

Abstract
Muscle wasting in chronic kidney disease (CKD) begins with impaired insulin/IGF-1 signaling, causing abnormal protein metabolism. In certain models of muscle atrophy, reduced satellite cell function contributes to atrophy, but how CKD affects satellite cell function is unknown. Here, we found that isolated satellite cells from mice with CKD had less MyoD, the master switch of satellite cell activation, and suppressed myotube formation compared with control mice. In vivo, CKD delayed the regeneration of injured muscle and decreased MyoD and myogenin expression, suggesting that CKD impairs proliferation and differentiation of satellite cells. In isolated satellite cells from control mice, IGF-1 increased the expression of myogenic genes through an Akt-dependent pathway. CKD impaired Akt phosphorylation in satellite cells after muscle injury. To test whether impaired IGF-1 signaling could be responsible for decreased satellite cell function in CKD, we created an inducible IGF-1 receptor knockout mouse and found impaired satellite cell function and muscle regeneration. In addition, both CKD and IGF-1 receptor knockout mice developed fibrosis in regenerating muscles. Taken together, impaired IGF-1 signaling in CKD not only leads to abnormal protein metabolism in muscle but also impairs satellite cell function and promotes fibrosis in regenerating muscle. These signaling pathways may hold potential therapeutic targets to reduce CKD-related muscle wasting.
Complications of chronic kidney disease (CKD) including acidosis, impaired insulin/IGF-1 signaling, and excess angiotensin II or IL-6, stimulate muscle wasting by increasing protein degradation and decreasing protein synthesis.1–6 The mechanisms changing protein metabolism include impaired insulin/IGF-1 signaling, which activates caspase-3 and the ubiquitin-proteasome system to increase muscle protein breakdown.3,4,7–10
Impaired activity of muscle progenitor or satellite cells also might contribute to CKD-induced muscle atrophy as it does in other catabolic conditions.11,12 Satellite cells are located beneath the basal lamina of myofibers and have at least two functions. First, they proliferate, becoming myoblasts, and then differentiate, forming new muscle fibers to repair injured muscle.13 Second, muscle homeostasis requires proliferation and differentiation of satellite cells for normal muscle growth.9 In mice with muscle atrophy from hindlimb suspension or in rodent models of aging, myopathy, or muscle denervation, the number and activity of satellite cells were found to be reduced, indicating that satellite cells are involved in maintaining muscle mass.11,12
After muscle injury, satellite cells are activated and express the MyoD and myogenin transcription factors, leading to cell proliferation and differentiation, respectively.14–16 When these cells express embryonic myosin heavy-chain protein (eMyHC), there is myotube formation. In muscle of mice with CKD, we found decreased expression of MyoD, myogenin, and eMyHC mRNAs, abnormalities that were corrected in muscle by chronic overloading as a model of resistance exercise.17 In contrast, treadmill running of mice with CKD (a model of endurance exercise) reduced muscle protein degradation but not the decrease in protein synthesis or low levels of MyoD, myogenin, or eMyHC mRNAs. The mechanisms for CKD-induced changes in satellite cell function have not been reported, so we evaluated whether defects in IGF-1 signaling impair satellite cell function because impaired insulin/IGF-1 signaling causes abnormal muscle protein metabolism.5,6 Our results identify another mechanism for the muscle atrophy that is induced by CKD.
Results
CKD Impairs Satellite Cell Function
Previously, we reported that CKD reduces MyoD and myogenin expression in muscle.17 By counting nuclei outside the dystrophin-stained sarcolemma, it seemed that there was a significant decrease in the progenitor cells. In these experiments, we used a more rigorous method of identifying satellite cells: Positive staining with Pax-7.18,19 Using this method, we counted the number of Pax-7–positive cells in muscle of 13 pairs of CKD and control mice; there was no statistically significant difference in the number of Pax-7–positive satellite cells in the two groups (Supplemental Figure 1A). The Pax-7 mRNA level, like Pax-7 staining, was slightly lower in CKD muscles, but, again, the difference was not statistically significant versus control (Figure 1A). There were, however, significantly reduced mRNAs of the myogenic markers MyoD, myogenin, and Myf-5 in gastrocnemius muscles of CKD mice versus values in control muscles (Figure 1A). We also studied isolated satellite cells from muscles of control and CKD mice.20 Satellite cells were identified as Pax-7– and Pax-3–positive cells (Supplemental Figure 1B). There was no difference in the terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling assay or activated caspase-3, indicating that apoptosis does not contribute substantially to CKD-induced satellite cell dysfunction (data not shown).
Figure 1.
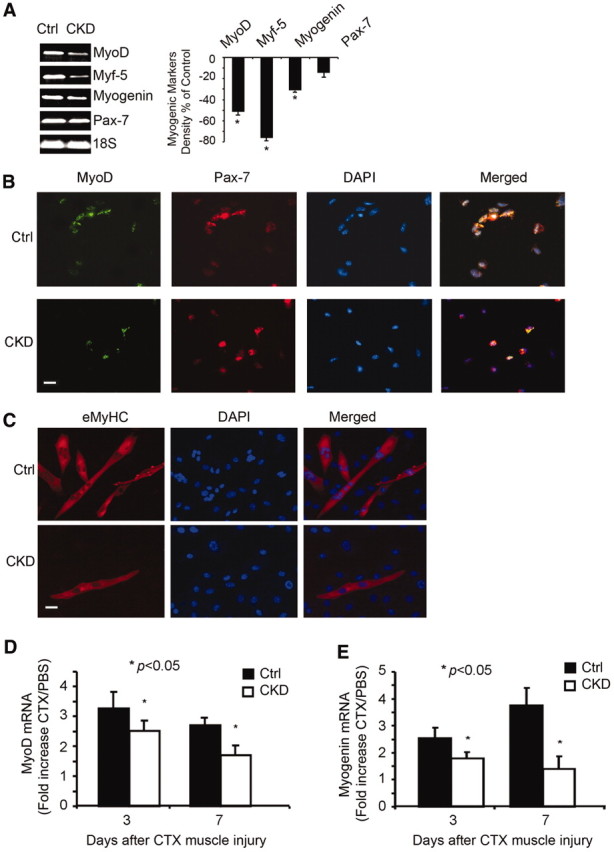
CKD impairs satellite cell activation. (A) mRNAs of myogenic markers (MyoD, myogenin, and Myf-5) are reduced in CKD mouse gastrocnemius muscles (left). Differences were quantified as percentage difference = [(CKD − control)/control] × 100 (right). Reverse transcriptase–PCR (RT-PCR) was repeated three times for each group of five mice (*P < 0.05, CKD versus control [Ctrl]). (B) Isolated satellite cells from muscles of CKD and control mice were cultured for 16 hours before immunostaining for MyoD (green), Pax-7 (red), and DAPI (blue). Yellow color in the merged picture indicates that Pax-7–positive cells expressed MyoD. Bar = 20 μm. (C) Satellite cells isolated as in B were cultured for 10 days before incubation in 2% horse serum to elicit differentiation. Immunostaining of eMyHC (red) identified myofibers, and DAPI (blue) detected nuclei. Bar = 20 μm. (D and E) For examination of satellite cell activation in vivo, satellite cells were isolated from CTX-injured muscles. Using RT-PCR, MyoD and myogenin mRNAs from satellite cells were measured and normalized to 18S mRNA. The ratio of satellite cell mRNAs from injured versus control muscles is shown (*P < 0.05 versus results from satellite cells of control mice; n = 5 mice per group).
When we plated the same number of satellite cells isolated from muscles of CKD and control mice, MyoD-positive cells from CKD mice were significantly lower compared with results from control mice (Figure 1B). CKD also reduced satellite cell proliferation, measured as bromodeoxyuridine (BrdU) incorporation (Supplemental Figure 1C). Differentiation of isolated satellite cells from CKD mice was decreased in cells, as identified by reduced immunostaining for eMyHC (Figure 1C). Thus, reduced MyoD expression and the lower number of eMyHC-positive cells demonstrate that CKD suppresses satellite cell activation and differentiation.
Third, we examined whether CKD impairs satellite cell activation in vivo using the standard stimulus of these cells: Cardiotoxin (CTX) injection into muscles.21,22 At 3 or 7 days after injury, isolated satellite cells from injured tibialis anterior (TA) and gastrocnemius muscles of CKD and control mice were assayed for mRNAs of myogenic genes. MyoD and myogenin mRNAs were decreased in satellite cells from muscle of CKD mice (Figure 1, D and E). To confirm that similar changes occurred in injured muscles from CKD and control mice, we examined mRNAs of MyoD and myogenin. After 3 days, mRNAs of MyoD and myogenin in injured muscles of CKD mice were significantly decreased versus results in control mice (Supplemental Figure 1, D and E). After 14 days, the trend persisted: MyoD and myogenin mRNAs in CKD muscles were significantly lower versus results from control mice.
CKD Reduces Muscle Regeneration
For an in vivo test of satellite cell function, we used muscle injury to determine whether CKD interfered with muscle regeneration.21,23,24 During the initial 72 hours, mononuclear cells accumulated in injured muscles of CKD and control mice. After 5 days, there were new myofibers (designated by central nuclei) in TA muscles of control mice, but in injured TA muscles of CKD mice, there were fewer new myofibers and more mononuclear cells versus results in control mice. At 7 days, most myofibers in injured muscles of control mice had central nuclei; mononuclear cells were found only between myofibers. In CKD mice, however, there were fewer new myofibers, myofibers were disorganized, and mononuclear cells persisted. After 14 days, control muscles were virtually normal and there were few mononuclear cells, whereas muscles from CKD mice exhibited expansion of the interstitial space, persistent mononuclear cell infiltration, and smaller new myofibers (Figure 2A). After 1 month, the size distribution of newly formed myofibers in injured muscles of CKD mice was shifted to smaller values versus results in control mice (Figure 2B). The average size of new myofibers in CKD muscles (505 μm2) was substantially smaller than in control muscles (1715 μm2; P < 0.001). Thus, CKD significantly impairs muscle regeneration.
Figure 2.

CKD suppresses muscle regeneration in vivo induced by injuring muscle to activate satellite cells. (A) Representative hematoxylin- and eosin-stained cross-cryosections of injured TA muscles were obtained from control and CKD mice. Regenerated myofibers were identified by their central nuclei. Injured muscles from CKD mice had increased infiltration of cells and slowed regeneration. Bar = 20 μm. (B) At 1 month after injury, fibers were immunostained for laminin, and the sizes of myofibers with central nuclei were measured. The histogram of the sizes of the myofibers with central nuclei indicates that CKD impaired regeneration. (C) At days 7 and 14 after injury, muscles were immunostained for macrophages (Mac-2 antibody; red) and DAPI for nuclei (blue). At both times, macrophages were increased in muscles of CKD mice. Bar = 50 μm. (D) Sequential changes in F4/80 mRNAs after CTX injury were detected by RT-PCR and normalized for 18S mRNA. The fold increase over PBS treatment confirmed that macrophages were more plentiful in muscles of CKD mice. (*P < 0.05 versus results in injured muscles of control mice; n = 5 mice in each group).
CKD Prolongs Inflammation in Injured Muscle
Previously, we found that muscle injury causes infiltration of neutrophils and macrophages with increased levels of cytokines and chemokines.25 To study how CKD influences macrophage and neutrophil infiltration, we immunostained muscle sections with anti–Mac-2 to identify macrophages and myeloperoxidase to identify neutrophils. One day after injury, muscles from both groups were infiltrated by neutrophils and macrophages, but by day 3, the macrophage infiltration in injured CKD muscles was more intense. The pattern persisted through day 7 or 14 (Figure 2C). High mRNA levels of the macrophage marker F4/80 confirmed the Mac-2 immunostaining results (Figure 2D). We also analyzed the expression of cytokines/chemokines in injured muscle: At 3 days after injury, cytokine and chemokine mRNAs in muscle of CKD mice were higher versus results in control mice (Table 1). These responses presumably reflect the more intense infiltration of macrophages in injured muscle of CKD mice.
Table 1.
Cytokine and chemokine change in injured muscle of CKD and control mice
| Parameter | Ratio of CKD to Control |
||||||
|---|---|---|---|---|---|---|---|
| MCP-1 | IL-6 | RANTES | TNF-α | MIP-1α | TCA-3 | MIP-1β | |
| No injury | 0.70 ± 0.10 | 1.30 ± 0.23 | 1.50 ± 0.07 | 2.05 ± 0.20 | 1.24 ± 0.06 | 1.40 ± 0.10 | 1.23 ± 0.20 |
| Injury day 3 | 6.40 ± 0.80 | 7.60 ± 0.90 | 4.20 ± 0.80 | 10.44 ± 1.10 | 2.17 ± 0.30 | 10.04 ± 0.90 | 2.50 ± 0.03 |
| Injury day 7 | 1.50 ± 0.03 | 2.50 ± 0.20 | 2.00 ± 0.50 | 5.01 ± 0.40 | 1.70 ± 1.80 | 3.40 ± 0.60 | 1.90 ± 0.40 |
MCP, monocyte chemoattractant protein; MIP, macrophage inflammatory protein; TCA-3, T cell activator-3.
CKD Impairs IGF-1/Insulin Signaling in Muscle and Satellite Cells
Defective insulin/IGF-1 signaling reduces Akt phosphorylation (p-Akt) in muscle, leading to CKD-induced muscle atrophy.6,26,27 In examining whether IGF-1 signaling influences satellite cell function, we isolated cells from control muscles and examined their responses to IGF-1 or its downstream product, Akt. IGF-1 increased the expression of MyoD, Myf-5, myogenin, and eMyHC in satellite cells; these responses were blocked by a phosphatidylinositol 3-kinase inhibitor, LY294002 (Figure 3). In satellite cells infected with an adenovirus expressing activated myristylated Akt (Akt-myr) or the inactive Akt (Akt-AAA; K179A/T308A/S473A), we found that Akt-myr mimics IGF-1 and activates myogenic gene expression. In contrast, Akt-AAA blocked this response. Thus, IGF-1 signaling activates satellite cell functions.28
Figure 3.

IGF-1 signaling pathway regulates MyoD, myogenin, and Myf-5 expression in satellite cells. Satellite cells were isolated from muscles of C57/BL6 mice, and 106 cells were plated on matrigel-coated six-well plates. After 5 days, they were infected with an adenovirus to express myristoylated Akt or the serine Akt mutant Akt-AAA; an adenovirus expressing GFP was used as a control. In other wells, cells were treated with 100 ng of IGF-1/ml or IGF-1 plus the phosphatidylinositol 3-kinase inhibitor LY294002 (10 μM). The cells were differentiated over 48 hours before being lysed and subjected to Western blotting using the indicated antibodies.
To examine whether CKD suppresses IGF-1–stimulated signaling in satellite cells after injury, we injected 500 ng of IGF-1 into injured TA muscles of CKD and control mice to stimulate Akt phosphorylation. After 15 minutes, muscle sections were obtained and immunostained for both p-Akt and myogenin (a marker of differentiating satellite cells). In muscle of CKD mice, there were fewer myogenin-positive cells and a lower degree of Akt phosphorylation in myogenin-positive cells (Supplemental Figure 2A). Because most Pax-7–positive cells were myogenin positive (Supplemental Figure 2B), we conclude that CKD impairs IGF-1/insulin signaling in satellite cells.
IGF-1 Receptor Modifies Satellite Cell Activation and Myofiber Formation
Because CKD suppresses IGF-1 signaling (Supplemental Figure 2) and muscle regeneration (Figure 2), we hypothesized that defects in the IGF-1 receptor (IGF-1R) signaling contribute to abnormal satellite cell function and regenerative capacity. To examine this hypothesis, we created conditional IGF-1R knockout mice (IGF-1R-KO; see the Concise Methods section). By Western blotting, IGF-1R expression was decreased in gastrocnemius muscle (Figure 4A); decreased IGF-1R was confirmed by immunostaining TA muscles and isolated satellite cells (Supplemental Figure 3, A and B). MyoD expression was sharply reduced in satellite cells from IGF-1R-KO mice, indicating that IGF-1 regulates MyoD expression (Supplemental Figure 3B). Likewise, in muscles of IGF-1R-KO mice, mRNAs of other myogenic markers were suppressed (Figure 4B). Next, we isolated satellite cells from IGF-1R-flox mice and infected them with an adenovirus expressing Cre to knockout the IGF-1R (Supplemental Figure 3C). Impaired satellite cell proliferation was measured by reduced immunostaining of Ki67 in IGF-1R-KO cells versus cells expressing IGF-1R (Ki67 is a marker of proliferation). Also, significantly fewer myotubes were formed by IGF-1R-KO satellite cells (Supplemental Figure 3D).
Figure 4.
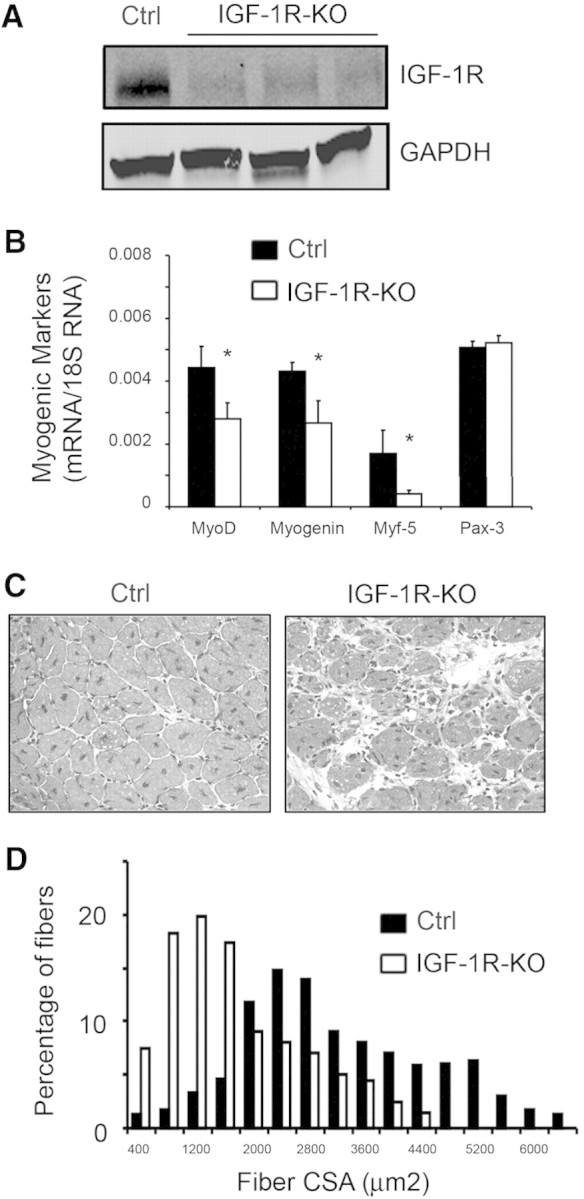
Knockout of IGF-1R impairs muscle regeneration stimulated by muscle injury. (A) Western blot to detect IGF-1R in gastrocnemius muscles from IGF-1R-flox (control) or three IGF-1R-KO mice. (B) mRNAs from gastrocnemius muscles of control and IGF-1R-KO mice were analyzed by RT-PCR for measurement of mRNAs of myogenic markers after correction for 18S mRNA (*P < 0.05 in IGF-1R-KO versus control; n = 3 mice in each group). (C) At 7 days after injury, new myofibers (central nuclei) were detected by hematoxylin and eosin staining. When compared with results in control muscles, muscles of IGF-1R-KO mice exhibited more cellular infiltration and slowed regeneration. (D) At 1 month after injury, a histogram of the percentage of myofibers of different sizes of regenerating myofibers showed that myofibers in muscle of IGF-1R-KO mice were shifted to smaller sizes.
To examine whether IGF-1R-KO slows muscle regeneration in vivo, we injured TA muscles of these mice, and 7 days later, muscle regeneration was reduced in IGF-1R-KO mice versus results in control, IGF-1R-flox mice (Figure 4C). After 1 month, the sizes of newly formed myofibers in IGF-1R-KO muscles were significantly smaller versus control muscles (Figure 4D). Thus, satellite cells lacking the IGF-1R exhibit decreased expression of myogenic factors and reduced regeneration capacity, as occurs in CKD mouse muscles. These results emphasize the important role of IGF-1R signaling in the activation and function of satellite cells.
Fibrosis Is Increased in Injured Muscle of CKD or IGF-1R-KO Mice
Others have reported that TGF-β1 is expressed during the development of fibrosis in injured muscle.29,30 We found that CKD increased TGF-β1 mRNA in injured muscle to a greater degree versus results from control mice. The difference was detected at 1 day after injury, and maximum values were present at day 3 followed by a gradual decrease to day 14 (Figure 5A). Injured muscles of CKD mice also had a sharp increase in collagen (Figure 5B).
Figure 5.

Fibrosis develops in muscles of CKD and IGF-1R-KO mice. (A) At various days after CTX injury, mRNAs of TGF-β1 (normalized for 18S) were higher in muscle of CKD mice. The fold increase of TGF-β1 mRNA in injured over uninjured muscles is shown (*different from values in control mice; n = 3 for each time period; P < 0.05). (B) At 1 month after injury, Sirius red was used to detect collagen accumulation in muscles of CKD and control mice. Bar = 20 μm. (C) At various days after CTX injury, mRNAs of TGF-β1 (normalized for 18S) were higher in muscle of IGF-1R-KO mice. The fold increase in TGF-β1 mRNA in injured over uninjured muscles is shown (*different from values in control mice; n = 3 for each time period; P < 0.05). (D) At 1 month after injury, Sirius red was used to detect collagen accumulation in muscles of IGF-1R-KO and control mice. Bar = 20 μm.
Muscle injury of IGF-1R-KO mice increased TGF-β1 mRNA two- to three-fold versus results in control mice (Figure 5C). At 1 month after injury, muscle collagen in IGF-1R-KO mice was sharply increased compared with control results (Figure 5D); therefore, muscle injury in CKD or IGF-1R-KO decreases satellite cell function and muscle regeneration but increases TGF-β1 expression and stimulates fibrosis.
Discussion
Previously, we found that CKD impairs muscle protein metabolism, producing atrophy. Impaired insulin/IGF-1 cell signaling and activation of proteolytic pathways caused these results.2,3 We now uncover a new mechanism for CKD-induced muscle atrophy: Impaired satellite cell function linked to decreased IGF-1R signaling. Others reported that activation, proliferation, and differentiation of satellite cells are necessary for maintenance of muscle mass11,12; our results show that CKD impairs these functions, reducing satellite cell proliferation and differentiation. We also identified how impaired IGF-1 signaling causes the abnormalities in satellite cell functions. Finally, CKD and IGF-1R-KO both increase TGF-β1 mRNA and fibrosis in injured muscles. Our results suggest that improving satellite cell function in CKD could be useful in strategies designed to ameliorate muscle atrophy.
We found that CKD reduced the expression of MyoD and myogenin transcription factors in intact and injured muscle (Figure 1A; Supplemental Figure 1, D and E). Reportedly, these transcription factors are present in myonuclei.31 To determine that our results reflect events in satellite cells, we isolated Pax-7–positive cells from muscle of CKD mice; they had depressed MyoD expression (Figure 1B). CKD also suppressed the mRNAs of myogenic factors in satellite cells obtained from injured muscle (Figure 1, D and E). This is relevant because muscle injury markedly stimulates MyoD and myogenin expression in satellite cells, leading to their cellular proliferation/differentiation.14–16,25 Thus, CKD depresses satellite cell function.
We examined the influence of CKD on satellite cells in vivo and in vitro. First, the expression of myogenic markers was reduced in muscle of CKD versus control mice (Figure 1A). Second, Pax-7–positive satellite cells isolated from muscles of CKD and control mice exhibited decreased proliferation on the basis of BrdU incorporation (Supplement Figure 1C). These cells also exhibited reduced MyoD expression and delayed differentiation into myotubes (Figure 1). Third, CKD suppressed MyoD and myogenin mRNAs in injured muscles and in satellite cells isolated from injured muscles (Figure 1, D and E; Supplemental Figure 1, D and E). Finally, CKD limited satellite cell regeneration, indicated by the smaller sizes of new myofibers (Figure 2, A and B).
Why does CKD suppress satellite cell function in regenerating muscle? Inflammation or CKD complications (e.g., high levels of angiotensin II or IL-6) could play a role.5,21,32 This is suggested because our results show that CKD prolongs mononuclear cell accumulation in muscle while increasing cytokine and chemokine expression (Figure 2B, Table 1). The increased F4/80 mRNA found in injured muscles of CKD mice confirmed the greater degree of macrophage infiltration (Figure 2D). Suppressed muscle regeneration and inflammation responses in injured CKD muscle could be related to impaired insulin/IGF-1 signaling because local expression of muscle IGF-1 was shown to downregulate proinflammatory cytokines and promote muscle regeneration selectively.21 Indeed, we found that CKD impairs IGF-1 signaling in satellite cells, leading to impaired muscle regeneration.
IGF-1 is involved in many developmental processes, including cell proliferation, differentiation, and regeneration.33–37 Musaro and colleagues37,38 demonstrated that overexpression of locally acting IGF-1 (mIGF-1) improves muscle function in mouse models of senescence, injury, myodystrophy, or amyotrophic lateral sclerosis. In muscle and satellite cells, we found that CKD impaired IGF-1R–mediated signaling (Supplemental Figure 2, A and B), and when we suppressed the IGF-1 receptor in mice (IGF-1R-KO), satellite cell activation and muscle regeneration were reduced as in CKD mice (Figure 4). In our studies of IGF-1R-KO mice, other hormones changing in response to IGF-1R-KO could affect our results; therefore, we isolated satellite cells from muscle of IGF-1R-flox mice and knocked out IGF-1R using a Cre-expressing adenovirus. In these cells, there was reduced proliferation and differentiation, as occurred in intact muscles (Supplemental Figure 3, C and D). The results point to a critical role for IGF-1R signaling in satellite cells.
Myostatin could be another mechanism causing CKD-induced responses, because it can suppress muscle growth.39,40 Increased myostatin in muscle of CKD animals is associated with muscle atrophy,41 and we find that inhibiting myostatin function in CKD mice can improve their muscle weight and protein metabolism (Zhang et al., manuscript in preparation). Besides myostatin, there is evidence that muscle repair could respond positively to endogenous stem cells.42 We have not examined whether circulating stem cells influence the maintenance of muscle mass, but this possibility would be interesting.
The development of fibrosis is a serious complication of impaired muscle regeneration. Fibrosis can fill spaces in injured muscles and connect injured muscle fragments, but it makes regaining muscle strength difficult because of impaired connective tissue contraction. Others reported that prolonged inflammatory responses can initiate fibrosis linked to increased expression of TGF-β1 and collagen deposition.43–45 In injured muscles, we found TGF-β1 mRNA increased, especially in CKD mice. Collagen deposition followed the same pattern as the TGF-β1 mRNA (Figure 5, A and B). We speculate that the collagen production in injured CKD muscles was due to the increase in TGF-β1 from macrophages accumulating in injured muscles (Figure 2C). Macrophages are profibrotic cells, and TGF-β1 is a key inducer of fibrosis in muscle and other tissues.46 Indeed, Warren et al.47 reported freeze injury of muscles in CCR2-null mice prolonged macrophage accumulation and increased fibrosis. Thus, impaired IGF-1 signaling in regenerating muscles of CKD mice led to fibrosis, because similar events occurred in IGF-1R-KO mice (e.g., increased TGF-β1 and collagen deposition in injured muscles; Figure 5). In fact, increased IGF-1 expression in injured muscle suppresses inflammation and reduces fibrosis.21,29,48 How frequently CKD causes muscle fibrosis is unknown, because only small series of patients with CKD have been tested for muscle fibrosis.49
We conclude that CKD reduces IGF-1R–mediated signaling in satellite cells, impairing their activation and contributing to CKD-induced muscle atrophy. Because catabolic conditions frequently cause impaired insulin/IGF-1 signaling, our results could apply to other conditions that cause muscle atrophy.
Concise Methods
Reagent and Antibodies
CTX was obtained from Calbiochem (La Jolla, CA) and an antibody against phospho-Akt (Ser473) from Cell Signaling Technology (Beverly, MA). Antibodies against IGF-1R, MHC, and Myf-5 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The MyoD antibody was from Vector Laboratories (Burlingame, CA), and antibodies against Pax-7, Pax-3, and eMyHC and myogenin were from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). The laminin antibody was from Sigma-Aldrich (St. Louis, MO), anti–Mac-2 was from Cedarlane Labs (Burlington, NC); anti-Ki67 and IGF-1 were from R&D System (Minneapolis, MN). The anti-myeloperoxidase antibody was from Abcam (Cambridge, MA). DMEM and FBS were from Cellgro Mediatech (Manassas, VA). BrdU Labeling and Detection Kit II was obtained from Roche Applied Science (Indianapolis, IN).
Muscle Regeneration Model
All animal experiments and procedures were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. Anesthetized male mice, 6 to 10 wk old, were studied after injection of 80 μl of 10 μM CTX in saline into one TA or gastrocnemius muscle using a 27-G needle; the contralateral muscle was injected with the same volume of PBS.
Satellite Cell Isolation
Muscles were digested with 0.2% Collagenase Type II (Worthington Biochemical, Lakewood, NJ) at 37°C for 30 minutes in DMEM with 1% penicillin/streptomycin. The mixture was passed through a 100-μm filter, and satellite cells were isolated by centrifugation at the interface between 40 and 70% Percoll.20 Satellite cells on matrigel-coated plates (BD Bioscience, San Jose, CA) were cultured in DMEM with 20% FBS, 1% penicillin/streptomycin mixture, 40 μg/ml gentamicin, and 2% chicken embryo extract. To differentiate cells into myotubes, we incubated them for 5 days in DMEM plus 2% horse serum.
Reverse Transcriptase–PCR
Total RNA was extracted from muscle using Trizol, and cDNAs were synthesized using the first-strand cDNA synthesis kit with oligo dT 12 to 18 primers (Invitrogen, Carlsbad, CA); real-time PCR was performed with the Opticon Real-Time PCR (MJ Research, Waltham, MA). Primers we used will be provided if requested. Relative mRNA expression levels were calculated from cycle threshold (Ct) values using 18S as the internal control [relative expression = 2(sample Ct − 18S Ct)].
Subtotal Nephrectomy
C57/BL6 mice underwent subtotal nephrectomy in two stages: Approximately 50% of the right kidney was removed; 7 days later, the left kidney was removed. The mice were fed a 12% protein diet for 2 weeks before beginning 40% protein chow.3 At 1 month after second surgery, mice with a blood urea nitrogen level >80 mg/dl were studied.
Generation of Mice with Inducible IGF-1R Knockout
Transgenic mice expressing the estrogen receptor-Cre (ER-Cre) were obtained from Jackson Laboratory (Bar Harbor, ME). After breeding with mice (C57BL6 background) containing the IGF-1R-flox (exon 3), those expressing both ER-Cre and IGF-1R-flox were identified by genotyping. At 3 months of age, IGF-1R-KO and control mice (IGF-1R-flox) were administered an intraperitoneal injection of 2 mg of tamoxifen (10 mg/ml in corn oil; Sigma-Aldrich) daily for 5 days, and IGF-1R deletion was confirmed by reverse transcriptase–PCR, Western blotting, and immunohistochemistry. Others studied mice after 7 days of tamoxifen to obtain more complete IGF-1R knockout as confirmed by reverse transcriptase–PCR50; we treated mice for 5 days to reduce systemic responses to IGF-1R knockout.
Histochemical and Immunohistochemical Staining
Serial frozen (−20°C) transverse cryosections (8 μm) from the midbelly of control and injured TA muscles were mounted on glass slides. Sections were air-dried and fixed in acetone for 10 minutes before staining with hematoxylin and eosin. Tissue collagen and fibrosis were stained using Sirius red (Sigma-Aldrich). MyoD, myogenin, and p-Akt were examined by standard immunohistochemical techniques.5
Imaging of Muscle Sections and Analyses
Images were obtained using a Nikon 80i upright microscope (Melville, NY). Myofiber sizes were measured after immunostaining muscle with anti-laminin using NIS-Elements Br 3.0 software (Nikon); the distribution of fiber sizes was calculated as described previously.5
Statistical Analysis
Results are expressed as means ± SEM. Significance testing was performed using one-way ANOVA followed by pair-wise comparisons using the Student-Newman-Keuls test. Statistical significance was set at P < 0.05. A minimum of three replicates were performed for each experimental condition.
Disclosures
None.
Supplementary Material
Acknowledgments
These studies were sponsored by National Institutes of Health grants R37 DK37175, R01 DK62828, and P50 DK64233 and a grant to L.Z. from Satellite Healthcare and Shanghai Science and Research funding (07QH14020).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental information for this article is available online at http://www.jasn.org/.
References
- 1. May RC, Kelly RA, Mitch WE: Metabolic acidosis stimulates protein degradation in rat muscle by a glucocorticoid-dependent mechanism. J Clin Invest 77: 614–621, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. May RC, Kelly RA, Mitch WE: Mechanisms for defects in muscle protein metabolism in rats with chronic uremia: Influence of metabolic acidosis. J Clin Invest 79: 1099–1103, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bailey JL, Wang X, England BK, Price SR, Ding X, Mitch WE: The acidosis of chronic renal failure activates muscle proteolysis in rats by augmenting transcription of genes encoding proteins of the ATP-dependent ubiquitin-proteasome pathway. J Clin Invest 97: 1447–1453, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Song YH, Li Y, Du J, Mitch WE, Rosenthal N, Delafontaine P: Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J Clin Invest 115: 451–458, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, Mitch WE: IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J Am Soc Nephrol 20: 604–612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bailey JL, Zheng B, Hu Z, Price SR, Mitch WE: Chronic kidney disease causes defects in signaling through the insulin receptor substrate/phosphatidylinositol 3-kinase/Akt pathway: Implications for muscle atrophy. J Am Soc Nephrol 17: 1388–1394, 2006 [DOI] [PubMed] [Google Scholar]
- 7. Lee SW, Dai G, Hu Z, Wang X, Du J, Mitch WE: Regulation of muscle protein degradation: Coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. J Am Soc Nephrol 15: 1537–1545, 2004 [DOI] [PubMed] [Google Scholar]
- 8. Wang X, Hu Z, Hu J, Du J, Mitch WE: Insulin resistance accelerates muscle protein degradation: Activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology 147: 4160–4168, 2006 [DOI] [PubMed] [Google Scholar]
- 9. Le Grand F, Rudnicki M: Satellite and stem cells in muscle growth and repair. Development 134: 3953–3957, 2007 [DOI] [PubMed] [Google Scholar]
- 10. Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ: The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14: 395–403, 2004 [DOI] [PubMed] [Google Scholar]
- 11. Mitchell PO, Pavlath GK: Skeletal muscle atrophy leads to loss and dysfunction of muscle precursor cells. Am J Physiol Cell Physiol 287: C1753–C1762, 2004 [DOI] [PubMed] [Google Scholar]
- 12. Jejurikar SS, Kuzon WM, Jr: Satellite cell depletion in degenerative skeletal muscle. Apoptosis 8: 573–578, 2003 [DOI] [PubMed] [Google Scholar]
- 13. Hawke TJ, Garry DJ: Myogenic satellite cells: Physiology to molecular biology. J Appl Physiol 91: 534–551, 2001 [DOI] [PubMed] [Google Scholar]
- 14. Sabourin LA, Girgis-Gabardo A, Seale P, Asakura A, Rudnicki MA: Reduced differentiation potential of primary MyoD−/− myogenic cells derived from adult skeletal muscle. J Cell Biol 144: 631–643, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smith CK, 2nd, Janney MJ, Allen RE: Temporal expression of myogenic regulatory genes during activation, proliferation, and differentiation of rat skeletal muscle satellite cells. J Cell Physiol 159: 379–385, 1994 [DOI] [PubMed] [Google Scholar]
- 16. Yablonka-Reuveni Z, Rivera AJ: Temporal expression of regulatory and structural muscle proteins during myogenesis of satellite cells on isolated adult rat fibers. Dev Biol 164: 588–603, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang XH, Du J, Klein JD, Bailey JL, Mitch WE: Exercise ameliorates chronic kidney disease-induced defects in muscle protein metabolism and progenitor cell function. Kidney Int 76: 751–759, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA: Pax7 is required for the specification of myogenic satellite cells. Cell 102: 777–786, 2000 [DOI] [PubMed] [Google Scholar]
- 19. Relaix F, Rocancourt D, Mansouri A, Buckingham M: A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature 435: 948–953, 2005 [DOI] [PubMed] [Google Scholar]
- 20. Morgan JE: Myogenicity in vitro and in vivo of mouse muscle cells separated on discontinuous Percoll gradients. J Neurol Sci 85: 197–207, 1988 [DOI] [PubMed] [Google Scholar]
- 21. Pelosi L, Giacinti C, Nardis C, Borsellino G, Rizzuto E, Nicoletti C, Wannenes F, Battistini L, Rosenthal N, Molinaro M, Musaro A: Local expression of IGF-1 accelerates muscle regeneration by rapidly modulating inflammatory cytokines and chemokines. FASEB J 21: 1393–1402, 2007 [DOI] [PubMed] [Google Scholar]
- 22. Musaro A, Giacinti C, Borsellino G, Dobrowolny G, Pelosi L, Cairns L, Ottolenghi S, Cossu G, Bernardi G, Battistini L, Molinaro M, Rosenthal N: Stem cell-mediated muscle regeneration is enhanced by local isoform of insulin-like growth factor 1. Proc Natl Acad Sci U S A 101: 1206–1210, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tiidus PM. Skeletal Muscle Damage and Repair, Champaign, IL, Human Kinetics, 2008 [Google Scholar]
- 24. Gulati AK: Pattern of skeletal muscle regeneration after reautotransplantation of regenerated muscle. J Embryol Exp Morphol 92: 1–10, 1986 [PubMed] [Google Scholar]
- 25. Zhang L, Ran L, Garcia GE, Wang XH, Han S, Du J, Mitch WE: Chemokine CXCL16 regulates neutrophil and macrophage infiltration into injured muscle promoting muscle regeneration. Am J Pathol 175: 2518–2527, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maloff BL, McCaleb ML, Lockwood DH: Cellular basis of insulin resistance in chronic uremia. Am J Physiol 245: E178–E184, 1983 [DOI] [PubMed] [Google Scholar]
- 27. Sun DF, Zheng Z, Tummala P, Oh J, Schaefer F, Rabkin R: Chronic uremia attenuates growth hormone-induced signal transduction in skeletal muscle. J Am Soc Nephrol 15: 2630–2636, 2004 [DOI] [PubMed] [Google Scholar]
- 28. Philippou A, Halapas A, Maridaki M, Koutsilieris M: Type I insulin-like growth factor receptor signaling in skeletal muscle regeneration and hypertrophy. J Musculoskelet Neuronal Interact 7: 208–218, 2007 [PubMed] [Google Scholar]
- 29. Sato K, Li Y, Foster W, Fukushima K, Badlani N, Adachi N, Usas A, Fu FH, Huard J: Improvement of muscle healing through enhancement of muscle regeneration and prevention of fibrosis. Muscle Nerve 28: 365–372, 2003 [DOI] [PubMed] [Google Scholar]
- 30. McCroskery S, Thomas M, Platt L, Hennebry A, Nishimura T, McLeay L, Sharma M, Kambadur R: Improved muscle healing through enhanced regeneration and reduced fibrosis in myostatin-null mice. J Cell Sci 118: 3531–3541, 2005 [DOI] [PubMed] [Google Scholar]
- 31. Hyatt JP, Roy RR, Baldwin KM, Edgerton VR: Nerve activity-independent regulation of skeletal muscle atrophy: Role of MyoD and myogenin in satellite cells and myonuclei. Am J Physiol Cell Physiol 285: C1161–C1173, 2003 [DOI] [PubMed] [Google Scholar]
- 32. Stenvinkel P, Ketteler M, Johnson RJ, Lindholm B, Pecoits-Filho R, Riella M, Heimburger O, Cederholm T, Girndt M: IL-10, IL-6, and TNF-alpha: Central factors in the altered cytokine network of uremia–the good, the bad, and the ugly. Kidney Int 67: 1216–1233, 2005 [DOI] [PubMed] [Google Scholar]
- 33. Kiepe D, Ciarmatori S, Hoeflich A, Wolf E, Tonshoff B: Insulin-like growth factor (IGF)-I stimulates cell proliferation and induces IGF binding protein (IGFBP)-3 and IGFBP-5 gene expression in cultured growth plate chondrocytes via distinct signaling pathways. Endocrinology 146: 3096–3104, 2005 [DOI] [PubMed] [Google Scholar]
- 34. Engert JC, Berglund EB, Rosenthal N: Proliferation precedes differentiation in IGF-I-stimulated myogenesis. J Cell Biol 135: 431–440, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A: Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev Biol 229: 141–162, 2001 [DOI] [PubMed] [Google Scholar]
- 36. Limesand KH, Barzen KA, Quissell DO, Anderson SM: Synergistic suppression of apoptosis in salivary acinar cells by IGF1 and EGF. Cell Death Differ 10: 345–355, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Musaro A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N: Localized IGF-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet 27: 195–200, 2001 [DOI] [PubMed] [Google Scholar]
- 38. Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L, Molinaro M, Rosenthal N, Musaro A: Muscle expression of a local IGF-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol 168: 193–199, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R: Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol 162: 1135–1147, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee SJ, McPherron AC: Myostatin and the control of skeletal muscle mass. Curr Opin Genet Dev 9: 604–607, 1999 [DOI] [PubMed] [Google Scholar]
- 41. Cheung WW, Rosengren S, Boyle DL, Mak RH: Modulation of melanocortin signaling ameliorates uremic cachexia. Kidney Int 74: 180–186, 2008 [DOI] [PubMed] [Google Scholar]
- 42. Sampaolesi M, Blot S, D'Antona G, Granger N, Tonlorenzi R, Innocenzi A, Mognol P, Thibaud JL, Galvez BG, Barthelemy I, Perani L, Mantero S, Guttinger M, Pansarasa O, Rinaldi C, Cusella De Angelis MG, Torrente Y, Bordignon C, Bottinelli R, Cossu G: Mesangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 444: 574–579, 2006 [DOI] [PubMed] [Google Scholar]
- 43. Hogaboam CM, Steinhauser ML, Chensue SW, Kunkel SL: Novel roles for chemokines and fibroblasts in interstitial fibrosis. Kidney Int 54: 2152–2159, 1998 [DOI] [PubMed] [Google Scholar]
- 44. Border WA, Noble NA: Transforming growth factor beta in tissue fibrosis. N Engl J Med 331: 1286–1292, 1994 [DOI] [PubMed] [Google Scholar]
- 45. Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, et al. : Transforming growth factor type beta: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A 83: 4167–4171, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wynn TA: Cellular and molecular mechanisms of fibrosis. J Pathol 214: 199–210, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Warren GL, Hulderman T, Mishra D, Gao X, Millecchia L, O'Farrell L, Kuziel WA, Simeonova PP: Chemokine receptor CCR2 involvement in skeletal muscle regeneration. FASEB J 19: 413–415, 2005 [DOI] [PubMed] [Google Scholar]
- 48. Sanz S, Pucilowska JB, Liu S, Rodriguez-Ortigosa CM, Lund PK, Brenner DA, Fuller CR, Simmons JG, Pardo A, Martinez-Chantar ML, Fagin JA, Prieto J: Expression of insulin-like growth factor I by activated hepatic stellate cells reduces fibrogenesis and enhances regeneration after liver injury. Gut 54: 134–141, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Levine JM, Taylor RA, Elman LB, Bird SJ, Lavi E, Stolzenberg ED, McGarvey ML, Asbury AK, Jimenez SA: Involvement of skeletal muscle in dialysis-associated systemic fibrosis (nephrogenic fibrosing dermopathy). Muscle Nerve 30: 569–577, 2004 [DOI] [PubMed] [Google Scholar]
- 50. Cheng J, Du J: Mechanical stretch simulates proliferation of venous smooth muscle cells through activation of the insulin-like growth factor-1 receptor. Arterioscler Thromb Vasc Biol 27: 1744–1751, 2007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.