

Abstract
Residual structure in the denatured state of a protein may contain clues about the early events in folding. We have simulated by molecular dynamics the denatured state of barnase, which has been studied by NMR spectroscopy. An ensemble of 104 structures was generated after 2 ns of unfolding and following for a further 2 ns. The ensemble was heterogeneous, but there was nonrandom, residual structure with persistent interactions. Helical structure in the C-terminal portion of helix α1 (residues 13–17) and in helix α2 as well as a turn and nonnative hydrophobic clustering between β3 and β4 were observed, consistent with NMR data. In addition, there were tertiary contacts between residues in α1 and the C-terminal portion of the β-sheet. The simulated structures allow the rudimentary NMR data to be fleshed out. The consistency between simulation and experiment inspires confidence in the methods. A description of the folding pathway of barnase from the denatured to the native state can be constructed by combining the simulation with experimental data from φ value analysis and NMR.
Keywords: protein folding, molecular dynamics, NMR
The denatured state of a protein is an ensemble of rapidly interconverting structures (1–3). It is only recently that structural information for the denatured state has become available at atomic resolution because of developments in high field NMR spectroscopy (4–18). These studies have focused on areas of persistent structure, with the aim of obtaining insight into the relationship between sequence, structure, and in particular, the mechanism of folding. The energy landscape available to a protein is complex, leading to the question of how proteins navigate it and fold on a reasonable, and often very fast, timescale. One hypothesis is that persistent structure under denaturing conditions may guide the folding of the polypeptide chain by limiting the conformational space available or even actively nucleating and promoting structure formation. To test this hypothesis, the structural attributes of the denatured state must be characterized for proteins for which detailed folding information is available.
Barnase is particularly well suited for such endeavors. It is a 110-residue ribonuclease from Bacillus amyloliquefaciens, whose folding/unfolding properties have been well characterized (19). Barnase is a single-domain protein with three helices (α1–α3) in the first half of the sequence followed by a five-stranded antiparallel β-sheet (20) (Fig. 1). The major core is formed by the packing of hydrophobic residues in α1 and the antiparallel β-sheet.
Figure 1.
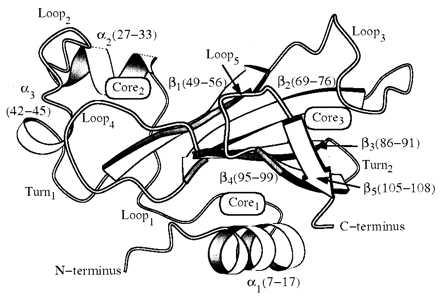
The NMR solution structure of barnase (20) with segments of secondary structure labeled.
The pH, temperature, and urea-denatured states of barnase have been studied by NMR spectroscopy (6, 8, 14). Structural parameters, such as chemical shifts, coupling constants, and nuclear Overhauser effect crosspeaks, suggest that there is residual structure in the regions corresponding to α1, α2, and β(3–4) of native barnase (6, 8). Unfortunately, transformation of these data into specific structural models is not possible. α1 and a portion of β(3–4) make significant interactions in the intermediate and transition states, as determined by the protein engineering method (21–23). The second helix, α2, remains at least partially helical under most denaturing conditions, yet it does not appear to be structured in either the intermediate or transition state (21–23). What then is the relationship between stable structures observed under denaturing conditions at equilibrium and the kinetic folding pathway and its associated structures?
We hope both to obtain a more detailed description of the denatured state and to gain insight into the potential importance of residual structure in folding by complementing the experimental picture with a molecular dynamics-generated ensemble of denatured conformations of barnase and the unfolding pathway. To this end, we simulated the thermal denaturation of barnase, whose early unfolding behavior has been the subject of previous unfolding simulations (24, 25). Here, the denatured ensemble is taken as the collection of structures late (2–4 ns) in an all atom, high temperature simulation of barnase in water. The structural properties of the denatured ensemble are presented and compared with experimental NMR data (6, 8, 14). The extent to which residual structure in the unfolded, denatured ensemble contributes to the kinetic pathway is discussed.
METHODS
An all atom representation was used for both the protein and solvent box. The initial starting structure was the average NMR solution structure (20). Protonation states of side chains were chosen to mimic low neutral pH (Lys, Arg, and His residues were positively charged, and Asp and Glu were negatively charged). The first 2 ns of this denaturation simulation at 498 K, as well as the control at 298 K, have been described (26). The solvent density was set to the experimental value for liquid water at 498 K (27), which corresponds approximately to a pressure of 26 atm (1 atm = 101.3 kPa). Extensive tests of the water model (F3C) as a function of temperature and pressure have been described (28). The denaturation simulation was continued to 4 ns to generate an ensemble of unfolded structures. Another simulation was also performed, and its denatured state is in broad agreement with the results presented here, and detailed comparison of the two ensembles will be presented elsewhere. Here, we invoke the ergodic hypothesis: the time average of a property for a single system (a single molecular dynamics trajectory) is the same as the average property taken over the members of an ensemble at a particular time (experiment).
RESULTS
We present a 4-ns denaturation simulation, beginning from the average NMR structure, of barnase at 498 K in water. This is a continuation of a simulation and full descriptions of the unfolding pathway, a control simulation at 298 K, and comparisons with experiments for the transition and intermediate states are presented elsewhere (26). Here, we concentrate on the properties of the protein late in the simulation in an attempt to model the denatured state of barnase.
The Denatured Ensemble.
Unfolding was identified by the loss in secondary structure, tertiary contacts, and the overall α-carbon (Cα) root-mean-square deviation from the initial NMR structure (Table 1). All properties monitored showed little change in their average behavior after 1.5–2 ns. Therefore, we use the 2- to 4-ns structures to represent the denatured state, which corresponds to an ensemble of 104 structures. The denatured state was compact with an average radius of gyration of 16 Å compared with an estimated value of 34 Å for a random coil (30) (Table 1, Fig. 2). The average solvent-accessible surface area for the structures making up the denatured state was ≈50% greater than the native state. The denatured ensemble was heterogeneous with an average root-mean-square deviation spread of >6 Å (Table 1). The loss of secondary structure was not evenly distributed among the α-helices and β-sheet. The overall helical content was approximately constant with 13 residues (or 12% of the protein), on average, adopting the appropriate backbone geometries (31) (Table 1). In contrast, only ≈5 residues retained β-structure, and protracted β-strands were not observed. The backbone was dynamic, with average fluctuations in the main chain dihedral angles of ≈40° (Table 1).
Table 1.
Overall properties of the denatured ensemble
| Property | NMR structure | Denatured state (MD)* |
|---|---|---|
| Radius of gyration, ņ | 13.4 | 15.9 (0.7) |
| 〈Cα RMSD〉, Ň | — | 11.3 (0.7) |
| 〈Pairwise RMSD〉, Ň | — | 6.4 (1.6) |
| Total % α§ | 19 | 12 (4) |
| Total % β§ | 36 | 5 (3) |
| 〈Δφ〉, ° | — | 38 (18) |
| 〈Δϕ〉, ° | — | 42 (19) |
| 〈SASA〉, Å2¶ | 6357 | 9513 (370) |
| 〈Trp-35 SASA〉, Å2 | 1 | 162 (35) |
| 〈Trp-71 SASA〉, Å2 | 1 | 142 (37) |
| 〈Trp-94 SASA〉, Å2 | 76 | 94 (41) |
The values for the denatured ensemble were calculated for the 2 to 4-ns time period of the 498 K simulation. Standard deviations are given in parentheses. MD, molecular dynamics.
Only the α-carbons were used for the calculation of the radius of gyration.
The Cα root-mean-square deviation (RMSD) from the starting structure was calculated after removal of translational and rotational motion. The average pairwise RMSD is calculated by comparing all structures in the denatured ensemble in a pairwise manner.
The secondary structure content is the fraction of residues (of a total of 107) that adopted repeating secondary structure (3 or more consecutive residues) based on whether the (φ, ϕ) values fall in the α- or β-region of conformational space (31).
The solvent-accessible surface area (SASA) was calculated by using the program naccess (32) using a 1.4-Å probe radius.
Figure 2.
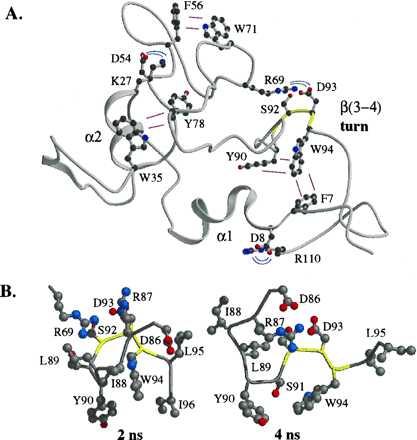
Representative hydrophobic clusters and electrostatic interactions in the denatured state of barnase. (A) The 3-ns structure is displayed. The backbone of the β(3–4) turn is colored yellow. (B) Representative side chain interactions in the β(3–4) turn are displayed for the 2- and 4-ns structures.
Residual Structure in the Denatured State.
α1 and α2 retained a significant amount of helical structure in the denatured ensemble. The average helix length was 6.4 (± 1.0) and 5.8 (± 0.5) residues for α1 and α2, respectively, compared with 11 and 7 for the native structure. Interestingly, α1 was partly unfolded at 2 ns, primarily at the N terminus (residues 7–12), but it regained most of its native hydrogen bonding network with time. The packing of hydrophobic residues between α1 and residues 88–89 increased as residues 7–13 were added to the helix (Fig. 3). The C terminus of the helix (residues 14–17) maintained a helical turn throughout the ensemble as judged by both backbone orientation and hydrogen bonding. Unlike α1, α2 was more ordered and yet made few contacts with other elements of structure (Fig. 2).
Figure 3.
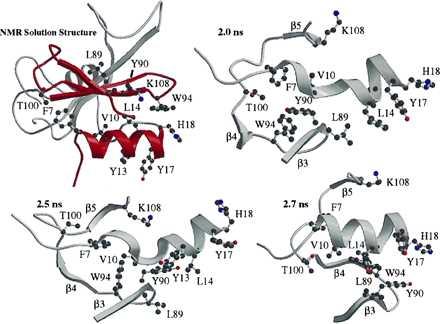
Side chain interactions between residues in α1 and the β-sheet region of the native protein that aid in the coil → helix transition from 2 to 2.5 ns.
Disruption of native secondary structure was extensive in the β-sheet region of denatured barnase (residues 50–110). The backbone dihedral angles of these residues sampled a large region of (φ, ϕ) space, exhibiting some preference for locally extended conformations but not strict repeating β-sheet structures. Many transient turns formed and punctuated the extended portions of the structure. In particular, residues in β3 and in a turn between β3 and β4 formed a nonnative cluster of hydrophobic residues in which Trp-94 figured prominently (Fig. 2), which resulted in low solvent accessibility compared with the other two tryptophans (Table 1). The hydrophobic cluster and electrostatic interactions (involving Arg-69, Asp-86, Arg-87, and Asp-93) stabilized the turn between Tyr-90 and Trp-94 (Fig. 2).
Dynamic Behavior of the Denatured State.
Given the variability in the structural properties of the ensemble, it is difficult to display in a comprehensive manner. A collection of five representative structures spanning the 2- to 4-ns time period is in Fig. 4. The structures are colored continuously from red at the N terminus to blue at the C terminus. Although the structures within the ensemble are heterogeneous, the overall topology is roughly similar to that of the native state. For example, α1 (reddish-orange) and portions of the β-sheet (green-blue) remained close in space. Further, residues 25–55, which include α2, α3, and the N-terminal portion of β1, behaved as a semiautonomous unit.
Figure 4.
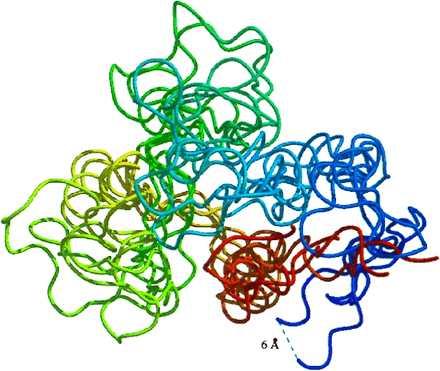
Snapshots from the denatured ensemble depicting motions of the protein backbone. The structures are colored from red at the N terminus (looking down the helix axis) to blue at the C terminus. The 2-, 2.5-, 3-, 3.5-, and 4-ns structures are displayed. The 6-Å separation labeled in the figure is shown merely to provide perspective.
DISCUSSION
Residual Structure: Comparison with Experiment.
Recent investigations of pH-, urea-, and temperature-denatured barnase have provided information regarding the structural and dynamic features of the denatured ensemble and suggest that some fraction of the denatured ensemble contains residual, nonrandom structure (6, 8, 14). Here, we attempt to obtain a deeper view of the residual structure by molecular dynamics simulations. The structures observed during the simulation were dynamic and interchanged between a multitude of conformations. However, there was nonrandom, residual structure. The most persistent interactions can be summarized as follows: (i) helical structure in the C-terminal portion of α1 (residues 13–17) and in α2; (ii) a turn and hydrophobic clustering between β3 and β4; and (iii) tertiary contacts between residues in α1 and the C-terminal portion of the β-sheet. Some of these features are illustrated in representative structures in Figs. 2 and 3.
The available structural experimental information for the denatured state of barnase, as summarized in Fig. 5, correlates with the regions of structure observed in the simulation. Significant deviations from random coil values for the amide and 15N chemical shifts occur in α1 and α2 and residues between β3 and β4 (Fig. 5), as do the strong sequential NH—NH (i → i + 1) nuclear Overhauser effect crosspeaks (6, 8, 14). These helical regions are observed directly in the simulation (Figs. 2 and 3). α1 underwent unfolding and refolding in the denatured state, although the C-terminal portion of the helix was stable throughout (Fig. 3). Interestingly, docking of side chain residues of the helix with residues in the β-sheet region of the protein aided in the folding of the helix even though the interactions were a mix of native and nonnative contacts because of the disorder of the β-structure. The main chain of β(3–4) appears to be the most structured portion of β-structure based on the NMR data (6, 8, 14). In addition, the side chains in this region appear to be partially structured, given the deviations of their side chain chemical shifts from random coil values (Fig. 5) and the strong sequential NH—NH nuclear Overhauser effect crosspeaks (6, 8, 14), yet interestingly these data are not consistent with native structure (8). The simulation provides a structural framework for interpretation of these results: residues 90–94 participate in a turn between β3 and β4 that is stabilized by a dynamic hydrophobic cluster (including many residues spanning β3 and β4, Fig. 2). Further, the cluster involved residues separated by >17 Å in the native state. Also, the native salt bridge between Asp-93 and Arg-69 aided in maintenance of the turn (Fig. 2). Experimental studies show the pKa of Asp-93 is decreased significantly in the denatured state, suggesting it is involved in a favorable electrostatic interaction (33). Despite these tertiary interactions, extension of the turn to form a β-hairpin did not occur, and the main chain adopted predominantly nonnative conformations, leading to low S values (discussed further below) (Fig. 5).
Figure 5.
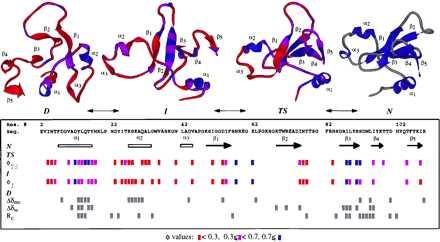
Summary of the available information on the major states along the folding pathway of barnase. The experimental φ values (21–23) are given in the box and colored according to their magnitude, from red (unstructured) to blue (structured). Chemical shift changes and Rc values suggestive of residual structure and/or regions of low mobility in the denatured state are marked in gray (6, 8, 14). A cut-off of 0.3 was used for the Rc values. Changes smaller in magnitude were considered to be within the noise. Structures from the simulation are given at the top of the figure. D is the 4-ns snapshot, I is the major intermediate state (the average structure from 470 to 730 ps) (26), TS represents the major transition state (the average structure between 135 and 140 ps), and N is the average NMR structure (20). The TS, I, and D structures are colored according to their S values, by using the same scale as for φ. Calculation of S values followed the procedure of Daggett et al. (37); the secondary structure component is based on (φ, ϕ) values, and the tertiary component is the ratio of contacts between nonneighboring residues relative to the extent of contacts in the native simulation (a cut-off of 5.4 and 4.6 Å was used for C—C and all other heavy atom pairs, respectively).
Residual Structure: Relevance to the Folding/Unfolding Pathway.
Both the NMR studies and the simulation provide evidence for residual structure in the denatured state of barnase. We have outlined the specifics of that structure, which in some cases is typified by tertiary interactions in the absence of secondary structure and in others is because of persistent but fluctuating secondary structure. Next, we consider the residual structure in light of the folding pathway of barnase. Both the major intermediate (I) and transition state (TS) have been investigated by φ value analysis (34–36). In this, the degree of formation of structure at a particular residue is inferred from changes in the energetics of kinetics and thermodynamics on mutation of that residue or surrounding ones. A φ value of 0 implies denatured-like structure and φ = 1, native-like structure at that position. Although structural attributes of the intermediate states during folding/unfolding are inferred just from the energetics by using this approach, there is a good correlation with the results of simulations that probe structure directly (24–26). Each residue in the putative models for these states may be assigned a structure index (S) for semi-quantitative comparison with φ (37). Thus, the approaches are quite complementary, and when the ensembles representing the transition or intermediate states in a simulation are in agreement with experiment, we assume that the structural models can be used in the interpretation of the experimental data. Therefore, for the purposes of discussing the possible role of residual structure in the folding/unfolding pathway of barnase, we consider the experimental and simulation results for the transition and intermediate states of barnase together.
The regions of residual structure in denatured barnase correspond to the moderately and highly structured portions of the intermediate and transition states fairly well, as determined by the protein engineering method (compare regions of residual structure in D to the magenta and blue φ values in Fig. 5). The C-terminal portion of α1, portions of the loop between β1 and β2, and several residues within β3 have φ values close to 1. In contrast, mutations throughout α2 do not appear to affect the stability of the intermediate or the transition state (23). Yet, the conclusion from the NMR studies is that residues within α2 are helical in the denatured state.
What then is the relationship between structure observed in denatured proteins and the deduced structural interactions in kinetic folding intermediates? It is not necessary for all structure observed in a denatured protein to lead to productive interactions along the folding pathway. Nonnative interactions that are sufficiently fluid can decrease the conformational search without excessive stabilization of partially folded or unfolded forms of the protein. Excessive stabilization of nonnative interactions slows down folding (36), as will excessive stabilization of native structure that is not formed in the transition state (38). This may be true even for the native structure seen in α2. In this regard, it is worth noting that while α2 is intact in the simulated denatured state, it is not present in the simulated intermediate state (Fig. 5). Whereas having α1 and α2 in the helical conformation in the denatured state presumably decreases the conformational search during folding, another important factor was contacts between loop 1 and β1 that served to “pinch off” the second domain, containing α2, α3, and loop 2 (Fig. 5). These interactions prevented gross extension of the first half of the protein. This region also displays lower mobility and/or residual structure by NMR (Fig. 5), in support of the idea that it is partially segregated from the rest of the protein.
The persistent structure seen in the denatured ensemble and intermediate and transition states, as suggested by both experiment and simulation, supports a folding pathway in which regions of local structure, primarily in the C terminus of α1 and the β(3–4) turn region, initiate early events in folding. This is in agreement with predictions of potential nucleation sites based on burial of hydrophobic surface area (39) and NMR studies of barnase peptide fragments (40). In other words, α1, core 1, and portions of the β-sheet fold about persistent initiation sites in the denatured ensemble until enough stabilizing interactions involving residues distant in sequence form a nucleation site (refs. 14 and 41; Fig. 5). Following initiation of folding, there is further consolidation of both secondary and tertiary structure, as reflected in the increase in the interaction energy, or φ values, and as observed in the simulation (Fig. 5).
An example of how such nonlocal interactions may not only stabilize but also induce the formation of secondary structure is illustrated in Fig. 3, which shows a coil to helix transition of α1 in the denatured state. An invariant one and one-half turns of helix were present in all of the denatured structures. However, the N-terminal portion of α1 only adopted significant amounts of helical structure in the presence of tertiary contacts with the β-sheet, in particular with the β(3–4) turn. Whereas this docking was not stable or persistent enough to lead to global refolding under these conditions, it did lead to repair of α1. Further, this process provides a description of a plausible structural mechanism for nucleation leading to consolidation of structure. In this regard, it is worth emphasizing that α1 and β(3–4) are structured in the denatured state but undergo conformational exchange. They are well established in the simulated intermediate state and are loosely coupled to neighboring strands. The packing of the other β-strands around this nucleus is essentially complete in the transition state (Fig. 5), but β(3–4) appears to be more important than α1 in propagation of the β-structure (Fig. 5), which is supported by fragment studies in which native-like structure persists on removal of α1 (42).
Acknowledgments
Figs. 1, 2, 3 were made by using molscript (43) and Figs. 4 and 5 with University of California, San Francisco MidasPlus (29). This work was supported by National Institutes of Health Grant GM 50789 (to V.D.).
References
- 1.Dobson C M. Curr Opin Struct Biol. 1992;2:6–12. [Google Scholar]
- 2.Wüthrich K. Curr Opin Struct Biol. 1994;4:93–99. [Google Scholar]
- 3.Shortle D R. Curr Opin Struct Biol. 1996;6:24–30. doi: 10.1016/s0959-440x(96)80091-1. [DOI] [PubMed] [Google Scholar]
- 4.Neri D, Billeter M, Wider G, Wüthrich K. Science. 1992;257:1559–1563. doi: 10.1126/science.1523410. [DOI] [PubMed] [Google Scholar]
- 5.Alexandrescu A T, Shortle D. J Mol Biol. 1994;242:527–546. doi: 10.1006/jmbi.1994.1598. [DOI] [PubMed] [Google Scholar]
- 6.Arcus V L, Vuilleumier S, Freund S M, Bycroft M, Fersht A R. Proc Natl Acad Sci USA. 1994;91:9412–9416. doi: 10.1073/pnas.91.20.9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Logan T M, Thériault Y, Fesik S W. J Mol Biol. 1994;236:637–648. doi: 10.1006/jmbi.1994.1173. [DOI] [PubMed] [Google Scholar]
- 8.Arcus V L, Vuilleumier S, Freund S M, Bycroft M, Fersht A R. J Mol Biol. 1995;254:305–321. doi: 10.1006/jmbi.1995.0618. [DOI] [PubMed] [Google Scholar]
- 9.Frank M K, Clore G M, Gronenborn A M. Protein Sci. 1995;4:2605–2615. doi: 10.1002/pro.5560041218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan H, Barbar E, Barany G, Woodward C. Biochemistry. 1995;34:13974–13981. doi: 10.1021/bi00043a002. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Shortle D. Biochemistry. 1995;34:15895–15905. doi: 10.1021/bi00049a004. [DOI] [PubMed] [Google Scholar]
- 12.Zhang O, Forman-Kay J D. Biochemistry. 1995;34:6784–6794. doi: 10.1021/bi00020a025. [DOI] [PubMed] [Google Scholar]
- 13.Zhang O, Forman-Kay J D. Biochemistry. 1995;36:3959–3970. doi: 10.1021/bi9627626. [DOI] [PubMed] [Google Scholar]
- 14.Freund S M V, Wong K, Fersht A R. Proc Natl Acad Sci USA. 1996;93:10600–10603. doi: 10.1073/pnas.93.20.10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Shortle D. Protein Sci. 1996;5:1898–1906. doi: 10.1002/pro.5560050916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong K B, Freund S M V, Fersht A R. J Mol Biol. 1996;259:805–818. doi: 10.1006/jmbi.1996.0359. [DOI] [PubMed] [Google Scholar]
- 17.Gillepsie J R, Shortle D. J Mol Biol. 1997;268:170–184. doi: 10.1006/jmbi.1997.0953. [DOI] [PubMed] [Google Scholar]
- 18.Schwalbe H, Fiebig K M, Buck M, Jones J A, Dobson C M. Biochemistry. 1997;36:8977–8991. doi: 10.1021/bi970049q. [DOI] [PubMed] [Google Scholar]
- 19.Fersht A R. FEBS Lett. 1993;325:5–16. doi: 10.1016/0014-5793(93)81405-o. [DOI] [PubMed] [Google Scholar]
- 20.Bycroft M, Ludvigsen S, Fersht A R, Poulsen F M. Biochemistry. 1991;30:8697–8701. doi: 10.1021/bi00099a030. [DOI] [PubMed] [Google Scholar]
- 21.Matouschek A, Serrano L, Fersht A R. J Mol Biol. 1992;224:819–835. doi: 10.1016/0022-2836(92)90564-z. [DOI] [PubMed] [Google Scholar]
- 22.Serrano L, Matouschek A, Fersht A R. J Mol Biol. 1992;224:805–818. doi: 10.1016/0022-2836(92)90563-y. [DOI] [PubMed] [Google Scholar]
- 23.Matthews J M, Fersht A R. Biochemistry. 1995;34:6805–6814. doi: 10.1021/bi00020a027. [DOI] [PubMed] [Google Scholar]
- 24.Caflisch A, Karplus M. J Mol Biol. 1995;252:672–708. doi: 10.1006/jmbi.1995.0528. [DOI] [PubMed] [Google Scholar]
- 25.Tirado-Rives J, Orozco M, Jorgensen W L. Biochemistry. 1997;36:7313–7329. doi: 10.1021/bi970096i. [DOI] [PubMed] [Google Scholar]
- 26.Li, A. & Daggett, V. (1998) J. Mol. Biol., in press.
- 27.Kell G S. J Chem Eng Data. 1967;12:66–68. [Google Scholar]
- 28.Levitt M, Hirshberg M, Sharon R, Laidig K E, Daggett V. J Phys Chem. 1997;101:5051–5061. [Google Scholar]
- 29.Ferrin T E, Huang C C, Jarvis L E, Langridge R. J Mol Graphics. 1988;6:13–27. [Google Scholar]
- 30.Miller W G, Goebel C V. Biochemistry. 1968;7:3925–3935. doi: 10.1021/bi00851a021. [DOI] [PubMed] [Google Scholar]
- 31.Daggett V, Kollman P A, Kuntz I D. Biopolymers. 1991;31:1115–1134. doi: 10.1002/bip.360310911. [DOI] [PubMed] [Google Scholar]
- 32.Hubbard S J, Thornton J M. naccess, Computer Program. London: University College; 1993. [Google Scholar]
- 33.Oliveberg M, Arcus V L, Fersht A R. Biochemistry. 1995;34:9424–9433. doi: 10.1021/bi00029a018. [DOI] [PubMed] [Google Scholar]
- 34.Matouschek A, Kellis J T, Jr, Serrano L, Fersht A R. Nature. 1989;342:122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 35.Fersht A R, Matouschek A, Serrano L. J Mol Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 36.Fersht A R. Curr Opin Struct Biol. 1995;5:79–84. doi: 10.1016/0959-440x(95)80012-p. [DOI] [PubMed] [Google Scholar]
- 37.Daggett V, Li A, Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1996;257:430–440. doi: 10.1006/jmbi.1996.0173. [DOI] [PubMed] [Google Scholar]
- 38.Lopez-Hernandez E, Serrano L. Folding & Design. 1996;1:43–55. [PubMed] [Google Scholar]
- 39.Moult J, Unger R. Biochemistry. 1991;30:3816–3824. doi: 10.1021/bi00230a003. [DOI] [PubMed] [Google Scholar]
- 40.Neira J L, Fersht A R. Folding & Design. 1996;1:231–241. doi: 10.1016/s1359-0278(96)00034-x. [DOI] [PubMed] [Google Scholar]
- 41.Fersht A R. Curr Opin Struct Biol. 1997;7:3–9. doi: 10.1016/s0959-440x(97)80002-4. [DOI] [PubMed] [Google Scholar]
- 42.Kippen A D, Sancho J, Fersht A R. Biochemistry. 1994;33:3778–3786. doi: 10.1021/bi00178a039. [DOI] [PubMed] [Google Scholar]
- 43.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]