

Abstract
Glycogen synthase kinase 3β (GSK3β), a serine/threonine protein kinase, is a key target of drug discovery in several diseases, including diabetes and Alzheimer disease. Because lithium, a potent inhibitor of GSK3β, causes nephrogenic diabetes insipidus, GSK3β may play a crucial role in regulating water homeostasis. We developed renal collecting duct-specific GSK3β knockout mice to determine whether deletion of GSK3β affects arginine vasopressin-dependent renal water reabsorption. Although only mildly polyuric under normal conditions, knockout mice exhibited an impaired urinary concentrating ability in response to water deprivation or treatment with a vasopressin analogue. The knockout mice had reduced levels of mRNA, protein, and membrane localization of the vasopressin-responsive water channel aquaporin 2 compared with wild-type mice. The knockout mice also expressed lower levels of pS256-AQP2, a phosphorylated form crucial for membrane trafficking. Levels of cAMP, a major regulator of aquaporin 2 expression and trafficking, were also lower in the knockout mice. Both GSK3β gene deletion and pharmacologic inhibition of GSK3β reduced adenylate cyclase activity. In summary, GSK3β inactivation or deletion reduces aquaporin 2 expression by modulating adenylate cyclase activity and cAMP generation, thereby impairing responses to vasopressin in the renal collecting duct.
Gycogen synthase kinase 3 (GSK3) is a family of ubiquitous serine/threonine protein kinases that consists of two isoforms, GSK3α and GSK3β, with GSK3β having a splice variant.1 GSK3 isoforms are structurally similar except for an additional glycine-rich N-terminal domain in the α isoform, and they have 98% sequence identity, excluding the C-terminus end.2 GSK3β activity has been implicated in diabetes, cancer, cell differentiation, and normal epithelial function.3 Recent studies using GSK3α knockout mice and skeletal muscle-specific GSK3β knockout mice have demonstrated improved glucose tolerance, elevated hepatic glycogen storage, and insulin sensitivity.4,5 On the basis of these and several other preclinical studies using inhibitors, GSK3 is currently considered a key target for drug discovery in diabetes and Alzheimer disease.3 However, recent studies using isoform-specific gene deletion demonstrated that GSK3β is a crucial regulator of embryonic cardiomyocyte proliferation and differentiation.6 Similarly, in the kidneys, inhibition of GSK3β has been suggested to cause a urinary concentrating defect.7–9 This view is formed by the observation that lithium (Li+), which is commonly used for treatment of bipolar disorder, inhibits GSK3 in the clinical therapeutic range and can cause renal toxicity.10 Patients on long-term Li+ treatment often have an irreversible and clinically important reduction in maximal urinary concentrating ability, which may lead to nephrogenic diabetes insipidus (NDI), and detectable impairment in renal concentrating ability has been reported in up to 50% patients.11–13 Since Li+ is a noncompetitive inhibitor of GSK3, loss of GSK3β activity could be a crucial factor in the impaired urine-concentrating ability after LiCl treatment.7,8 Our previous studies demonstrated that in mice, inhibition of GSK3β could play a crucial role in LiCl-induced NDI.8 Similarly, proteomic analysis of renal collecting ducts (CDs) from LiCl-treated rats suggested that GSK3β inactivation could be important for the development of Li+-induced NDI.7 These findings indicated that GSK3β might be crucial for urine concentration by the kidneys.
Regulation of water balance by the kidney is one of its fundamental homeostatic functions and is tightly controlled by arginine vasopressin (AVP). In response to an antidiuretic stimulus, AVP regulates both expression and trafficking of aquaporin 2 (AQP2), resulting in increased water reabsorption.14 In the CDs, AVP binds to AVP type-2 receptor (V2R), activating adenylate cyclase and increasing intracellular cAMP levels. cAMP-mediated activation of protein kinase A (PKA) or exchange protein directly activated by cAMP increases AQP2 transcription15–17 and trafficking to the apical plasma membrane.18 A drastic reduction of AQP2 expression has been associated with LiCl-induced NDI in rats.19–21 Although LiCl has been shown to directly inhibit adenylate cyclase,22–24 it remains controversial whether the reduction in AQP2 is mediated by Li+-induced inhibition of cAMP production.19,25,26 Our previous studies showed that inhibition of GSK3 by LiCl up-regulates renal medullary interstitial cyclooxygenase 2 (COX2) and prostaglandin E2 (PGE2),8 which is known to antagonize AVP-mediated water reabsorption in renal CDs.27,28 Although Li+ is a potent inhibitor of GSK3β, the Li+-induced urinary concentrating defect is associated with diverse mechanisms, making it difficult to decipher the role of GSK3β in AVP-mediated water reabsorption in renal CDs. Therefore, in this study we developed renal CD-specific GSK3β-null mice to examine the role of GSK3β in renal CD responsiveness to the antidiuretic actions of AVP. Therefore, we developed renal CD-specific GSK3β-null mice to examine the role of GSK3β in renal CD responsiveness to the antidiuretic actions of AVP. The maximal urinary concentrating ability in response to water deprivation or vasopressin treatment was significantly compromised in the knockout mice (KO) compared with wild-type (WT) mice. Consistent with this, AQP2 expression and apical membrane localization were reduced in the KO mice, possibly because of lower adenylate cyclase activity and lower intracellular cAMP levels in the presence of 3-isobutyl-1-methylxanthine (IBMX) when compared with WT mice. This is the first demonstration that GSK3β could regulate adenylate cyclase activity, which could possibly be the reason for low AQP2 abundance and reduced urine-concentrating ability in the KO mice.
Results
Generating the Renal CD-Specific GSK3β-Null Mice
To determine the role of GSK3β in renal water reabsorption, we generated renal CD-specific GSK3β KO mice. Deletion of GSK3β in the CD was confirmed by immunohistochemical staining of kidney sections for GSK3β (Figure 1A). Western blot analysis of acutely isolated inner medullary CDs obtained from renal inner medulla and papillae (Figure 1B) showed reduction of GSK3β expression in the KO mice compared with WT, whereas the GSK3α levels remained unchanged (Figure 1B). We confirmed Cre-loxP recombination by PCR of genomic DNA obtained from acutely isolated renal CD (Figure 1C). Mice used for the study were homozygous for floxed GSK3β and carried the HoxB7 Cre gene (KO). Controls were sex-matched littermates homozygous for floxed GSK3β but without Cre (WT).
Figure 1.

These images show the generation of the renal CD-specific GSK3β KO mice. (A) Immunostaining for GSK3β in renal papillae of WT and KO mice. Original magnification, ×20. (B) Western blot analysis shows reduced GSK3β and not GSK3α levels in acutely isolated CDs from the renal inner medulla and papilla. #P < 0.0001. (C) Agarose gel showing PCR products of genomic DNA obtained from renal CDs. KO mice show smaller bands compared with the WT mice (floxed), owing to recombination resulting in deletion of GSK3β.
Overall, adult KO mice were healthy, fertile, and similar in size and weight to their WT littermates. We detected no significant difference in their kidney weights or gross morphology as detected by hematoxylin and eosin staining. Under normal conditions, KO mice showed a modest 56% increase in 24-hour urine output compared with the WT mice (KO mice: 0.080 ± 0.02 ml/g body wt per day versus WT: 0.051 ± 0.015 ml/g body wt per day, P < 0.01, n = 6 in each group). Consistent with this mild polyuria, water consumption in KO mice was moderately higher compared with WT mice (KO mice: 0.25 ± 0.1 ml/g body wt per day versus WT: 0.2 ± 0.07 ml/g body wt per day, P < 0.05, n = 6 in each group). Urine osmolality was numerically lower but not statistically significant in the KO mice compared with the WT mice (KO mice: 1662 ± 285 mOsm/kg H2O versus WT: 2354 ± 324 mOsm/kg H2O, n = 6 in each group). The urine sodium levels were comparable (KO mice: 0.178 ± 0.05 mmol/24-hour urine versus WT 0.16 ± 0.03 mmol/24-hour urine, n = 6 in each group).
KO Mice Have Reduced Ability to Concentrate Urine after Water Deprivation
To determine whether CD-specific GSK3β deletion affected the maximal urinary concentrating capacity, we collected spot urine samples at baseline and after 18-hour water deprivation (free access to food but not water). When water deprived, both WT and KO mice initiated an antidiuretic response. However, the urinary osmolality of the 18-hour water-deprived KO mice was 32% lower than the WT mice (KO: 2898 ± 332 mOsm/kg H2O versus WT 4185 ± 265 mOsm/kg H2O, P < 0.0001, n = 8 in each group) (Figure 2).
Figure 2.

GSK3β gene deletion reduced urine osmolality after water deprivation. Urine osmolality values of urine collected during 18 hours of water deprivation. *P < 0.001, n = 8.
KO Mice Have Reduced AQP2 mRNA and Protein Levels in Response to Water Deprivation
To examine whether the lower urine-concentrating capacity in the KO mice was due to reduced AQP2 expression, we measured renal AQP2 mRNA and protein levels in WT and KO mice. Under normal conditions, the KO mice tended to have higher AQP2 mRNA levels compared with WT mice as shown by quantitative RT-PCR, although this difference did not reach statistical significance (Figure 3A). Similarly, semiquantitative immunoblotting showed 20% higher levels of AQP2 in the renal papilla and inner medulla of KO compared with WT mice at baseline (Figure 3B). However, water deprivation increased AQP2 mRNA by 6.6-fold in the WT mice compared with their baseline, whereas in the KO mice, the increase was less than 2-fold (Figure 3A). Consistent with this, after water deprivation, the total abundance of AQP2 protein levels in the KO mice was significantly lower than in the WT group (Figure 3B). Quantification of the AQP2 bands showed that the glycosylated and nonglycosylated protein levels were 55% and 58% lower in the KO mice compared with the WT mice, indicating that a blunted increase in AQP2 expression could be a major cause for the lower urine osmolality in the water-deprived KO mice.
Figure 3.

GSK3β gene deletion reduced renal AQP2 levels. (A) Quantitative real-time PCR of renal mRNA levels under basal conditions or after 18-hour water deprivation. #P < 0.0001. (B) Western blot analysis for AQP2 expression in the renal papillae at baseline or in water-deprived mice. 55 μg of protein was loaded for basal and 40 μg for water-deprived samples. Densitometry of AQP2 band density. *P < 0.001, #P < 0.0001.
We further determined the distribution of AQP2 and phosphorylated AQP2 in the kidney sections of water-deprived mice by immunohistochemistry. In WT mice, intense staining for AQP2, predominantly at the apical membrane of principal cells, was observed both in the medullary and cortical CDs (Figure 4, A and B). In the KO mice, AQP2 staining in both medulla and cortex was significantly reduced and appeared more diffuse and cytoplasmic (Figure 4, C and D). Although AQP2 appeared to localize apically in a few CDs in the KO mouse cortex, as shown in Figure 4D, it was significantly lower than the WT mice. Because phosphorylation of the COOH-terminal tail at serine 256 of AQP2 (pS256-AQP2) has been shown to be critical for AQP2 accumulation on the apical plasma membrane, we immunostained the kidney sections of the water-deprived mice for pS269-AQP2. In the WT mice, staining for pS269-AQP2 was mostly localized to the apical membrane of the CDs in the medulla as well as cortex (Figure 4, E and F). In the KO mice, we detected low levels of pS269-AQP2, mostly cytoplasmic, in the medulla (Figure 4G). In the cortex, the pS269-AQP2 staining and apical localization were significantly lower than in the WT mice (Figure 4H). The results indicated that the KO mice have reduced AQP2 expression and apical localization in response to water deprivation, indicating that GSK3β gene deletion may affect both AVP-mediated expression and apical targeting of AQP2. In contrast, quantitative real-time PCR carried out for AVP type 2 receptor (V2 receptor) revealed no difference between the WT and KO mice subjected to water restriction (data not shown).
Figure 4.
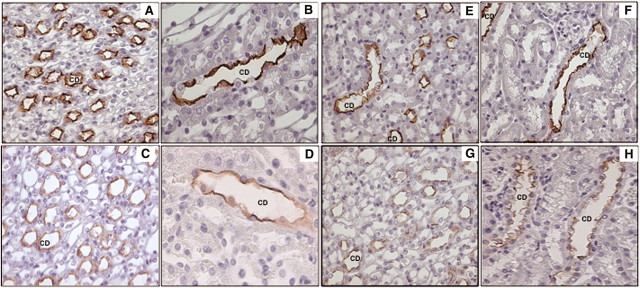
Water-deprived KO and WT mouse kidneys were immunostained for AQP2 and pS256-AQP2. AQP2 labeling in the CDs of inner medulla (A) or cortex (B) of WT mice shows intense labeling and apical localization compared with the inner medulla (C) or cortex (D) of KO mice. pS256-AQP2 labeling of inner medulla (E) or cortex (F) of WT mice shows apical labeling, whereas in the inner medulla (G) or cortex (H) of KO mice, labeling is less intense and more cytoplasmic. Original magnification, ×40.
KO Mice Exhibit a Blunted Response to dDAVP
To examine whether the urinary-concentrating defect in KO mice could be attributed to decreased functional responsiveness to hydro-osmotic effects of AVP, we administered a single intraperitoneal dose of dDAVP, a synthetic analogue of AVP, to the WT and KO mice. In the WT mice, dDAVP treatment increased urine osmolality by 63%, whereas in the KO mice, only a 19% increase was observed (Figure 5A). The Δ for increase in urinary osmolality was significantly greater in WT mice compared with KO mice (Figure 5B). Semiquantitative immunoblotting for pS269-AQP2 levels in the homogenate obtained from the renal inner medulla and papilla of mice treated with dDAVP for 1 hour revealed that in the KO mice, the pS269-AQP2 levels were significantly lower than in the WT mice (Figure 5C).
Figure 5.

GSK3β gene deletion reduced dDAVP-induced urine-concentrating capacity in mice. (a) WT and KO mice were injected with dDAVP (1 μM/kg body wt; intraperitoneally) and spot urine collected. *P < 0.001; n = 8. (b) Δ change in urine osmolality of dDAVP-treated mice compared with basal level. (c) pS256-AQP2 levels in the inner medulla and papilla of WT and KO mice 1 hour after dDAVP treatment. @P < 0.01.
Renal cAMP Levels in Response to dDAVP or Forskolin Are Lower in KO Mice
It is well known that acute increases in AVP levels can induce the expression of AQP2 by a cAMP-dependent pathway.15,29 To examine whether GSK3β activity is important for AVP-mediated cAMP generation, we analyzed urine cAMP levels in 18-hour water-restricted WT and KO mice to determine whether the reduced AQP2 expression in the latter was due to decreased cAMP levels. In KO mice, urine cAMP levels were 30% lower compared with WT mice (WT: 807 ± 150 pmol/24 hours versus KO: 569 ± 139 pmol/24 hours, P < 0.05, n = 8 in each group) (Figure 6A). To examine intracellular cAMP levels, the renal papilla and inner medulla were isolated from WT and KO mice and stimulated in vitro with either vehicle or ligand in the presence of IBMX, a phosphodiesterase inhibitor.30 At baseline, cAMP levels in the KO group were 37% lower than the WT, a value that was not statistically significant (Figure 6B). Stimulation with dDAVP (1 μM) nearly doubled cAMP levels in WT, whereas in the KO group, the increase in cAMP levels was less than half of the WT (WT: 1360 ± 300 pmol/mg protein versus KO: 650 ± 350 pmol/mg protein, P < 0.001, n = 8 in each group) (Figure 6B). To examine whether the lower cAMP levels in the KO group were due to a postreceptor mechanism, we tested the effect of forskolin, a direct activator of adenylate cyclase. Similar to dDAVP, forskolin (1 μM) induced a 3.5-fold increase in cAMP levels in WT, whereas the KO group showed only a marginal increase (WT: 2476 ± 437 pmol/mg protein versus KO: 698 ± 467 pmol/mg protein, P < 0.001, n = 8 in each group) (Figure 6B).
Figure 6.
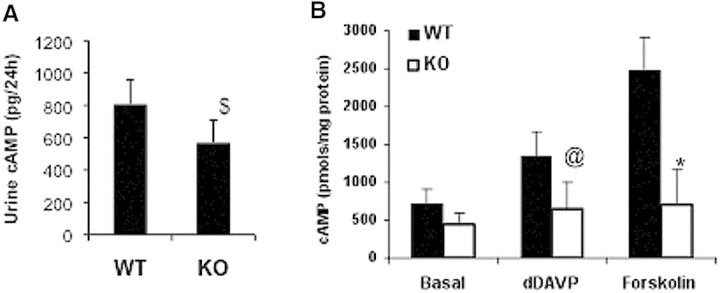
Renal cAMP levels are reduced in KO mice. (A) cAMP levels were measured in the urine of WT and KO mice that were water deprived for 18 hours. $P < 0.05. (B) cAMP levels in renal papillae. Renal papillae were isolated, finely minced, and treated for 20 minutes with IBMX followed by dDAVP (1 μM) or forskolin (1 μM) for an additional 10 minutes. @P < 0.01; *P < 0.001; WT versus KO mice; n = 8.
To examine the effect of GSK3 inhibition on cAMP generation, we studied the effect of SB216763, a small-molecule inhibitor of both GSK3α and GSK3β isoforms, in renal papillae isolated from normal C57BL/6J mouse kidney. Treatment with dDAVP increased cAMP levels by more than 3-fold in the vehicle-treated group, whereas a less than 2-fold increase was noted in SB216763-treated mice (vehicle treated: 1461 ± 250 pmol/mg protein versus SB216763 treated: 864 ± 218 pmol/mg protein, P < 0.01, n = 7 in each group). Forskolin-induced cAMP levels in the SB216763-treated group were only half of those observed in the vehicle-treated group (vehicle: 1311 ± 190 pmol/mg protein versus SB216763: 656 ± 151 pmol/mg protein, P < 0.001, n = 7 in each group) (Figure 7A). To confirm further the role of GSK3 in cAMP generation, we measured forskolin-induced cAMP levels in inner medullary collecting duct cells (IMCD) in culture pretreated with vehicle or SB216763. Forskolin induced a dose-dependent increase in cAMP levels in both the vehicle-treated as well as SB216763-treated cells. However, in the SB216763-treated cells, the cAMP generation induced by 1 μM forskolin was 44% lower than the vehicle-treated cells (vehicle: 653 ± 132 pmol/mg protein versus SB216763: 366 ± 68 pmol/mg protein, n = 6) (Figure 7B).
Figure 7.
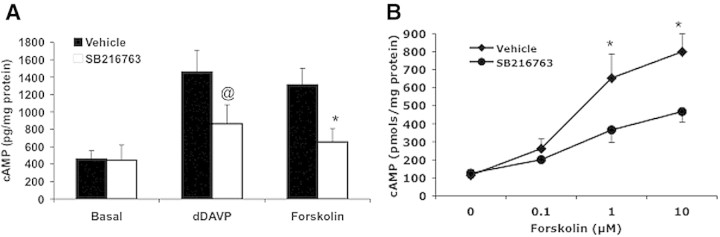
GSK3 inhibition reduced cAMP generation in WT C57BL/6J mice and cultured IMCD3 cells. cAMP levels in response to dDAVP (1 μM) or forskolin (1 μM) were measured in the renal papillae of normal C57BL/6J mice (A) or IMCD cells (B) pretreated with vehicle or SB216763 (5 μM). Mice n = 7; IMCD cells n = 6. @P < 0.01; *P < 0.001 vehicle versus SB216763.
KO Mice Have Reduced Renal Adenylate Cyclase Activity
To examine whether GSK3β gene deletion affected the hormonal and nonhormonal activation of adenylate cyclase, we directly measured its activity in the intact membrane preparations obtained from the renal inner medulla and papilla of WT and KO mice. When membrane preparations were stimulated with dDAVP (1 μM), the adenylate cyclase activity in the KO mice was 45% lower than in the similarly treated WT group (WT, 112 ± 7 pmol/min per mg protein versus 62 ± 12 pmol/min per mg protein, P < 0.001, n = 5) (Figure 8A). Forskolin treatment dose-dependently increased adenylate cyclase activity in the WT mice, with a 4.5-fold increase at 1 μM and a 9.5-fold increase at 10 μM forskolin treatment (Figure 8B). Compared with WT mice, the adenylate cyclase activity at forskolin concentrations of 1 and 10 μM were 30% and 38% lower, respectively. The forskolin concentration for half-maximal activation of adenylate cyclase was ∼5-fold higher in the KO mice compared with the WT mice. The effect of GSK3β inhibition by SB216763 on adenylate cyclase activity was measured in the membrane preparations obtained from wild-type C57Bl/6J mice. Pretreatment with SB216763 caused a concentration-dependent inhibition of forskolin-stimulated adenylate cyclase activity (Figure 8C). Fifty percent inhibition of adenylate cyclase occurred at an SB216763 concentration of ∼0.1 μM. These results indicate that GSK3β inhibition or deletion can reduce dDAVP-induced adenylate cyclase activity. To examine whether the KO mice had lower expression of adenylate cyclase, we carried out semiquantitative immunoblotting on homogenates obtained from the inner medulla and papilla of 18-hour water-deprived WT and KO mice. Expression levels of adenylate cyclases 3 and 6, the predominantly expressed isoforms in the mouse kidney, were not significantly different in the KO mice than WT (Figure 8D).
Figure 8.

GSK3β gene deletion or inhibition reduced adenylate cyclase activity. (A) Adenylate cyclase activity in response to dDAVP was determined in the intact membrane preparations obtained from renal inner medulla and papillae of WT or KO mice (*P < 0.001 WT versus KO mice treated with dDAVP; n = 5) or (B) forskolin (P < 0.0001; WT versus KO mice, determined by ANOVA; n = 5). (C) Adenylate cyclase activity in the membrane preparation of the renal papillae of WT mice treated with GSK3β inhibitor SB216763. (D) Adenylate cyclase 3 or 5/6 expression in renal medullary lysates of water-deprived WT and KO mice.
The Relative Number of Principal and Intercalated Cells in the Renal CD of the KO Mice Did Not Change
GSK3 inhibition has been shown to increase proliferation of cells in vitro31 and in vivo32. Similarly, LiCl treatment for 4 weeks progressively induced increased proliferation of principal cells in the cortical collecting duct cells and IMCD,33 with an increased number of intercalated cells.19,34 Hence, we investigated whether the decrease in AQP2 expression in the KO mice is due to disruption of AVP-induced signaling and not due to morphologic changes in the CD. Tissue sections were labeled for the B1 subunit of the vacuolar [H+]ATPase, a known marker for intercalated cells, or AQP4, a marker for CD principal cells. Immunohistochemical staining did not detect any significant difference in the number of intercalated cells between the WT and KO mice (Supplemental Information).
Discussion
In the current study, we have identified a role for GSK3β in regulating the renal CD response to AVP. This is the first direct evidence that tissue-specific gene deletion of GSK3β in renal CDs can lead to impaired AVP-responsive AQP2 expression and urine concentration in mice. We determined that reduced adenylate cyclase activity and concomitant decrease in cAMP generation likely underlie the reduced AQP2 expression and sensitivity to AVP in KO mice. Our data clearly demonstrate a distinct role for GSK3β in renal water reabsorption.
AVP is a major physiologic regulator of renal water excretion.35 Although previous studies have examined the role of a number of kinases in AVP-induced AQP2 expression and trafficking,7,36 the role of GSK3β in regulation of renal water homeostasis has not previously been examined. LiCl, a GSK3β inhibitor, is known to reduce AQP2 expression and cause NDI, although the mechanism is still incompletely understood. Our previous studies examined the role of GSK3β in LiCl-induced COX2 expression in the renal medullary interstitial cells, found adjacent to IMCDs. We demonstrated that within 5 days of LiCl treatment, GSK3β inhibition leads to increased COX2 in the renal medullary interstitial cells and increased PGE2 levels,8 which could act in a paracrine fashion on the IMCD cells to antagonize AVP action. Interestingly, in a similar study, no increase in COX2 or PGE2 levels was observed in rats treated with LiCl during 4 weeks.37 This brings into focus the possibility that the cellular response to Li+ depends on the length of LiCl treatment, as suggested by Nielsen et al.,7 which in the short term affects signaling and results in low AQP2 abundance, whereas prolonged treatment results in structural changes. Most studies of Li+-induced NDI have been carried out in rats or mice treated with LiCl for up to 40 days, whereas polyuria is noted within the first 7 days of LiCl treatment in mice8,38 and rats.7,33 Hence, the basic underlying mechanism that causes the initial disruption of urine concentration in these animals is not clear. In our previous study of Li+-induced NDI in mice, the decrease in urine osmolality and increase in urine volume in the first week of LiCl treatment corresponded with a time-dependent decline in renal GSK3β activity.8 Similarly, proteomic analysis to investigate the cause for low AQP2 expression in mice treated with LiCl for 7 days revealed increased levels of phosphorylated GSK3β (inactive form) in isolated renal CDs.7 Hence, the CD-specific inhibition of GSK3β could be important for the early changes in signaling in Li+-induced NDI.
To investigate whether GSK3β plays a crucial role in water reabsorption by the renal CDs, we generated a CD-specific GSK3β-null mouse. Although the KO mice did not exhibit the same degree of increase in basal urine output seen in LiCl-treated mice,8 the maximal urinary-concentrating capacity in response to 18-hour water deprivation in these mice failed to reach levels attained by the WT mice, indicating a decreased urinary-concentrating ability in response to AVP. AVP-induced AQP2 expression in the renal CD is modulated mainly at the levels of transcription and posttranscriptional modification.30,39 Previous studies of Li+-induced NDI have demonstrated decreased expression and apical targeting of AQP2 in rats,21,40,41 as well as decreased urine AQP2 excretion in humans.42 Our studies with KO mice indicated an important role for GSK3β activity to modulate AQP2 transcription. At baseline, AQP2 mRNA as well as protein levels in the KO mice were higher than WT mice, possibly as a compensatory mechanism to maintain normal water reabsorption. However, after 18 hours of water deprivation, the increase in AQP2 mRNA and protein levels in KO mice was significantly lower than WT mice, corresponding to a blunting of increases in urine osmolality and indicating a role for GSK3β in regulating AQP2 expression. Because water restriction is a potent stimulus for AVP secretion, we examined whether lower urine-concentrating ability in the KO mice was due to reduced response to AVP. When AVP levels in the mice were clamped at a supraphysiologic level by administration of dDAVP, the increase in urine osmolality in the KO mice was only half that of WT mice. The reduced urine osmolality in the water-deprived KO mice was accompanied by lower levels of pAQP2 and decreased apical membrane targeting of AQP2. Reduced levels of pAQP2 in the KO mice treated with dDAVP for 1 hour suggested that GSK3β deletion could also reduce trafficking of AQP2.
Earlier studies carried out in isolated CDs or kidneys of LiCl-treated rats had attributed the decrease in AQP2 expression to reduced AVP-dependent adenylate cyclase activity19,25,26 and cAMP generation.12,25,43,44 This is consistent with the presence of a cAMP-responsive element in the 5′ untranslated region of the AQP2 gene15,45 and with the demonstration that mice with inherently low cAMP levels have low expression of AQP2.46 However, Li et al.30 found no significant decrease in dDAVP-induced cAMP levels in LiCl-treated cultured cortical CD cells or Brattleboro rats, thus inferring that LiCl-induced decreased AQP2 expression is dissociated from adenylate cyclase activity. To examine the mechanism by which GSK3β gene deletion decreased AQP2 expression, we examined renal cAMP levels in the KO mice. To our knowledge, there is no previous evidence suggesting that GSK3β is involved in regulation of cAMP levels. We observed that the urine cAMP levels in water-deprived KO mice were significantly lower than WT mice. Because urine cAMP levels might not reliably reflect the renal CD cAMP levels, we measured intracellular cAMP levels in the renal inner medulla and papilla, which consists mostly of IMCDs and interstitial cells. In in vitro studies, the response of freshly isolated KO mice renal papillary tissue to dDAVP was less than half that of the WT mice, suggesting that GSK3β activity in the renal CD is important for dDAVP-induced cAMP generation. Because the above study was carried out in the presence of IBMX, which inhibits phosphodiesterase activity, the results also suggested that there could be a difference in adenylate cyclase activity. The activation of adenylate cyclase involves complex interaction of guanosine triphosphate-binding proteins and corresponding receptors that signal for stimulation and inhibition, respectively, from the hormone-occupied receptor (V2R) to the catalytic unit. When forskolin was used to directly activate adenylate cyclase, cAMP levels in KO mice were 3.5-fold lower than WT mice. The results were confirmed using a GSK3 inhibitor, which also reduced adenylate cyclase activity in normal C57BL/6J renal membrane preparations. Because forskolin is a direct activator of adenylate cyclase, the results indicated that at least part of the reduced AVP-induced cAMP accumulation occurs via post-V2 receptor mechanisms, suggesting that GSK3β could regulate signaling in response to AVP at the level of adenylate cyclase.
To confirm our finding that GSK3β deletion reduces cAMP generation, we directly measured adenylate cyclase activity in renal medullary membrane preparations. dDAVP- and forskolin-induced activities of adenylate cyclase were both significantly lower in the KO mice compared with the WT mice. These results were confirmed by studies using the GSK3β inhibitor SB216763. Our studies thus indicate that GSK3β activity is required for robust AVP signaling in the renal CD via regulation of adenylate cyclase activity. Nine isoforms of adenylate cyclase are expressed in the kidneys, of which mRNA levels of isoforms 2 to 7 are detectable in the renal IMCDs.47 We did not find any significant difference in the expression levels of adenylate cyclase isoforms 3 and 6, which are the most abundant isoforms in the CDs. Because in the KO mice, basal adenylate cyclase activity did not differ significantly from the WT mice, GSK3β deletion does not seem to impair the adenylate cyclase complex at the catalytic subunit or the guanyl nucleotide-coupling protein. Instead, because forskolin primarily acts to stimulate the catalytic subunit of adenylate cyclase, loss of GSK3β activity might affect ligand-mediated activation of the enzyme. We have detected conserved GSK3β phosphorylation sites on the C1 and C2 catalytic domains of adenylate cyclases 5 and 6. Further studies will be required to elucidate the mechanism by which GSK3β regulates adenylate cyclase activity.
It is evident from our studies that GSK3β deletion or inhibition reduces sensitivity to the hydro-osmotic effects of AVP, resembling the AVP-resistant urinary-concentrating defect in LiCl-treated mice. However, the KO mice in our study were not overtly polyuric under normal conditions. It is known that Li+-induced urinary-concentrating defect is associated with diverse mechanisms, including hypercalcemia,48 which can by itself cause NDI,49 increased proliferation of intercalated cells at the expense of AQP2-expressing principal cells of the CDs,33 increased renal PGE2 production that can promote Li+-induced diuresis50 and dysregulation of key molecular determinants of AQP2 expression and trafficking.51 It is interesting to note that the KO mice did not show structural changes, like increased number of intercalated cells as observed in chronic LiCl-treated rats. This could be because the KO mice have intact GSK3α. Nevertheless, the reduced adenylate cyclase activity, cAMP levels, and lower abundance and trafficking of AQP2 in the KO mice in response to AVP indicates that GSK3β is involved in signaling regulating AVP-induced increases in AQP2 and urine concentration. These studies suggest a mechanism of dysregulation of water homeostasis that may occur in clinical disorders like NDI, nephritic syndrome, cirrhosis, and a number of cardiac conditions, including congestive heart failure.
In summary, we have identified GSK3β as an important regulator of AVP-mediated water reabsorption by renal CDs. GSK3β inhibition or deletion could reduce AVP-induced adenylate cyclase activity, leading to reduced cAMP generation, resulting in low AQP2 expression and ultimately inhibition of CD water reabsorption. These results suggest that GSK3β might function as a physiologic regulator of AVP action in the renal CD.
Concise Methods
Generating the KO Mice
Mice with CD-specific deletion of GSK3β gene were generated by breeding mice containing the loxP-flanked (floxed) GSK3β gene5 with HoxB7-Cre mice (Jackson Labs).52 Female offspring, heterozygous for both HoxB7 Cre and floxed GSK3β, were mated with males homozygous for floxed GSK3β. Mice with floxed GSK3β genotype were identified by PCR using extracted tail DNA. Sense primer was 5′-GGGGCAACCTTAATTTCATT-3′ and antisense was 5′-AAAACCCTGCCAACACAAAG-3′. HoxB7-Cre genotype was determined by PCR using tail DNA. Sense primer 5′-GGTCACGTGGTCAGAAGAGG-3′ and antisense primer (5′-CTCATCACTCGTTGCATCGA-3′) were used.
Metabolic Cage Studies
All animal experiments were performed in accordance with the guidelines of the Vanderbilt University Institutional Animal Care and Use Committee. Mice were housed in metabolic cages (Hatteras Instruments, Cary, NC) and acclimatized for 5 days before the water-deprivation study. For dDAVP studies, mice were temporarily housed in small restriction chambers and given a single intraperitoneal injection of dDAVP (Sigma, St. Louis, MO) (1 μM/kg body wt). Spot urine samples were collected over 96-well plates, collected separately (two to three samples per mouse, depending on output) for 4 hours, and the highest urine osmolality attained by each was determined. Urine osmolality was measured using Osmett II (Precision Systems, Natick, MA).
Measurement of cAMP
To measure intracellular cAMP levels, renal papillae were directly used for studies as described previously,30 with some modifications. Tissue samples were minced and suspended in Krebs ringer buffer that contained 0.001% DNase I, prewarmed to 37°C. Tissues were incubated for 20 minutes in IBMX at 37°C, followed by the addition of 1 μM dDAVP or 1 μM forskolin for 10 min at 37°C in a total reaction volume of 1 ml. When indicated, tissue samples were preincubated with SB216763 for a total of 30 minutes before treatment with ligand. After reaction, tissue pellets were lysed by adding 0.1 N HCl and were incubated for 1 hour at room temperature, followed by centrifugation at >10,000 × g for 10 minutes. Supernatants were directly assayed for cAMP. Similarly, IMCD cells grown on transwells were pretreated with SB216763 for 2 hours, followed by 20-minute incubation with IBMX and varying concentrations of forskolin for 10 minutes. Urine samples were diluted in enzyme immunoassay buffer and assayed directly. Total cell protein was measured by bicinchoninic acid assay (Sigma). cAMP content was measured using a nonradioactive enzyme immunoassay kit (Cayman Chemical, Ann Arbor, MI), expressed in pmol/ml, and normalized to total protein and expressed as pmol of cAMP/μg of protein.
Quantitative Real-Time RT-PCR
Quantitative real-time RT-PCR on RNA isolated from whole-kidney samples was carried out as described previously.53 The 18S, AQP2, and V2 probes used were 4319413E, Mm00437575_m1, and Mm00517071_m1, respectively (Applied Biosystems, Foster City, CA)
Adenylate Cyclase Activity
Membrane preparation was obtained as described previously.54 Renal papillae were homogenized in ice-cold homogenizing buffer (50 mmol/L Tris-HCl, pH 8.0, containing 1 mmol/L EDTA, 0.2 mmol/L phenylmethyl sulfonyl fluoride, and 250 mmol/L sucrose) in a Dounce homogenizer and centrifuged to remove intact cells and nuclei. Supernatant was centrifuged at 50,000 rpm for 1 hour, and resulting pellet was used as membrane preparation. Adenylate cyclase activity was measured as the rate of conversion of [α-32P] ATP to [α-32P] cAMP. The method of Salomon et al.,55 described in detail by Kelly et al.56 was followed. Adenylate cyclase activity was expressed as pmol of cAMP/mg of protein per minute.
Western Blot Analysis
Generally, 40 μg of protein was loaded on a 12%, and in some cases 4% to 20%, SDS-PAGE minigel, and immunoblotting was carried out as described before.8 For measurement of basal AQP2 (Figure 3B) and 1-hour dDAVP-treated mice (Figure 5C), 55 μg of protein was loaded to visualize the nonglycosylated band. Monoclonal antibody for GSK3β and polyclonal antibody for GSK3α were obtained from BD-Transduction Laboratories and Cell Signaling Technology, respectively. Polyclonal antibodies for AQP2 and pS256-AQP2 were a gift from Dr. Robert Fenton (Water and Salt Research Center, Denmark). Polyclonal adenylate cyclase 3 and adenylate cyclase 5/6 antibodies were obtained from Santa Cruz Biotechnology.
Immunohistochemistry
The kidney sections were prepared and immunostained as described earlier.8 Rabbit polyclonal antibody for GSK3β (Abcam Inc., Cambridge, MA) was used, and antibody for H+ATPase was a gift from Dr. Rikke Nørregaard (Water and Salt Research Center, Denmark). Polyclonal AQP4 antibody was obtained from Alomone Labs Limited (Israel).
Statistical Analysis
Comparisons between WT and KO mice were analyzed by the unpaired t test. Comparisons of multiple points (urine osmolality, cAMP accumulation, and adenylate cyclase activity) were made using one-way ANOVA with the Bonferroni correction. P < 0.05 was taken as significant. Data were expressed as means ± SD.
Disclosures
None.
Acknowledgments
We thank Anastasia Golovin for technical assistance. These studies were supported by the American Society of Nephrology Carl W. Gottschalk Research Scholar Grant to R.R.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1. Mukai F, Ishiguro K, Sano Y, Fujita SC: Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3beta. J Neurochem 81: 1073–1083, 2002 [DOI] [PubMed] [Google Scholar]
- 2. Doble BW, Woodgett JR: GSK-3: Tricks of the trade for a multi-tasking kinase. J Cell Sci 116: 1175–1186, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Woodgett JR: Physiological roles of glycogen synthase kinase-3: Potential as a therapeutic target for diabetes and other disorders. Curr Drug Targets Immune Endocr Metabol Disord 3: 281–290, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR: Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab 6: 329–337, 2007 [DOI] [PubMed] [Google Scholar]
- 5. Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR: Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol 28: 6314–6328, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kerkela R, Kockeritz L, Macaulay K, Zhou J, Doble BW, Beahm C, Greytak S, Woulfe K, Trivedi CM, Woodgett JR, Epstein JA, Force T, Huggins GS: Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest 118: 3609–3618, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nielsen J, Hoffert JD, Knepper MA, Agre P, Nielsen S, Fenton RA: Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: mechanisms for aquaporin 2 down-regulation and cellular proliferation. Proc Natl Acad Sci U S A 105: 3634–3639, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rao R, Zhang MZ, Zhao M, Cai H, Harris RC, Breyer MD, Hao CM: Lithium treatment inhibits renal GSK-3 activity and promotes cyclooxygenase 2-dependent polyuria. Am J Physiol Renal Physiol 288: F642–F649, 2005 [DOI] [PubMed] [Google Scholar]
- 9. Woodgett JR, Force T: Unique and overlapping functions of GSK-3 isoforms in cellular differentiation, proliferation, and cardiovascular development. J Biol Chem 284: 9643–9647, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klein PS, Melton DA: A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A 93: 8455–8459, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bendz H, Sjodin I, Aurell M: Renal function on and off lithium in patients treated with lithium for 15 years or more. A controlled, prospective lithium-withdrawal study. Nephrol Dial Transplant 11: 457–460, 1996 [PubMed] [Google Scholar]
- 12. Boton R, Gaviria M, Batlle DC: Prevalence, pathogenesis, and treatment of renal dysfunction associated with chronic lithium therapy. Am J Kidney Dis 10: 329–345, 1987 [DOI] [PubMed] [Google Scholar]
- 13. Livingstone C, Rampes H: Lithium: A review of its metabolic adverse effects. J Psychopharmacol 20: 347–355, 2006 [DOI] [PubMed] [Google Scholar]
- 14. Nielsen S, Frokiaer J, Marples D, Kwon TH, Agre P, Knepper MA: Aquaporins in the kidney: From molecules to medicine. Physiol Rev 82: 205–244, 2002 [DOI] [PubMed] [Google Scholar]
- 15. Matsumura Y, Uchida S, Rai T, Sasaki S, Marumo F: Transcriptional regulation of aquaporin-2 water channel gene by cAMP. J Am Soc Nephrol 8: 861–867, 1997 [DOI] [PubMed] [Google Scholar]
- 16. Umenishi F, Narikiyo T, Vandewalle A, Schrier RW: cAMP regulates vasopressin-induced AQP2 expression via protein kinase A-independent pathway. Biochim Biophys Acta 1758: 1100–1105, 2006 [DOI] [PubMed] [Google Scholar]
- 17. Yasui M, Zelenin SM, Celsi G, Aperia A: Adenylate cyclase-coupled vasopressin receptor activates AQP2 promoter via a dual effect on CRE and AP1 elements. Am J Physiol 272: F443–F450, 1997 [DOI] [PubMed] [Google Scholar]
- 18. Fushimi K, Sasaki S, Marumo F: Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol Chem 272: 14800–14804, 1997 [DOI] [PubMed] [Google Scholar]
- 19. Christensen BM, Marples D, Kim YH, Wang W, Frokiaer J, Nielsen S: Changes in cellular composition of kidney collecting duct cells in rats with lithium-induced NDI. Am J Physiol Cell Physiol 286: C952–C964, 2004 [DOI] [PubMed] [Google Scholar]
- 20. Kwon TH, Laursen UH, Marples D, Maunsbach AB, Knepper MA, Frokiaer J, Nielsen S: Altered expression of renal AQPs and Na(+) transporters in rats with lithium-induced NDI. Am J Physiol Renal Physiol 279: F552–F564, 2000 [DOI] [PubMed] [Google Scholar]
- 21. Marples D, Christensen S, Christensen EI, Ottosen PD, Nielsen S: Lithium-induced downregulation of aquaporin-2 water channel expression in rat kidney medulla. J Clin Invest 95: 1838–1845, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goldberg H, Clayman P, Skorecki K: Mechanism of Li inhibition of vasopressin-sensitive adenylate cyclase in cultured renal epithelial cells. Am J Physiol 255: F995–F1002, 1988 [DOI] [PubMed] [Google Scholar]
- 23. Jackson BA, Edwards RM, Dousa TP: Lithium-induced polyuria: Effect of lithium on adenylate cyclase and adenosine 3′,5′-monophosphate phosphodiesterase in medullary ascending limb of Henle's loop and in medullary collecting tubules. Endocrinology 107: 1693–1698, 1980 [DOI] [PubMed] [Google Scholar]
- 24. Yamaki M, Kusano E, Tetsuka T, Takeda S, Homma S, Murayama N, Asano Y: Cellular mechanism of lithium-induced nephrogenic diabetes insipidus in rats. Am J Physiol 261: F505–F511, 1991 [DOI] [PubMed] [Google Scholar]
- 25. Christensen S, Kusano E, Yusufi AN, Murayama N, Dousa TP: Pathogenesis of nephrogenic diabetes insipidus due to chronic administration of lithium in rats. J Clin Invest 75: 1869–1879, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Timmer RT, Sands JM: Lithium intoxication. J Am Soc Nephrol 10: 666–674, 1999 [DOI] [PubMed] [Google Scholar]
- 27. Hebert RL: Cellular signalling of PGE2 and its selective receptor analogue sulprostone in rabbit cortical collecting duct. Prostaglandins Leukot Essent Fatty Acids 51: 147–155, 1994 [DOI] [PubMed] [Google Scholar]
- 28. Hebert RL, Jacobson HR, Fredin D, Breyer MD: Evidence that separate PGE2 receptors modulate water and sodium transport in rabbit cortical collecting duct. Am J Physiol 265: F643–F650, 1993 [DOI] [PubMed] [Google Scholar]
- 29. Hasler U, Mordasini D, Bens M, Bianchi M, Cluzeaud F, Rousselot M, Vandewalle A, Feraille E, Martin PY: Long term regulation of aquaporin-2 expression in vasopressin-responsive renal collecting duct principal cells. J Biol Chem 277: 10379–10386, 2002 [DOI] [PubMed] [Google Scholar]
- 30. Li Y, Shaw S, Kamsteeg EJ, Vandewalle A, Deen PM: Development of lithium-induced nephrogenic diabetes insipidus is dissociated from adenylyl cyclase activity. J Am Soc Nephrol 17: 1063–1072, 2006 [DOI] [PubMed] [Google Scholar]
- 31. Rao AS, Kremenevskaja N, Resch J, Brabant G: Lithium stimulates proliferation in cultured thyrocytes by activating Wnt/beta-catenin signalling. Eur J Endocrinol 153: 929–938, 2005 [DOI] [PubMed] [Google Scholar]
- 32. Salas TR, Reddy SA, Clifford JL, Davis RJ, Kikuchi A, Lippman SM, Menter DG: Alleviating the suppression of glycogen synthase kinase-3beta by Akt leads to the phosphorylation of cAMP-response element-binding protein and its transactivation in intact cell nuclei. J Biol Chem 278: 41338–41346, 2003 [DOI] [PubMed] [Google Scholar]
- 33. Christensen BM, Kim YH, Kwon TH, Nielsen S: Lithium treatment induces a marked proliferation of primarily principal cells in rat kidney inner medullary collecting duct. Am J Physiol Renal Physiol 291: F39–F48, 2006 [DOI] [PubMed] [Google Scholar]
- 34. Kim YH, Kwon TH, Christensen BM, Nielsen J, Wall SM, Madsen KM, Frokiaer J, Nielsen S: Altered expression of renal acid-base transporters in rats with lithium-induced NDI. Am J Physiol Renal Physiol 285: F1244–F1257, 2003 [DOI] [PubMed] [Google Scholar]
- 35. Verbalis JG: Vasopressin V2 receptor antagonists. J Mol Endocrinol 29: 1–9, 2002 [DOI] [PubMed] [Google Scholar]
- 36. Sasaki S, Noda Y: Aquaporin-2 protein dynamics within the cell. Curr Opin Nephrol Hypertens 16: 348–352, 2007 [DOI] [PubMed] [Google Scholar]
- 37. Kotnik P, Nielsen J, Kwon TH, Krzisnik C, Frokiaer J, Nielsen S: Altered expression of COX-1, COX-2, and mPGES in rats with nephrogenic and central diabetes insipidus. Am J Physiol Renal Physiol 288: F1053–F1068, 2005 [DOI] [PubMed] [Google Scholar]
- 38. Jia Z, Wang H, Yang T: Mice lacking mPGES-1 are resistant to lithium-induced polyuria. Am J Physiol Renal Physiol 297: 1689–1696, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hasler U, Nielsen S, Feraille E, Martin PY: Posttranscriptional control of aquaporin-2 abundance by vasopressin in renal collecting duct principal cells. Am J Physiol Renal Physiol 290: F177–F187, 2006 [DOI] [PubMed] [Google Scholar]
- 40. Marples D, Frokiaer J, Knepper MA, Nielsen S: Disordered water channel expression and distribution in acquired nephrogenic diabetes insipidus. Proc Assoc Am Physicians 110: 401–406, 1998 [PubMed] [Google Scholar]
- 41. Promeneur D, Kwon TH, Frokiaer J, Knepper MA, Nielsen S: Vasopressin V(2)-receptor-dependent regulation of AQP2 expression in Brattleboro rats. Am J Physiol Renal Physiol 279: F370–F382, 2000 [DOI] [PubMed] [Google Scholar]
- 42. Walker RJ, Weggery S, Bedford JJ, McDonald FJ, Ellis G, Leader JP: Lithium-induced reduction in urinary concentrating ability and urinary aquaporin 2 (AQP2) excretion in healthy volunteers. Kidney Int 67: 291–294, 2005 [DOI] [PubMed] [Google Scholar]
- 43. Cogan E, Abramow M: Inhibition by lithium of the hydroosmotic action of vasopressin in the isolated perfused cortical collecting tubule of the rabbit. J Clin Invest 77: 1507–1514, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cogan E, Svoboda M, Abramow M: Mechanisms of lithium-vasopressin interaction in rabbit cortical collecting tubule. Am J Physiol 252: F1080–F1087, 1987 [DOI] [PubMed] [Google Scholar]
- 45. Hozawa S, Holtzman EJ, Ausiello DA: cAMP motifs regulating transcription in the aquaporin 2 gene. Am J Physiol 270: C1695–C1702, 1996 [DOI] [PubMed] [Google Scholar]
- 46. Frokiaer J, Marples D, Valtin H, Morris JF, Knepper MA, Nielsen S: Low aquaporin-2 levels in polyuric DI +/+ severe mice with constitutively high cAMP-phosphodiesterase activity. Am J Physiol 276: F179–F190, 1999 [DOI] [PubMed] [Google Scholar]
- 47. Hoffert JD, Chou CL, Fenton RA, Knepper MA: Calmodulin is required for vasopressin-stimulated increase in cyclic AMP production in inner medullary collecting duct. J Biol Chem 280: 13624–13630, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Robben JH, Knoers NV, Deen PM: Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol 291: F257–F270, 2006 [DOI] [PubMed] [Google Scholar]
- 49. Procino G, Carmosino M, Tamma G, Gouraud S, Laera A, Riccardi D, Svelto M, Valenti G: Extracellular calcium antagonizes forskolin-induced aquaporin 2 trafficking in collecting duct cells. Kidney Int 66: 2245–2255, 2004 [DOI] [PubMed] [Google Scholar]
- 50. Sugawara M, Hashimoto K, Ota Z: Involvement of prostaglandin E2, cAMP, and vasopressin in lithium-induced polyuria. Am J Physiol 254: R863–R869, 1988 [DOI] [PubMed] [Google Scholar]
- 51. Nielsen J, Kwon TH, Christensen BM, Frokiaer J, Nielsen S: Dysregulation of renal aquaporins and epithelial sodium channel in lithium-induced nephrogenic diabetes insipidus. Semin Nephrol 28: 227–244, 2008 [DOI] [PubMed] [Google Scholar]
- 52. Yu J, Carroll TJ, McMahon AP: Sonic hedgehog regulates proliferation and differentiation of mesenchymal cells in the mouse metanephric kidney. Development 129: 5301–5312, 2002 [DOI] [PubMed] [Google Scholar]
- 53. Rao R, Redha R, Macias-Perez I, Su Y, Hao C, Zent R, Breyer MD, Pozzi A: Prostaglandin E2-EP4 receptor promotes endothelial cell migration via ERK activation and angiogenesis in vivo. J Biol Chem 282: 16959–16968, 2007 [DOI] [PubMed] [Google Scholar]
- 54. Kim SW, Jeon YS, Lee JU, Kang DG, Kook H, Ahn KY, Kim SZ, Cho KW, Kim NH, Han JS, Choi KC: Diminished adenylate cyclase activity and aquaporin 2 expression in acute renal failure rats. Kidney Int 57: 1643–1650, 2000 [DOI] [PubMed] [Google Scholar]
- 55. Salomon Y, Londos C, Rodbell M: A highly sensitive adenylate cyclase assay. Anal Biochem 58: 541–548, 1974 [DOI] [PubMed] [Google Scholar]
- 56. Kelly JJ, Stevens T, Thompson J, Seifert R: Adenylyl and guanylyl cyclase assays. Curr Protoc Pharmacol 2.2.1–2.2.21, 2005 [DOI] [PubMed] [Google Scholar]