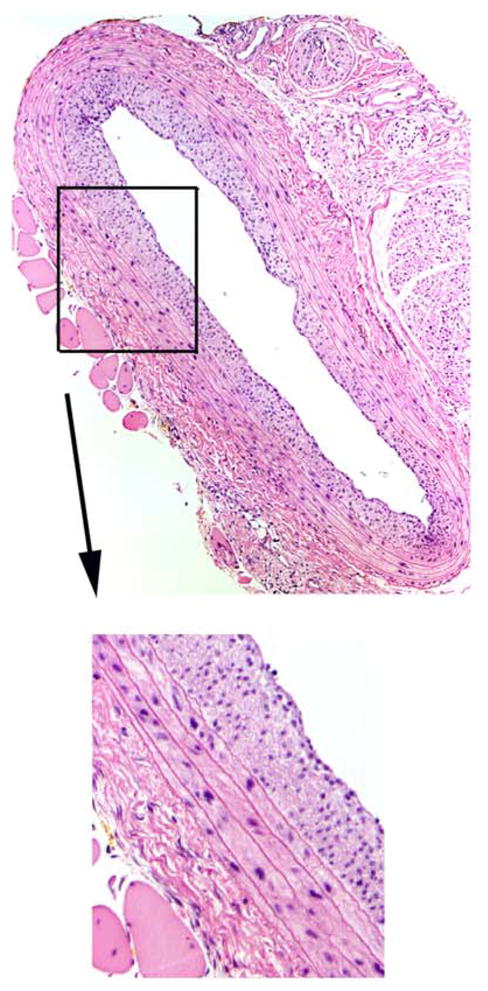
A cross-section of a rat balloon-injured carotid artery 2 weeks post-injury stained with hematoxylin and eosin (H&E) clearly depicts cellular-rich neointima development. Magnification is 100x for the large photomicrograph.
The most commonly used and instructive staining procedure employed by the author for rat carotid artery balloon injury studies is a combination of a hematoxylin stain using an iron mordant along with an acid fuchsin/picric acid solution. This staining procedure is termed Verhoeff’s elastic tissue stain with Van Gieson counterstain (VVG) and is used for routine histomorphometry and image analysis vital to animal vascular injury studies. Ferric chloride (10%) is included in the hematoxylin solution along with Lugol’s iodine and a 2% ferric chloride differentiation step. Elastin fibers stain intensely black and other cellular components are easily detectable, thus allowing for precise quantitation of routine morphometric parameters as discussed in Section 3.8. below.
Verhoeff’s elastic tissue stain with Van Gieson counterstain:
-
Verhoeff’s solution:
4% alcoholic hematoxylin (20 ml), 10% aqueous ferric chloride (8 ml), 2 g. Lugol’s iodine, 4 g. potassium iodine, 100 ml distilled water (8 ml)
-
Van Gieson solution:
1% aqueous solution of acid fuchsin (10 ml), saturated aqueous solution of picric acid (200 ml)
-
Protocol:
deparaffinize and rehydrate tissues to distilled water
stain for 20 min or until fibers are blue/black in Verhoeff’s solution (see Note 26)
wash in slow running warm tap water (see Note 27)
differentiate in 2% ferric chloride until background is clear
wash in slow running warm tap water (see Note 27)
place in 95% alcohol to remove iodine stain
wash in slow running warm tap water ≤ 5 min (see Note 27)
counterstain in Van Gieson solution for 3 to 5 min
dehydrate through serial solutions of graded alcohols, clear with xylene, dry, and coverslip.
Results from VVG staining are intensely blue/black elastic fibers, blue-to-black nuclei (based on variables for hematoxylin staining), and red collagen with associated tissues (fat, nerves) staining yellow. Several photomicrographs of VVG-stained cross-sections of a rat balloon-injured carotid arteries are shown in Figure 5 (see Figs. 6 and 7 in Chapter of this book series for additional VVG-stained photomicrographs of rat injured carotid arteries). Figure 5 also illustrates several common problems associated with the rat carotid artery balloon injury model and associated histomorphometry.
Figure 5.
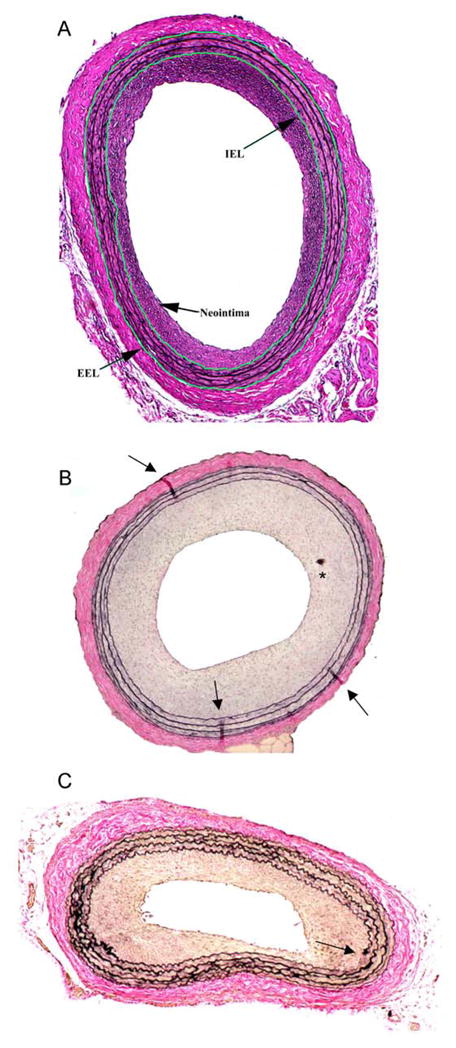
Variations in Verhoeff-van Gieson-stained cross-sections of rat balloon-injured carotid arteries 2 weeks post-injury. A depicts a concentric, elastin-rich neointima with the internal elastic laminae (IEL) and external elastic lamina (EEL) digitally traced and clearly defined. A hearty adventitial layer is clearly present. B shows a robust and concentric neointima containing artefacts (tissue folds, tissue debris) from improper histology (most likely problems associated with sectioning and/or slide preparation). Arrowheads indicate tissue folds; * indicates tissue debris deposited on the slide. C illustrates a non-concentric neointima (diminished in lower right corner) as a result of improper ballooning during surgery. Highly corrugated internal and external elastic lamina suggest that perfuse-fixation was not performed at an adequate distending pressure of the animal. Also evident is a break in the elastin staining of the internal elastic lamina (indicated by arrowhead). Magnification is 100x for all photomicrographs.
Figure 6.
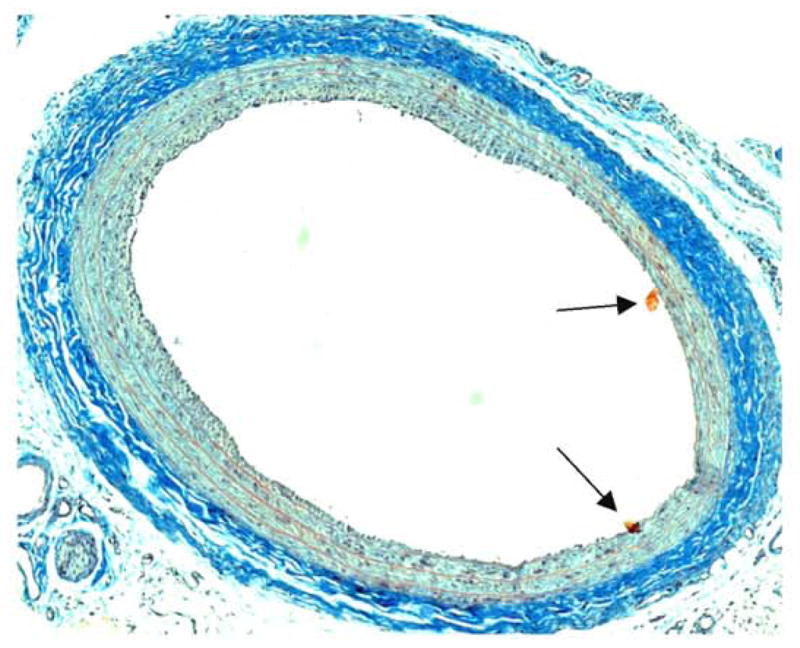
A cross-section of a rat balloon-injured carotid artery 2 weeks post-injury stained with Masson’s trichrome technique. Robust adventitial collagen is stained deep blue. Several histological artefacts (possibly floating tissue debris) are shown (arrowheads). Note the minimal neointima development (compared to Figs. 4, 5), implying an impaired injury response in this animal. Magnification is 100x.
Figure 7.
Photos of rat balloon-injured carotid arteries at various times post-injury treated with Evans blue dye (re-drawn from Ref. 7). Tissues were treated with Evans blue (0.5 ml of a 5% solution) in situ 10 min prior to sacrifice, perfusion-fixed, harvested intact, split longitudinally, and pinned out on a silicon-padded dish for analysis. A shows a contralateral uninjured right carotid artery 2 weeks following injury on the left carotid artery. Absence of Evans blue staining indicates an intact endothelial layer. B shows an injured left carotid artery 30 min post-injury, and complete loss of the endothelial lining is indicated by complete Evans blue staining of the sub-endothelial matrix along the entire length of the vessel. C and D illustrate two carotid arteries with partial endothelial regrowth from the border zones 2 weeks post-injury. White (unstained) regions at the proximal and distal ends of these vessels are indicated by arrowheads and suggest that endothelial cells have partially regenerated into the central injured section at this time point.
Several highly used staining procedures preferentially color connective tissue in the vasculature, namely collagen, fibrin, and elastin. Connective tissue forms a scaffolding architecture around cellular components in the vessel wall and provides support to these tissues. Connective tissue can also house various cellular components including fibroblasts, mast cells, adipose cells, histiocytes, reticular cells, and bone cells (1). A common technique used in analyzing collagen and extracellular matrix (ECM) components in rat carotid arteries is Masson’s trichrome stain. This procedure colors nuclei blue/black, collagen and other sulfated muco-substances blue to blue/green, and cytoplasm, muscle, and various ECM components including fibrin red. An illustration of a Masson’s trichrome-stained rat balloon-injured carotid artery is shown in Figure 6, and a brief outline of this procedure is listed below (1).
Masson’s trichrome:
-
Solutions:
acid fuchsin (0.5 g.), glacial acetic acid (0.5 ml), distilled water (100 ml)
phosphomolybdic acid (1.0 g.), distilled water (100 ml)
methyl blue (2.0 g.), glacial acetic acid (2.5 ml), distilled water (100 ml)
-
Protocol:
deparaffinize and rehydrate tissues to distilled water
wash in slow running tap water
stain nuclei with celestin blue-hematoxlyin (see Note 30)
differentiate with 1% acid alcohol
wash well in slow running tap water
stain with solution A for 5 min
rinse in distilled water
treat with solution B for 5 min
drain but do not rinse
stain with solution C for 3–5 min
rinse in distilled water
treat with 1% acetic acid for 2 min
dehydrate through serial solutions of graded alcohols, clear with xylene, dry, and coverslip.
3.8. Microscopic and morphological evaluation
Following detailed and well-differentiated staining of vascular tissues, they are now ready to be carefully examined, qualified, and quantified under microscopy. Generally speaking, most of the histomorphometry protocols used for routine examination of rat arterial tissues use trans-illumination under light microscopy. Phase contrast, interference, polarized light, fluorescence, electron, and confocal microscopy are alternate more advanced means by which evaluation of prepared tissue samples can take place. These procedures are highly dependent upon an operating image analysis system, complete with a light microscope, attached camera with an interface to a computer, and an image analysis software program. The basic goal of image analysis is to generate data which describes aspects of the tissue specimen on the microscope slide. The objective here is to describe, both in terms of qualitative characteristics as well as precise quantitative measurements, anatomical parameters of each vessel cross-section. Specific parameters suitable for analysis for rat carotid artery injury studies include the perimeters (lengths) of the lumen, the internal elastic lamina (IEL), and the external elastic lamina (EEL), areas inside the lumen, IEL, and EEL (these are all measured directly), and areas of the medial wall and neointima (these are calculated). Additionally, thickness of the neointima and/or medial wall can be measured directly if so desired as an additional means to estimate arterial remodeling. These parameters can also be quantitated for the adventitial layer if desired.
For standard morphologic assessment of stained slides under microscopy, the author primarily employs VVG-stained cover-slipped sections. The microscope to be used should be prepared and “calibrated” ahead of time in order to center the light path and to achieve the best trans-illumination of the specimen with optimal resolution. The eyepieces should be adjusted to compensate for variation between individual users as well. Calibration of the image analysis program must also be completed ahead of time based on the instructions for each particular software program. This usually involves use of a stage micrometer to calibrate distances and lengths and is dependent upon the magnification of each separate objective used. Using an ultra-fine point marker, make a mark around each vessel directly on the cover-slip of each microscope slide in order to simplify location when the slide is on the microscope stage and under an objective. Initially using a low-powered objective, locate the vessel cross-section and then move to progressively higher-powered objectives until suitable magnification is achieved. The author performs most of his analyses using a 10x objective (total magnification 100x) for gross vessel inspection and a 40x objective (total magnification 400x) for more detailed examination. The author also utilizes an oil immersion 100x objective (1000x magnification) for meticulous scrutiny of stained specimens. First, it is recommended to scan all vessel cross-sections in a given batch to rule out specimens that cannot be used for a variety of reasons (broken or ruffled tissues, inadequately stained sections, artefacts that could directly hinder interpretation, etc.; see Notes 20, 22, 25, 29). Make detailed notes regarding all specimens that are removed from experimental analysis and the reason(s) why they will not be included. Digitally capture and save all remaining images (“image acquisition”) that will be used for qualification/quantitation and locate these in appropriate file(s) on the computer. Once these images have been saved, then proceed with specimen qualification and/or data quantitation of the morphological variables according to the instructions pertinent to the software of choice.
For tissue specimen analyses, both qualitative subjective interpretation and quantitative objective measurements should be performed. Notes should be made for characteristics of each vessel cross-section including the nature of the neointima (concentricity, degree, consistency, elastic- versus cellular composition, staining intensity of a particular dye, integrity of luminal lining, etc.), the medial wall (disrupted laminal layers, broken or fragmented tissue, staining), and the adventitia as well as the presence of any artefacts that might be present (see Notes 20, 22, 25, 29). As mentioned, specific quantifiable parameters suitable for analysis of rat arterial tissues include perimeters of and areas inside the lumen, IEL, and EEL. Conversely, if only perimeters lengths are measured then areas can be calculated: area = π[(perimeter/2 π)2] (5). Based on these data, parameters for the medial wall and neointima can then be directly determined (i.e., area inside the EEL – area inside the IEL = medial wall area; area inside the IEL – lumen area = neointimal area). Measurements of neointimal, medial, and/or adventitial thickness can also be performed. As stated earlier in this chapter, the exact subjective and/or objective measurements to be performed for each group of tissues are entirely dependent upon the goals of the research study and are at the discretion of the investigator(s).
3.9. Endothelial cell regeneration
This section stands alone as a separate protocol that can be used to measure the extent of endothelial cell regrowth from the border zones of the injured area (to estimate endothelial regeneration) and can also serve as an indication of the success of the injury in the rat carotid artery if performed shortly after injury (5,6,7). This method does not involve many of the procedures detailed above but instead utilizes pre-treatment of the animal with Evans blue dye, a sub-endothelial matrix- and basement membrane-specific stain, a short time prior to sacrifice (10–60 min) followed by perfusion-fixation of the animal as described.
Prepare the Evans blue dye ahead of time according to concentration desired (see Note 9). Intravenous injections in rat tail vessels (or other suitable site for vascular access if so desired) are required for this protocol, so an individual experienced in performing such techniques is recommended. At the desired time for examination of endothelial cell function (as determined by the investigator), anesthetize the animal as routine. Once a surgical plane of anesthesia is achieved, immerse the entire tail of the rat (all the way up to the testis) in very hot water for several minutes. This serves to vasodilate the tail vein making intravenous access easier. Quickly dry the tail and lay the animal supine on the operating table. The area for injection will appear darkened underneath the skin as the vein has become dilated. With a slight angle to a needle (attached to a syringe containing Evans blue dye), make an injection through the skin into the tail vein and gently withdraw the plunger to make sure venous access is achieved (blood will draw into the syringe). Slowly inject the desired quantity of Evans blue dye (see Note 9). The entire body of the animal will gradually turn a dark blue, thus ensuring success in the administration. After the desired time (10–60 min following injection), perfuse-fix the animal as described above. Immediately following harvesting of the entire lengths of both the left (injured) and right carotid arteries all the way from the bifurcation to the aortic arch (for the left carotid) and from the bifurcation to the innominate artery (for the right carotid), maintaining proper orientation lay them on a dissecting dish (coated with silicone and containing a small volume of fixative) and cut each in half longitudinally using fine forceps and micro-scissors. Pin out the entire lengths of the cut arteries on the silicone pad and perform quantitation of the following areas: unstained proximal and distal portions (indicating an intact endothelial layer), the stained central portion of the vessel (the denuded section), and the entire vessel. Figure 7 illustrates results from an Evans blue endothelial cell regrowth assay on rat carotid arteries at both acute and 2-week time points (redrawn from Ref. 7).
5. Conclusions
For animal vascular injury studies that employ histologic and morphometric analyses, investigators must first prepare their work plan according to their desired research goals keeping in mind specifics and individual requirements for tissue fixation, processing, embedding, sectioning, staining, and microscopy. Each of these inherent steps has many variables that can directly impact the success of otherwise routine procedures. It is suggested that the investigators utilize control samples of tissue in order to optimize these protocols for any given project before using valuable experimental specimens. The rat carotid artery balloon injury model is a useful procedure that allows investigation into many cellular, molecular, physical, and chemical mechanisms involved in the vascular injury response. Success of this model, however, is entirely dependent upon consistent, reproducible, and scientifically accurate histomorphometry, with the ultimate goal of discerning valuable scientific data from control and/or treated animal tissues. The background, protocols, and special considerations included in this chapter aim to provide a solid basis for successful and routine performance of many of these procedures; nevertheless, individual preferences, practical experiences, and research goals may dictate alternate courses of action.
Acknowledgments
Work for this review was supported by NHLBI grant HL-59868 and by a grant from the American Heart Association.
Footnotes
An anesthetic to sedate the animal prior to sacrifice should be chosen consistent with the scientific goals of the experimental plan and in accordance with the guidelines of the institutional animal care and use committee. The choice of anesthetic should not cause physiological interference with specific endpoints in the study as determined by the investigator(s).
A variety of fixatives can be used as determined by the investigator(s) that account for differences in tissue architecture and composition. As in Note 1, these should not interfere with specific endpoints that will be analyzed in the study. Fixatives can generally be classified as coagulative or cross-linking. Coagulative fixatives work by establishing a penetrable network in tissues thus allowing for penetration of processing solutions and embedding media. These include mercuric chloride and picric acid (a common reagent in many stains and dyes). Cross-linking fixatives operate by linking and stabilizing proteins in the tissue, thus creating a “gel” that limits denaturation of the tissue and enhances solution penetration. These include the commonly used fixatives formaldehyde, glutaraldehyde, and potassium dichromate. A comprehensive list of suitable fixatives for use in various animal vascular studies is available (3). Good choices for general fixatives for use in histomorphometry in rat arterial injury studies are formaldehyde or 10% neutral buffered formalin (pH 6.8). These agents fix by cross-linking proteins, particularly lysine residues, but do so in a manner that maintains antigenicity and therefore are consistent with immunostaining techniques. These agents work slowly but penetrate tissues well and are recommended for use by the author. Zinc formalin (formaldehyde) maintains good tissue morphometry and preserves epitopes for prolonged periods of time, thus allowing immunostaining protocols to be performed at a later date. Zinc formalin also eliminates the need for laborious and risky antigen recovery protocols. A 2% solution of buffered glutaraldehyde penetrates tissues poorly but fixes them quickly, gives good nuclear and cytoplasmic differentiation, and can be used for electron microscopy analysis; however, glutaraldehyde causes deformation of α-helix protein structures and is not recommended for immunostaining. Alternate choices for fixatives include mercurials such as B-5 or Zenker’s solution, alcohols, picrates including Bouin’s solution, or oxidizing agents.
A number of factors will affect the fixation process for vascular tissues. Fixation is optimized at neutral pH between 6.0 and 8.0. Prolonged duration of surgical techniques or other interventions that cause tissue hypoxia prior to fixation will lower the pH of those tissues, so adequate buffering capacity of the fixative will prevent excessive acidity. Penetration of tissues depends on the diffusibility for each individual fixative and on the thickness of the tissue to be fixed. Formalin and alcohols penetrate the best while glutaraldehyde penetrates poorly. The ratio of volume of fixative to tissue weight is important for optimum fixation and is recommended at 10:1 (although 20:1 has also been suggested (3)). If large amounts of tissues are being fixed, then replacing used or “de-fixed” fixative with new solution will minimize the volume needed. Mild agitation of the tissue in fixative will also enhance fixation. Temperature of the preparation is directly proportional to the degree of fixation as long as the increasing temperatures do not interfere or disrupt the tissue specimens. Heated formalin is often the initial step in automated tissue processing procedures. The concentration of the fixative should be adjusted to the lowest level achievable where fixation is still possible, as higher concentrations can adversely affect tissues. Examples include formalin at 10% and glutaraldehyde between 0.25 and 4%. Finally, the duration of time between obtaining the tissue and fixation is very important. Fresh tissues will fix better than ones that have dried too much following removal from the carcass. With increasing time away from the carcass, artefacts can be introduced, cellular organelles and molecular/cellular signals can be lost, epitopes can be masked, and nuclear shrinkage and condensation can occur. These potential biases are controlled for when using in situ perfusion-fixation procedures included here as long as the time from sacrifice to perfusing the tissues with fixative is kept brief.
In order to remove blood and other circulating factors from the tissues of interest, use of a warmed saline (PBS) solution perfused at mean arterial pressure is recommended. Alternately, if the investigator chooses to dilate the vasculature, a suitable vasodilator solution (that does not interfere with the scientific questions being asked) can be used for perfusion. In the lab of the author, warmed PBS solution perfused at mean arterial pressure completely removes blood from the vessel lumen and completely dilates the carotid arteries (noticed as a “non-corrugated” appearance of the elastic laminae).
The author recommends using an intravascular over-the-needle Teflon catheter (20 g. × 2″ Terumo Surflo I.V. catheter, National Health Resources, LLC) for trans-cardial perfusion-fixation instead of a similar sized needle for a very important reason. By using only a needle, the surgeon takes a chance that once cardiac puncture is achieved, the sharp bevel of the needle will advance without resistance through any tissues it encounters. Based on the angle and direction of the needle being inserted, this can often miss the aortic valve and aorta and puncture through adjacent areas of the base of the heart, likely the left atrium. When this happens, the needle must be withdrawn and additional attempts made to advance the needle directly into the aorta. However, this will leave puncture holes in the heart through which the perfusion fluids will travel, thus decreasing the pressure and the amount of fluid perfusing the carotid vasculature. By using an over-the-needle catheter, once cardiac puncture is achieved the surgeon carefully withdraws the needle leaving the flexible catheter in place inside the heart. This then can be advanced easily through the aortic valve and into the aorta. If resistance is met with the catheter tip, it will not penetrate that tissue and can be withdrawn slightly and insertion into the aorta can be attempted again. Once the catheter is inserted into the aorta (evidenced visually through the aortic wall), it can be attached to the luer-lock end of the 2-way stopcock and perfusion can continue.
Clearing of tissues is vital for adequate removal of remnant dehydrant (alcohol, acetone, etc.) and for proper infiltration of the embedding medium into the tissue. The most common clearing agent for vascular tissues is xylene, but toluene also works well but may be cost-limited. Other clearing agents that could be used for these tissues include chloroform (slow action, health hazard), methyl salicylate (oil of wintergreen; expensive), limolene derivatives, or complexes of long-chain aliphatic hydrocarbons (less forgiving with poorly-fixed, severely dehydrated, or sectioned tissues).
The most common embedding agent used for histology is paraffin, a waxy crystalline substance comprised of a complex mixture of hydrocarbons with a density similar to most normal tissue. Paraffins can differ in their melting points (40–70°C), a consideration for hardness in relation to tissue type (a high melting point makes a harder paraffin block than one with a lower melting point). In general, paraffin is inexpensive, easy to handle, and straight-forward in terms of embedding and sectioning. During processing, vacuum and pressure is applied to the tissues to enhance penetration of the paraffin embedding medium. Several commonly used paraffins include TissuePrep, Paramat, or Paraplast (contains plasticizers for ease in sectioning). Other embedding agents that could be used include various plastic resins (methyl or glycol methacrylate, araldite, epon). These agents solidify particularly hard and are especially useful for cutting very thin sections; however, they require specialized (i.e., expensive) reagents for dehydration and clearing and specialized microtomy procedures. For practical purposes, use of plastic resins for embedding may be “overkill” for routine rodent vascular studies.
Alcohol, small quantities of detergent, or other suitable agents can be added to the flotation bath to reduce surface tension and to allow the cut wax sections to flatten out with ease.
Various concentrations and/or doses of Evans blue dye are reported in the literature for performing endothelial cell regeneration assays in the rat injured carotid artery. Original articles reported 50–60 mg/kg dose of Evans blue given intravenously 30–60 minutes prior to perfusion-fixation (5, 6), yet the author has successfully used 0.5 ml of a 5% Evans blue solution in saline 10 min prior to sacrifice (7; see Fig. 7).
For purposes of the perfusion-fixation protocol, in standard normotensive laboratory rats mean arterial pressure (MAP) is estimated to be approximately 100 mmHg. A measured value for rat diastolic blood pressure (DBP) is 90 mmHg while a range for rat systolic blood pressure (SBP) is 116–180 mmHg (8). In this protocol perfusion is performed at approximate mean arterial pressure in order to replicate distending pressure under normal conditions and to fix the tissues at this normal pressure. If hypertensive animals are to be used and if the investigator(s) wishes to perfuse at their respective elevated pressures, or if for some reason the investigator chooses to perfuse at a pressure different than the MAP, then calculation of mean arterial pressure on an animal-to-animal basis should be performed (MAP = [(2 × DBP) + SBP]/3).
The author chooses to set up this apparatus on a lab bench top with the tubing draped to the floor (for a total height of ~53.5 inches; see Figs. 1, 2). In this manner, when the perfusion step is conducted the investigator places the animal carcass on the floor (in a surgical pan) in order to maintain columns of fluid equivalent to a MAP of 100 mmHg. However, if the investigator wishes this apparatus can be setup on a top shelf in a lab (with the animal carcass on a lower bench top) whereby columns of fluid at the appropriate heights can still be maintained for perfusion at MAP.
Be cautious with the 3-way stopcock during opening and closing. When the switch in a 3-way stopcock is turned in a specific direction, both columns of fluid will become open to each other and the levels of fluid in each section of tubing will equilibrate. This will result in mixing of the fixative and PBS (or dilator solution) and will render those solutions useless. Practice with opening and closing the 3-way stopcock in order to ascertain proper switching to open either the PBS or the fixative (but not both at the same time!).
These volumes of fluid are suggested when performing a perfusion-fixation protocol for only the carotid vasculature as outlined in this chapter. This method involves clamping the thoracic aorta to prevent perfusion of the lower body, and thus only the upper thorax, neck, and head regions are perfused in this protocol. If an investigator chooses to perfuse the entire body, then larger volumes of fluid will be needed (estimated ~120–150 ml) in order to achieve adequate perfusion and fixation of all tissues.
In this step it is imperative that PBS (or vasodilator solution) flow through the tissues first, followed by fixative. This will result in adequate removal of blood from the vasculature and will keep the vessels expanded at approximate MAP. Followed immediately by fixative, the vessels will be fixed at an expanded open caliber at MAP without interference of blood components in the lumen. In order to achieve this, make sure that the fluid in the short section of “common” tubing is filled with PBS (or vasodilator), and that the first stopcock that will be opened for perfusion allows only PBS to flow through the system, followed then by fixative.
If adequate perfusion does not take place (estimated by lack of flow through the system and/or improper outflow), the surgeon needs to re-evaluate the adequacy of the catheter insertion to make sure it is inside the aortic lumen and also to make sure that the tip of the catheter is not pushed up against the inside wall of the aorta, thus preventing fluid movement through it. The surgeon should also check the outflow incision in the right atrium and if this is not adequate, make additional cuts in the tissue to increase the area for outflow. One other factor that could exist to reduce or prevent outflow is the formation of a large thrombus at the incision site on the right atrium. Gently swab the area around the right atrium to remove any clots possibly located there.
If air bubbles enter the vasculature of the animal during perfusion, they can get lodged at certain sites which could cause problems during the sectioning procedures. More often than not, air bubbles trapped in the carotid arteries will be removed during processing; however, if air should remain trapped inside the lumen of a carotid artery, then during sectioning this could cause crushing or disintegration of the tissue by the microtome blade.
Immediately following perfusion-fixation, gently remove the carcass from the surgical tray and place nearby on an absorbant pad or towels. Carefully empty contents of the surgical tray (mostly PBS or vasodilator solution, fixative, and blood) into an appropriately labeled waste container and cap the container. Wash the tray to remove residual fixative.
It has been suggested by others that to ensure complete fixation for animal vascular tissues, excised specimens should be post-fixed for at least 24 hours prior to transferring the samples to 70% alcohol (3). This author routinely post-fixes rat carotid artery samples for 4 hours with success; however, longer post-fixation times can be used at the discretion of the investigator(s). The investigator(s) though must be aware of potential problems associated with prolonged fixation times (1).
For a routine rat carotid artery sample, it can be placed directly in a pre-labeled embedding cassette for processing. However, if the investigator wishes (especially important for small samples like mouse vessels), the sample can be first placed inside a fine porous paper (Kimwipe) which is folded several times and then placed inside the cassette.
If vessel cross-sections are desired to be analyzed under microscopy, then it is imperative that the vessel be in perfect vertical orientation during the embedding procedure. If the vessel is embedded slanted or otherwise not vertical, then during microtome cutting the vessel cross-sections will appear skewed and asymmetrical. These cannot be correctly analyzed for morphometry due to inherent biases in the tissue. If this is noted for a particular embedded vessel, then the investigator must re-embed the tissue and prepare a new block. To do this first lay the embedding block (containing the vessel) on the heating block of the paraffin embedder in order to melt away most of the wax. When the wax has sufficiently melted away from the tissue, either carefully re-orient the tissue so it is vertical or completely remove the tissue from the block and prepare a new block. Either way, once the vessel is properly aligned inside the embedding block, re-fill the block with liquid paraffin and let harden as routine.
A dull blade during microtome sectioning can destroy the tissue and render it useless even if it is re-embedded in a new block. In fact, tissues can even be pulled out of a paraffin block if the blade is too dull! Replacing dull or heavily used blades is essential for proper sectioning and preservation of important experimental tissues. Knives can either be of a standard thick metal variety which needs custom sharpening (and are expensive too) or of the disposable kind (inexpensive and suitable for most histology specimens).
It is important to have properly fixed, cleared, and embedded tissues or artefacts can be introduced during the sectioning step. Several common artefacts that become apparent during sectioning include tearing or ripping of the wax section after cutting, the appearance of holes in the sections, and folding or wrinkling of the tissues and/or wax section. Insufficient dehydration prior to clearing (during processing), inadequate infiltration with paraffin (during processing and embedding), and/or the presence of air bubbles in the tissues can all lead to problems evidenced during sectioning. Tissue processing cycles should allow sufficient time for tissue dehydration, with at least one step involving incubation in 100% ethanol. Covering or sealing all solutions from the ambient air also helps to avoid tissue rehydration, especially in humid environments. During in situ perfusion-fixation and tissue harvesting, make all necessary steps to avoid introduction of air into the vasculature as the presence of air inside the vessel lumen will surely cause destruction of the tissue during sectioning.
For the studies described in this chapter, universal 76 × 25 mm slides with thickness 1.0–1.2 mm are recommended. These are cost-effective and do not break easily. More expensive coated or frosted slides or slides with polished edges can be used if desired by the investigator.
Following staining and cover-slipping, slides can be stored for long periods of time unless a decaying-type of stain has been used. Most light stains will slowly deteriorate over time, thus decreasing clarity of the stain and ease of visualization. Fluorescent stains or radioactive treatments have inherent limits to the length of time that they can be analyzed, as their treatments involve a natural decay that will lessen the stain with time.
Commonly-used fixative pigments including those associated with formalin, if not adequately removed from the tissues during the processing and/or embedding steps, can render artefacts in the samples under acidic conditions that can be observed during microscopic evaluation of the stained tissues. Formalin residues commonly appear as brown or brownish-black anisotropic (birefringent) deposits in tissues that have been fixed in acidic hematoxylin. In order to remove these pigment residues use of a saturated alcoholic picric acid solution on unstained tissues is recommended. This is included in the protocol described for Verhoeff’s elastic tissue stain with Van Gieson counterstain in this chapter.
Determining optimal staining times for hematoxylin in routine procedures is complex and dependent upon several variables. These include the type of hematoxylin used (Mayer’s 10–20 min; Ehrlich’s 20–45 min), the age of the stain (intensity decreases with age), the intensity of use of the stain, progressive versus regressive procedures (based on staining depth), pre- and post-treatment of tissues including factors for processing, clearing, embedding, and staining, and personal preference of the investigator(s). It is recommended by the author that control samples of tissues be used with various incubation times until an optimal staining time is achieved.
A weak alkali solution such as standard tap water or 0.05% ammonia water is used to differentiate these stains in a process termed “bluing”. The use of warm water is suggested to further enhance the intensity of elastin fiber staining. This is an essential step in the differentiation of the dye and in conversion of the initial stain to its final colored product.
Pre-treatment of tissues with 1% potassium permanganate for 5 min followed by oxalic acid improves the sharpness and intensity of elastin fiber staining in this protocol.
Use of a Lugol’s iodine/sodium thiosulfate sequence has been suggested to remove mercury pigment artefacts in tissues fixed with a mercury-based fixative (1). Mercury residue is usually evidenced as a brown/black extracellular crystal. Iodine is the classic method for removing this pigment, followed by bleaching in a weak sodium thiosulfate (hypo) solution.
The celestin-blue aluminum hematoxylin nuclear stain is recommended for use in conjunction with Masson’s trichrome technique. This method uses deparaffinized and rehydrated tissues which are stained with celestine blue solution (celestine blue B (2.5 g.), ferric ammonium sulfate (25 g.), glycerin (70 ml), distilled water (500 ml)) for 5 min followed by rinsing in tap water. Tissues are then stained in an alum hematoxylin of choice for 5 min and washed in tap water until blue color sets. The remaining staining procedure is then followed.
References
- 1.Bancroft JD, Gamble M. Theory and practice of histological techniques. 5. Churchill Livingstone: Harcourt Publishers Limited; 2002. [Google Scholar]
- 2.Junqueira LC, Carneiro J, Kelley RO. Basic histology. 8. Appleton & Lange: A Simon & Schuster Company; 1995. [Google Scholar]
- 3.Seifert P, Rogers C, Edelman ER. Histological and immunohistological methods for vascular animal model studies. In: Simon D, Rogers C, editors. Contemporary Cardiology: Vascular Disease and Injury: Preclinical Research. Humana Press, Inc; 2001. [Google Scholar]
- 4.Tulis DA, Durante W, Peyton KJ, Chapman GB, Evans AJ, Schafer AI. YC-1, a benzyl indazole derivative, stimulates vascular cGMP and inhibits neointima formation. Biochem Biophys Res Commun. 2000;279:646–652. doi: 10.1006/bbrc.2000.3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clowes AW, Reidy MA, Clowes MM. Mechanisms of stenosis after arterial injury. Lab Invest. 1983;49:208–215. [PubMed] [Google Scholar]
- 6.Clowes AW, Clowes MM, Reidy MA. Kinetics of cellular proliferation after arterial injury. III Endothelial and smooth muscle growth in chronically denuded vessels. Lab Invest. 1986;54:295–303. [PubMed] [Google Scholar]
- 7.Tulis DA, Durante W, Peyton KJ, Evans AJ, Schafer AI. Heme oxygenase-1 attenuates vascular remodeling following balloon injury in rat carotid arteries. Atherosclerosis. 2001;155:113–122. doi: 10.1016/s0021-9150(00)00552-9. [DOI] [PubMed] [Google Scholar]
- 8.Waynforth HB, Flecknell PA, editors. Experimental and surgical technique in the rat. 2. Academic Press, A Harcourt Science and Technology Company; 2001. [Google Scholar]