

Abstract
Juxtaglomerular cells are highly specialized myoepithelioid granulated cells located in the glomerular afferent arterioles. These cells synthesize and release renin, which distinguishes them from other cells. How these cells maintain their identity, restricted localization, and fate is unknown and is fundamental to the control of BP and homeostasis of fluid and electrolytes. Because microRNAs may control cell fate via temporal and spatial gene regulation, we generated mice with a conditional deletion of Dicer, the RNase III endonuclease that produces mature microRNAs in cells of the renin lineage. Deletion of Dicer severely reduced the number of juxtaglomerular cells, decreased expression of the renin genes (Ren1 and Ren2), lowered plasma renin concentration, and decreased BP. As a consequence of the disappearance of renin-producing cells, the kidneys developed striking vascular abnormalities and prominent striped fibrosis. We conclude that microRNAs maintain the renin-producing juxtaglomerular cells and the morphologic integrity and function of the kidney.
In adult mammals, juxtaglomerular (JG) cells synthesize, store, and release renin, the key regulatory enzyme for the control of BP and fluid electrolyte homeostasis. JG cells are strategically located in the wall of the afferent arterioles at the entrance to the glomerulus to respond to signals from adjacent structures; however, in early embryonic development, renin cells are broadly distributed along intrarenal arteries, in the glomerular mesangium, and in a few developing tubules. With maturation, the cells become progressively restricted to the classical adult JG localization. Lineage studies showed that this progressive restriction is achieved by differentiation of renin cells into other cell types such as vascular smooth muscle, mesangial, and a subset of tubular cells.1 Understanding how these cells acquire and maintain their phenotype is of fundamental biologic importance; however, the mechanisms that maintain the identity of the renin-synthesizing cell have not been fully identified.
Endogenous microRNAs (miRs) are a group of small noncoding RNAs that are 18 to 22 nucleotides in length and exhibit temporal and spatial (tissue/cell) specificity, suggesting their involvement in tissue morphogenesis, developmental timing, and cell differentiation.2–6 The generation of miRs is a complex, multistep process that involves the sequential cleavage of a primary miR (pri-miR)—approximately 100 to 1000 nucleotides in length—by two protein complexes involving RNase III endonucleases: Drosha-DGCR8 in the nucleus and Dicer in the cytoplasm. It has been suggested that miRs mediate gene regulation by a number of posttranscriptional events: degradation of target mRNAs, translational repression, and deadenylation accompanied by mRNA decay.2–4,7 Recently, an effect on chromatin remodeling was also described.8
Because Dicer-null embryos die in utero at embryonic day 7.5, revealing the crucial role of this enzyme and therefore miRs in early development,9 conditional deletion of Dicer has proved to be of great value to demonstrate a requirement for miRs in the development and/or function of several tissues and organs.4,10–12 Within the kidney, Dicer was recently deleted in podocytes, resulting in normal development of podocytes but glomerular disease in adult life.13–15 Those experiments highlight the importance of miRs in tissue development and/or function. The requirement for miRs for the specification and maintenance of cell identity seems to vary among different cell types.16 For instance, conditional deletion studies of Dicer in skeletal muscle indicated that miRs are required for normal embryonic development,17 whereas, in podocytes, the embryonic development is not affected but miRs are crucial for the maintenance of the filtration barrier in adult life.13–15 Conversely, in renal proximal tubular cells, Dicer deficiency does not affect tubular morphology and seems to be protective against ischemic renal injury.18
The role of miRs in the maintenance of JG cells has not been explored. To address this question, we generated mice with a conditional deletion of Dicer specifically and exclusively in renin-expressing cells (by expression of a knock-in allele of cre-recombinase inserted in the Ren1 locus), resulting in selective ablation of miR generation only in renin cells and their descendants. Deletion of Dicer resulted in severe reduction in the number of JG cells accompanied by decreased expression of the Ren1 and Ren2 genes, decreased plasma renin concentration (PRC), decreased BP, abnormal renal function, and striking nephrovascular abnormalities including striped corticomedullary fibrosis.
Results
To assess the effectiveness of the deletion of Dicer in renin cells, we performed in situ hybridization for miR-145, which is expressed in vascular smooth muscle cells (SMCs) and in JG cells. miR-145 serves as a marker for the presence of Dicer; its absence confirms loss of Dicer activity. In control mice, miR-145 is expressed in the JG area (JGA) and in arterioles (Figure 1, A, C, and E). Expression of miR-145 in Dicer conditional knockout (Dicer cKO) mice is completely abolished in JG cells (Figure 1B) and in SMCs of the afferent arteriole (Figure 1F). Because in larger arteries not all of the SMCs originate from renin cells, some do not express cre-recombinase; therefore, in larger arterioles, there is lack of expression of miR-145 in most SMCs, whereas some SMCs unrelated to the renin lineage maintain their expression (Figure 1D).
Figure 1.
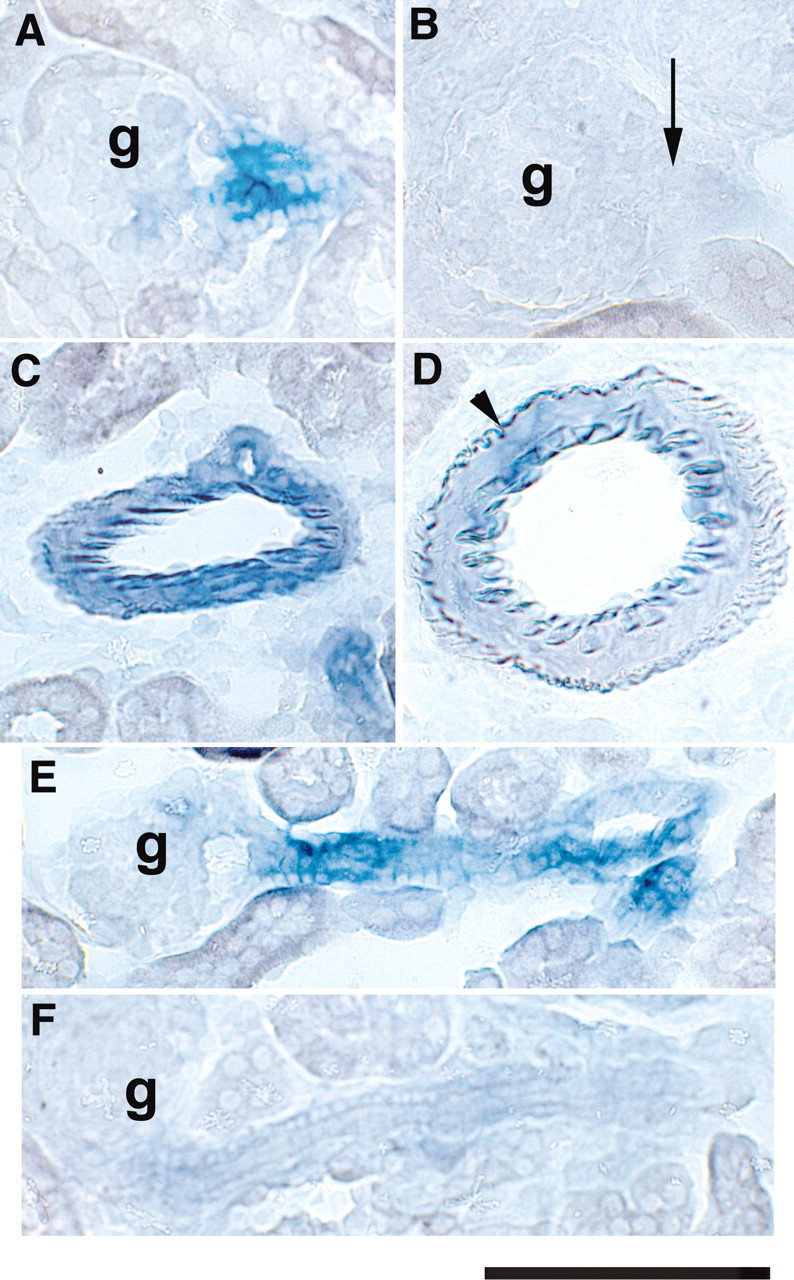
In situ hybridization for miR-145 is shown. (A) Normal expression of miR-145 in JG cells at the entrance to the glomerulus. (B) Expression of miR-145 in JG cells is abolished in Dicer cKO kidneys (arrow). (C) In control mice, miR-145 is expressed throughout the smooth muscle layers of an intrarenal artery. (D) Dicer cKO kidney shows decreased expression of miR-145 in a large vessel. Expression persists in a few cells (arrowhead). (E) Expression of miR-145 along the afferent arteriole of a control kidney. (F) Lack of miR-145 along the afferent arteriole of a Dicer cKO kidney. g, glomerulus. Bar = 100 μm.
Severe Depletion of JG Cells in Dicer cKO Mice
Figure 2A shows the distribution of JG cells within the kidney using renin as the only known marker of JG cells. Whereas in control mice JG cells are present in normal number, the kidneys of Dicer cKO mice were practically devoid of JG cells. The percentage of glomeruli containing renin-positive JGAs in control mice was 33.26 ± 4.37%, whereas in Dicer cKO mice, the percentage fell to 1.43 ± 0.35%. In addition, in Dicer cKO mice, the remaining cells within the few renin-positive JGAs were fewer in number, thinner, and smaller than in the controls. As expected, the marked reduction in the number of renin-positive cells was accompanied by a reduction in renin mRNA levels as judged by allele-specific quantitative reverse transcriptase–PCR (qRT-PCR) analysis comparing Dicer cKO and control mice each with one Ren2 and one Ren1c allele (Dicer cKO and Dicer control 2; Table 1, Figure 2B). Loss of Dicer caused an 82% reduction in Ren1 and an 89% reduction in Ren2 mRNA. In addition, PRC was reduced by 96% (Figure 2C).
Figure 2.

Renin expression and PRC in mice with conditional deletion of Dicer in renin cells are shown. (A) Renin immunostaining. Control mice have the normal endowment of renin-positive cells (brown); however, Dicer cKO mice lack renin-positive cells. *Glomeruli. Bar = 200 μm. (B) qRT-PCR shows reduced levels of both Ren1 and Ren2 renin mRNA in the cKO deletion samples compared with controls. (C) PRC determined by angiotensin I (AngI) generation. Data are means ± SEM. ***P < 0.001, **P < 0.01, *P < 0.05 versus control.
Table 1.
Renin genes, renin distribution, and renal morphology in Dicer cKO and control mice
| Denomination | Genotype | Renin Genes | Renin Distribution | Renal Morphology |
|---|---|---|---|---|
| Dicer cKO | Dicerflox/Δ;Ren1dcre/+ | 1Ren2+1Ren1c | Abnormal | Abnormal |
| Dicer cKO | Dicerflox/flox;Ren1dcre/+ | 1Ren2+1Ren1c | Abnormal | Abnormal |
| Dicer control 1 | Dicerflox/Δ;Ren1c+/+ | 2Ren1c | Normal | Normal |
| Dicer control 2 | Dicerflox/+;Ren1dcre/+ | 1Ren2+1Ren1c | Normal | Normal |
| Dicer control 3 | Dicerflox/+;Ren1c+/+ | 2Ren1c | Normal | Normal |
| Additional control | Ren1dcre/+ | 1Ren2+1Ren1c | Normal | Normal |
| Additional control | Ren1c+/− | 1Ren1c | Normal | Normal |
| Additional control | Ren1c+/+ | 2Ren1c | Normal | Normal |
| Additional control | Ren1dRen2+/+ | 2Ren1d+2Ren2 | Normal | Normal |
Decreased BP in Dicer cKO Mice
Consistent with the decrease in renin mRNA and circulating renin, Dicer cKO mice exhibited a 15-mmHg decrease in systolic BP when measured by tail cuff (Figure 3A). We also measured arterial BP using radiotelemetry. Survival of Dicerflox/Δ;Ren1dcre/+ mice was compromised by insertion of the radiotelemeter. Consequently, we used Dicerflox/flox;Ren1dcre/+ mice and predicted a less severe phenotype when two floxed alleles were present. Indeed, histologic analysis suggested that mice with two Dicer floxed alleles had a more modest phenotype than mice with one floxed and one null allele. Nevertheless, Dicer cKO mice exhibited a significant decrease (9 to 10 mmHg) in systolic, diastolic, and mean arterial BP without apparent change in the overall diurnal/nocturnal BP pattern (Figure 3B).
Figure 3.

Arterial BP measurements of Dicer cKO mice are shown. (A) Tail-cuff measurements of systolic BP (SBP) from Dicerflox/Δ;Ren1dcre/+ mice. **P < 0.01 versus control (n = 3 per group). (B) Radiotelemetry measurements of mean arterial BP (MAP) from Dicerflox/flox;Ren1dcre/+ mice. *P < 0.05 versus control (cKO, n = 6; control, n = 7).
Severe Renal Abnormalities in Dicer cKO Mice
Dicer cKO mice were born at the expected Mendelian frequency and survived to adulthood. At 2 to 3 months of age, the body weight and kidney weight were significantly decreased (Supplemental Figure S1, A and B). In addition, the kidney weigh–body weight ratio was significantly decreased, suggesting an even more pronounced effect on kidney than on somatic growth (Supplemental Figure S1C). Basic blood chemistry measurements showed that Dicer cKO mice had increased blood urea nitrogen and creatinine (Supplemental Figure S1, D and E).
The marked depletion of JG cells was accompanied also by several renal alterations. The kidney surface was hard and irregular as a result of cortical depressions (Figure 4). As evidenced by Masson's trichrome staining, the indentations in the kidney surface corresponded to the shrinkage of multiple corticomedullary bands of scarring containing collagen fibers in the interstitium (Figure 4). The scarred areas were interspersed with areas of less affected tubules and glomeruli, resulting in striped fibrosis. Within these fibrotic areas, arterioles were barely recognizable, tubular structures were missing, and many glomeruli had crescents (Figure 4).
Figure 4.
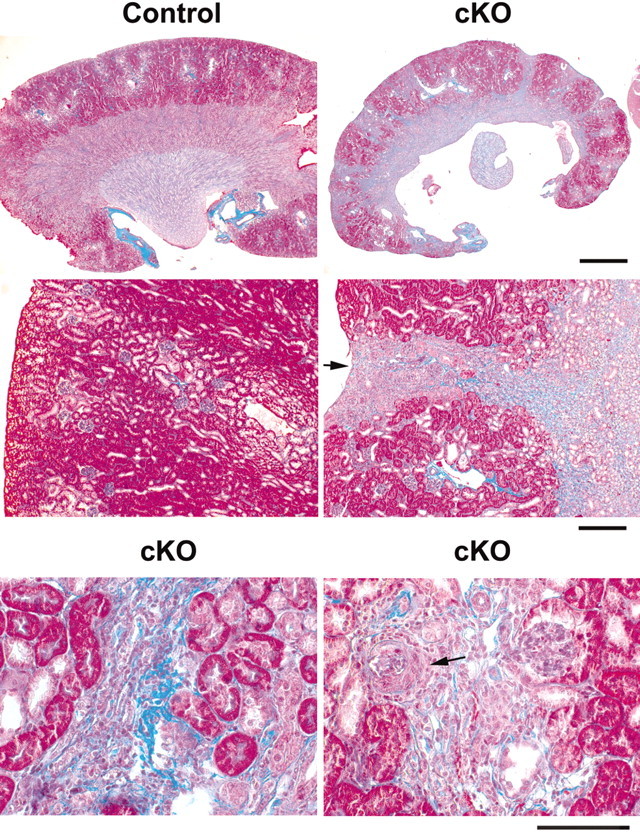
Masson's trichrome staining of Dicer cKO kidney sections is shown. Interstitial fibrosis is shown by the blue colored staining of the collagen fibers. (Top) Low magnification shows that the mutant kidney is smaller than the control and presents striped fibrosis extending from the cortex to the medulla. (Middle, left) Normal cortex in the control. (Middle, right) A scarred area (striped fibrosis) in the Dicer cKO coincides with the depression of the kidney surface (arrow). (Bottom, left) Interstitial fibrosis involves an artery that has lost its boundaries, and its structure is barely recognizable. (Bottom, right) Area with increased fibrosis shows an abnormal glomerulus with a crescent (arrow) and loss of tubular structures. Bars = 1 mm (top), 200 μm (middle), and 100 μm (bottom).
Periodic acid-Schiff (PAS) staining revealed that in addition to the cortical areas of depression, the corticomedullary separation was ill-defined. Within the stripes of fibrosis, there was crowding of sclerotic glomeruli around abnormally formed or missing blood vessels (Supplemental Figure S2).
Immunostaining for α-smooth muscle actin (α-SMA) showed in the interstitium an increased expression of α-SMA (Supplemental Figure S3). In the affected areas, there were dilated tubules surrounded by fibrosis and a decrease in the number of vessels. Some vessels showed an increase in the number of cells in the adventitial layer (perivascular fibroplasia), maintaining a thin layer of SMCs. Sclerotic and cystic glomeruli were also surrounded by cells expressing α-SMA.
Additional alterations in the kidney included thyroidization of some tubuli with the presence of protein casts within the tubular lumens (Supplemental Figure S4A), collapsed glomeruli with thickening of the tubular basement membrane (Supplemental Figure S4B), and glomerulosclerosis with increased periglomerular interstitial cells (Supplemental Figure S4C). The Bowman's capsule showed an increase in the size and in the number of the epithelial cells, which were in some cases arranged in several cell layers rather than the single layer of cells that is observed in normal animals (Supplemental Figure S4, D through F). The epithelial cells of Bowman's capsule, proximal tubules, and epithelioid areas surrounded by interstitial cells (Supplemental Figure S4, B through G) had large nuclei and multiple nucleoli and also showed mitotic figures (Supplemental Figure S4G). There was also flattening of the macula densa (Supplemental Figure S4D). As assessed by PAS staining and α-SMA immunostaining, larger intrarenal arteries were thicker as a result of a marked perivascular fibroplasia (massive enlargement of the adventitial layer) and not as a result of an increase in the arterial smooth muscle layers (Supplemental Figure S4, H and I).
Discussion
This study shows that the enzyme Dicer, whose primary role is to generate mature miRs,19–23 is crucial for the maintenance of the JG cell. The severe reduction in the number of JG cells is accompanied by (1) decreased Ren1 and Ren2 mRNA levels, (2) decreased circulating renin, (3) reduced BP, and (4) severe renal nephrovascular disease and striped fibrosis.
The striking decrease in the number of JG cells in Dicer cKO mice indicates that Dicer and consequently mature miRs are indispensable for the maintenance of the renin cell. Because no obvious cell death (apoptosis or necrosis; data not shown) was associated with the loss of JG cells, we speculate that lack of Dicer resulted in a loss of the JG cell phenotype as evidenced by lack of renin expression and abnormal morphology of the remaining JG cells. It is well accepted that the acquisition and maintenance of a cell's identity is the result of the balanced expression and repression of hundreds of genes. Significant progress has been made regarding some of the pathways that control the acquisition of the identity of the renin cells24–27; however, the role of potential repressors such as miRs in the maintenance of the JG cell identity has not been previously considered. Given that Dicer processes hundreds of miRs and each one of them has the potential to influence the activity of thousands of other genes, it is likely that the diminished number of JG cells is the consequence of the orchestrated balanced action of the numerous genes that control and maintain this cell's phenotype. The results of this study indicate that miRs regulate crucial mechanisms for the maintenance of the JG cell identity. Additional studies will be necessary to determine the molecular network responsible for this striking effect.
Lack of JG cells was accompanied by decreased renin gene expression and circulating renin with the resultant drop in arterial BP. We previously showed a direct relationship between the number of cells that synthesize renin and the levels of renin mRNA and circulating renin.28 Thus, we hypothesize that the decreased circulating renin and renin gene expression found in this study are attributable to the significant drop in the number of JG cells.
Deletion of Dicer in cells from the renin lineage resulted also in unique renal abnormalities. The renal abnormalities included a peculiar striped pattern of interstitial fibrosis involving both cortical and medullary regions of the kidney. The fibrotic bands alternated with better preserved areas (skipped zones). Within the fibrotic bands were noticeable alterations in the vasculature ranging from near replacement of the arterioles by interstitial cells within the stripes to distorted arterioles affected by fibroplasia. The mechanisms underlying the vascular alteration are likely multiple. It has been shown that renin cells are precursors for a subset of renal arteriolar SMCs1; therefore, the vascular abnormalities may indicate that lack of mature miRs in arteriolar SMCs, which express renin in early life, may have impaired the normal differentiation and/or maintenance of the renal arteriolar tree. It is important to recognize that the lack of renin per se, which is known to be involved in nephrovascular development,29,30 could not account solely for the observed vascular alterations. Indeed, deletion of any of the genes encoding components of the renin-angiotensin system (RAS; renin, angiotensin-converting enzyme, angiotensinogen, and combined deletion of both AT1A and AT1B receptors)31–36 have resulted in renal vascular abnormalities strikingly different from the ones described in this study with marked hypertrophy of the SMCs of the arterial tree; however, some of the pathologic findings such as tubulointerstitial fibrosis (in the more affected areas) and medullary atrophy resemble the ones observed in other RAS KO mice and are probably due to the lack of angiotensin II. Moreover, many of the abnormalities observed in Dicer cKO mice are unique and different from the ones found in Ren1c−/− mice and in all of the other deletions of the RAS components. The Ren1c−/− mice previously described33 have different degrees of hydronephrosis not present in the Dicer cKO mice, diffuse interstitial fibrosis (rather than striped fibrosis), glomerulosclerosis, and hypertrophy of the medial layer of the intrarenal arterioles with marked inflammatory perivascular infiltration. In the Dicer cKO mice, the kidney has well-demarcated radial bands of scarring in the territories where the blood vessels traverse the kidney. In addition, the renal vasculature in cKO mice did not show thickening of the arterial smooth muscle layer but instead showed perivascular adventitial fibroplasia. That the arterioles are not hypertrophied (as observed in the Ren1c−/− and in the deletion of all of the other components of the RAS) suggests that the effect is due either to a decrease in the number of renin cells (and not lack of renin per se) or to abnormal differentiation of SMCs. In fact, we previously described that targeted ablation of renin cells—with diphtheria toxin—although leading to marked alterations in kidney structure similar to the ones described for the deletion of the RAS genes, seemed to protect the arterioles from hypertrophy, supporting the notion that other factors in renin cells may have a hyperplasic effect in SMCs of the renal arterial tree.37 Further support for this comes from recent studies on SMC development in mice lacking certain miRs.38
It is likely that a combination of the mechanisms described herein have contributed to the vascular abnormalities that in turn were accompanied with this peculiar striped fibrosis. Although additional studies will be needed to ascertain the mechanisms underlying the arteriolar abnormalities that occur when Dicer is deleted in renin cells, the striped fibrotic pattern of renal injury deserves some comment. This lesion has been described with the prolonged use of immunosuppressants of the calcineurin inhibitor class (tacrolimus and cyclosporine), in hyperuricemia and hyperhomocysteinemia.39–41 Whereas the underlying cause in the pathogenesis of cyclosporine and tacrolimus nephropathy seems to be sustained renal vasoconstriction and renal ischemia leading to tubulointerstitial fibrosis, there is also an increase in the number of renin-expressing cells along the afferent arterioles. The increase in renin augments angiotensin II levels that, in turn, stimulate secretion of TGF-β, thereby contributing to the fibrosis. In the cyclosporine toxicity model, the increase in the number of renin-expressing cells is accompanied by hypertension rather than the lower BP that we observed in our Dicer cKO model. Hyperuricemia also induces activation of the RAS and hypertension, leading to renal fibrosis. Conversely, the striped renal fibrosis observed in hyperhomocysteinemia is not associated with hypertension and/or activation of the RAS; however, it results in arterial and arteriolar wall thickening, leading to hypoperfusion. In the Dicer cKO mouse model described here, the resulting interstitial striped fibrosis is not associated with hypertension, activation of the RAS, or thickening of the SMC layers of the arteriolar wall, and it is nonetheless the likely result of abnormal vessel structure and hypotension leading to hypoperfusion, renal ischemia, and subsequent fibrosis (Figure 5). In addition to the alterations in the kidney interstitium and the renal vasculature, deletion of Dicer was associated with glomerular and tubular abnormalities, including cystic or collapsed glomeruli, glomerulosclerosis, hypercellularity of the Bowman's capsule, abnormal glomerular tubular junctions, and proximal tubular atrophy with tubular dilation and cast formation. These findings are not surprising, because renin cells give rise to mesangial cells, epithelial cells of Bowman's capsule, and a subset of proximal tubule cells, and therefore it is reasonable to assume that lack of Dicer in these cells may have led to loss of epithelial and mesangial descendants, resulting in the aforementioned collapsed/cystic glomeruli and atrophic tubuli. The lack of immunostaining for α-SMA inside otherwise affected glomeruli supports the notion that a depletion of mesangial cells may have occurred and could also underlie the glomerular pathology.
Figure 5.

Possible mechanisms for the pathogenesis of striped renal fibrosis in Dicer cKO mice are shown.
In summary, the results suggest that Dicer and miRs are crucial for the maintenance of renin cells and the morphologic integrity and function of the kidney. The study reveals a potential new mechanism for the regulation and maintenance of the myoepithelioid renin-producing JG cell identity and opens the inquiry for the identification of miR–gene networks required for specification and/or maintenance of renin cells, vascular morphogenesis, and preservation of renal architecture and function. This mouse model will be valuable for studying the mechanisms of renal fibrosis independent of hypertension and/or activation of the RAS.
Concise Methods
Generation of Dicer cKO mice
To study the role of Dicer in renin-expressing cells, we followed two breeding schemes described in Figure 6. Ren1dcre/+ mice1 were first crossed to DicerΔ/+ mice (generated using β-actin cre) to produce DicerΔ/+;Ren1dcre/+ mice, which were then bred to Dicerflox/flox mice42 to obtain Dicerflox/Δ;Ren1dcre/+ experimental and Dicerflox/Δ;Ren1c+/+ control mice (Figure 6A). We also studied Dicerflox/flox;Ren1dcre/+ mice generated as shown in Figure 1B. The three mouse strains were maintained on a C57BL/6J background.
Figure 6.

Breeding scheme to generate cKO of Dicer in renin cells is shown. (A) Strategy using DicerΔ/+ and Dicerflox/flox mice. An additional control (control 3) with both Dicer alleles intact is not depicted (Dicerflox/+;Ren1c+/+). (B) Using Dicerflox/flox mice, additional genotypes Dicerflox/flox;Ren1c+/+, Dicer+/+;Ren1c+/+, and Dicer+/+;Ren1dcre/cre are not shown.
Mice from the experimental group harbor either two floxed Dicer alleles or one floxed Dicer allele and one null (Δ) Dicer allele. The experimental mice also carry a knock-in of cre-recombinase into the Ren1d allele. In these mice, cre-recombinase expression under control of the Ren1d locus results in deletion of Dicer from the renin lineage (we refer to this group as Dicer cKO, and, unless otherwise indicated, the results shown are based on Dicerflox/Δ;Ren1dcre/+ mice). Dicer function in control groups described in Figure 6 is retained, because at least one normal Dicer allele is always present: In control 1 (Dicerflox/Δ;Ren1c+/+) from one floxed allele (without cre-recombinase), in control 2 (Dicerflox/+;Ren1dcre/+) from one wild-type allele, and in control 3 (Dicerflox/+;Ren1c+/+) from both alleles (one floxed and one wild-type). Depending on the strain from which they were derived, mice can have either one renin gene (Ren1c from the C57BL6 strain, Dicerflox mice) or two renin genes (Ren2 Ren1d from the 129J/Sv strain, Ren1dcre knock-in mice). Any mice carrying the Ren1dcre allele also have a Ren2 allele, because the targeting to produce this mouse was performed in embryonic stem cells containing both Ren1d and Ren2.1 The identity of the renin genes present in the mice of each genotype is shown in Table 1. The cKO mice have one Ren2 gene and one Ren1c gene. Controls 1 and 3 have two Ren1c alleles, and control 2 has the same renin alleles as the cKO mice. In addition, Table 1 identifies the renin genes in several other control strains. The normal pattern of renin distribution in the kidney as well as a normal renal morphology is observed in all of the controls generated from the Dicer crosses (this study) and in all of the additional controls listed in Table 1.1,33,43
Because there was also no significant statistical difference in any of the parameters evaluated among the various controls (controls 1, 2, and 3; Table 1) the three groups were combined unless otherwise specified. All procedures were performed following the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Virginia and University of Iowa Animal Care and Use Committee.
In Situ Hybridization
Kidney frozen sections (7 μm) were hybridized at 37°C overnight with 40 nM specific digoxigenin-labeled locked nucleic acid probe to mouse miR-145 (Exiqon, Woburn, MA) and washed in 0.2× SSC three times for 30 minutes at 40°C and then one time for 5 minutes at room temperature. The anti–digoxigenin-AP antibody (Roche Diagnostics Corp., Indianapolis, IN) and the BM Purple AP substrate (Roche Diagnostics Corp.) were used following the manufacturer's protocols.
Histologic Analysis and Immunostaining
Paraffin sections of kidneys were stained with hematoxylin, PAS reagent, and Masson's trichrome and immunostained for renin and α-SMA as described previously (see Supplemental Methods sections).1,25
RNA Extraction and qRT-PCR Analysis
RNA extraction and qRT-PCR analysis were carried out with established methods (see Supplemental Methods section).
Blood Chemistry and PRC
Blood chemistry and determination of PRC (by addition of exogenous substrate) were performed as described previously.37,43
Tail-Cuff and Intra-arterial BP Measurements
Mice were trained for tail-cuff measurements for 7 days followed by daily recording for an additional 7 days as we previously reported. For each day's recording, there were 10 unrecorded cuff inflations and 30 recorded inflations. An average of at least 20 successful recordings were used for data analysis. For radiotelemetry, mice were anesthetized with ketamine:xylazine (87.5 mg:12.5 mg/kg) and implanted with radiotelemeter catheters (Model PA-C10; Data Sciences Int.) into the left carotid artery for direct day/night measurement of arterial pressure (AP) and heart rate (HR). Then, the transmitter was placed subcutaneously into the right flank. Ten days were allowed for recovery, and the AP and HR were continuously recorded for 10 days (sampling every 5 minutes with 10-second intervals). The AP and HR group averages were determined by the average from consecutive 10-day recordings of each individual.
Statistical Analysis
Data are expressed as means ± SEM. Statistical significance was determined by t test and ANOVA (to compare all of the various control groups). P < 0.05 was considered significant.
Disclosures
None.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants DK75481 to M.L.S.S.L., HL066242 to R.A.G., and HL48058 and HL84207 to C.D.S.
Part of this work was presented at the 63rd High Blood Pressure Research Conference, September 23 through 26, 2008; Chicago, IL; and published in abstract form.
We greatly thank the technical assistance of Kimberly Hilsen-Durette and Debbie Davis for the mouse work and of Gulser Gurocak for the renin concentration assays and the CHRC Molecular Biology Core.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental information for this article is available online at http://www.jasn.org/.
References
- 1.Sequeira Lopez ML, Pentz ES, Nomasa T, Smithies O, Gomez RA: Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev Cell 6: 719–728, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Song L, Tuan RS: MicroRNAs and cell differentiation in mammalian development. Birth Defects Res C Embryo Today 78: 140–149, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Bartel DP: MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116: 281–297, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Stefani G, Slack FJ: Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol 9: 219–230, 2008 [DOI] [PubMed] [Google Scholar]
- 5.Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, Impey S: A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci U S A 102: 16426–16431, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenfeld N, Aharonov R, Meiri E, Rosenwald S, Spector Y, Zepeniuk M, Benjamin H, Shabes N, Tabak S, Levy A, Lebanony D, Goren Y, Silberschein E, Targan N, Ben-Ari A, Gilad S, Sion-Vardy N, Tobar A, Feinmesser M, Kharenko O, Nativ O, Nass D, Perelman M, Yosepovich A, Shalmon B, Polak-Charcon S, Fridman E, Avniel A, Bentwich I, Bentwich Z, Cohen D, Chajut A, Barshack I: MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol 26: 462–469, 2008 [DOI] [PubMed] [Google Scholar]
- 7.He L, Hannon GJ: MicroRNAs: Small RNAs with a big role in gene regulation. Nat Rev Genet 5: 522–531, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez S, Pisano DG, Serrano M: Mechanistic principles of chromatin remodeling guided by siRNAs and miRNAs. Cell Cycle 7: 2601–2608, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ: Dicer is essential for mouse development. Nat Genet 35: 215–217, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Chen JF, Murchison EP, Tang R, Callis TE, Tatsuguchi M, Deng Z, Rojas M, Hammond SM, Schneider MD, Selzman CH, Meissner G, Patterson C, Hannon GJ, Wang DZ: Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc Natl Acad Sci U S A 105: 2111–2116, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damiani D, Alexander JJ, O'Rourke JR, McManus M, Jadhav AP, Cepko CL, Hauswirth WW, Harfe BD, Strettoi E: Dicer inactivation leads to progressive functional and structural degeneration of the mouse retina. J Neurosci 28: 4878–4887, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papaioannou MD, Pitetti JL, Ro S, Park C, Aubry F, Schaad O, Vejnar CE, Kuhne F, Descombes P, Zdobnov EM, McManus MT, Guillou F, Harfe BD, Yan W, Jegou B, Nef S: Sertoli cell Dicer is essential for spermatogenesis in mice. Dev Biol 326: 250–259, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho J, Ng KH, Rosen S, Dostal A, Gregory RI, Kreidberg JA: Podocyte-specific loss of functional microRNAs leads to rapid glomerular and tubular injury. J Am Soc Nephrol 19: 2069–2075, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harvey SJ, Jarad G, Cunningham J, Goldberg S, Schermer B, Harfe BD, McManus MT, Benzing T, Miner JH: Podocyte-specific deletion of Dicer alters cytoskeletal dynamics and causes glomerular disease. J Am Soc Nephrol 19: 2150–2158, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi S, Yu L, Chiu C, Sun Y, Chen J, Khitrov G, Merkenschlager M, Holzman LB, Zhang W, Mundel P, Bottinger EP: Podocyte-selective deletion of Dicer induces proteinuria and glomerulosclerosis. J Am Soc Nephrol 19: 2159–2169, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saal S, Harvey SJ: MicroRNAs and the kidney: Coming of age. Curr Opin Nephrol Hypertens 18: 317–323, 2009 [DOI] [PubMed] [Google Scholar]
- 17.O'Rourke JR, Georges SA, Seay HR, Tapscott SJ, McManus MT, Goldhamer DJ, Swanson MS, Harfe BD: Essential role for Dicer during skeletal muscle development. Dev Biol 311: 359–368, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei O, Bhatt K, Mi QS, Haase V, Dong Z: Targeted deletion of Dicer in proximal tubular cells protects against ischemic renal injury and renal failure [Abstract]. J Am Soc Nephrol 19: 25A, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K: Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev 19: 489–501, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tam OH, Aravin AA, Stein P, Girard A, Murchison EP, Cheloufi S, Hodges E, Anger M, Sachidanandam R, Schultz RM, Hannon GJ: Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature 453: 534–538, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, Chiba H, Kohara Y, Kono T, Nakano T, Surani MA, Sakaki Y, Sasaki H: Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 453: 539–543, 2008 [DOI] [PubMed] [Google Scholar]
- 22.Calabrese JM, Seila AC, Yeo GW, Sharp PA: RNA sequence analysis defines Dicer's role in mouse embryonic stem cells. Proc Natl Acad Sci U S A 104: 18097–18102, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yi R, Pasolli HA, Landthaler M, Hafner M, Ojo T, Sheridan R, Sander C, O'Carroll D, Stoffel M, Tuschl T, Fuchs E: DGCR8-dependent microRNA biogenesis is essential for skin development. Proc Natl Acad Sci U S A 106: 498–502, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pentz ES, Lopez ML, Cordaillat M, Gomez RA: Identity of the renin cell is mediated by cAMP and chromatin remodeling: An in vitro model for studying cell recruitment and plasticity. Am J Physiol Heart Circ Physiol 294: H699–H707, 2008 [DOI] [PubMed] [Google Scholar]
- 25.Gomez RA, Pentz ES, Jin X, Cordaillat M, Sequeira Lopez ML: CBP and p300 are essential for renin cell identity and morphological integrity of the kidney. Am J Physiol Heart Circ Physiol 296: H1255–H1262, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pan L, Black TA, Shi Q, Jones CA, Petrovic N, Loudon J, Kane C, Sigmund CD, Gross KW: Critical roles of a cyclic AMP responsive element and an E-box in regulation of mouse renin gene expression. J Biol Chem 276: 45530–45538, 2001 [DOI] [PubMed] [Google Scholar]
- 27.Chen L, Kim SM, Oppermann M, Faulhaber-Walter R, Huang Y, Mizel D, Chen M, Lopez ML, Weinstein LS, Gomez RA, Briggs JP, Schnermann J: Regulation of renin in mice with Cre recombinase-mediated deletion of G protein Gsalpha in juxtaglomerular cells. Am J Physiol Renal Physiol 292: F27–F37, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Kim HS, Maeda N, Oh GT, Fernandez LG, Gomez RA, Smithies O: Homeostasis in mice with genetically decreased angiotensinogen is primarily by an increased number of renin-producing cells. J Biol Chem 274: 14210–14217, 1999 [DOI] [PubMed] [Google Scholar]
- 29.Sequeira Lopez ML, Gomez RA: The role of angiotensin II in kidney embryogenesis and kidney abnormalities. Curr Opin Nephrol Hypertens 13: 117–122, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Reddi V, Zaglul A, Pentz ES, Gomez RA: Renin-expressing cells are associated with branching of the developing kidney vasculature. J Am Soc Nephrol 9: 63–71, 1998 [DOI] [PubMed] [Google Scholar]
- 31.Kim HS, Krege JH, Kluckman KD, Hagaman JR, Hodgin JB, Best CF, Jennette JC, Coffman TM, Maeda N, Smithies O: Genetic control of blood pressure and the angiotensinogen locus. Proc Natl Acad Sci U S A 92: 2735–2739, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yanai K, Saito T, Kakinuma Y, Kon Y, Hirota K, Taniguchi-Yanai K, Nishijo N, Shigematsu Y, Horiguchi H, Kasuya Y, Sugiyama F, Yagami K, Murakami K, Fukamizu A: Renin-dependent cardiovascular functions and renin-independent blood-brain barrier functions revealed by renin-deficient mice. J Biol Chem 275: 5–8, 2000 [DOI] [PubMed] [Google Scholar]
- 33.Takahashi N, Lopez ML, Cowhig JE, Jr, Taylor MA, Hatada T, Riggs E, Lee G, Gomez RA, Kim HS, Smithies O: Ren1c homozygous null mice are hypotensive and polyuric, but heterozygotes are indistinguishable from wild-type. J Am Soc Nephrol 16: 125–132, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Hilgers KF, Reddi V, Krege JH, Smithies O, Gomez RA: Aberrant renal vascular morphology and renin expression in mutant mice lacking angiotensin-converting enzyme. Hypertension 29: 216–221, 1997 [DOI] [PubMed] [Google Scholar]
- 35.Oliverio MI, Kim HS, Ito M, Le T, Audoly L, Best CF, Hiller S, Kluckman K, Maeda N, Smithies O, Coffman TM: Reduced growth, abnormal kidney structure, and type 2 (AT2) angiotensin receptor-mediated blood pressure regulation in mice lacking both AT1A and AT1B receptors for angiotensin II. Proc Natl Acad Sci U S A 95: 15496–15501, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsuchida S, Matsusaka T, Chen X, Okubo S, Niimura F, Nishimura H, Fogo A, Utsunomiya H, Inagami T, Ichikawa I: Murine double nullizygotes of the angiotensin type 1A and 1B receptor genes duplicate severe abnormal phenotypes of angiotensinogen nullizygotes. J Clin Invest 101: 755–760, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pentz ES, Moyano MA, Thornhill BA, Sequeira Lopez ML, Gomez RA: Ablation of renin-expressing juxtaglomerular cells results in a distinct kidney phenotype. Am J Physiol Regul Integr Comp Physiol 286: R474–R483, 2004 [DOI] [PubMed] [Google Scholar]
- 38.Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L, Braun T: Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest 119: 2634–2647, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vanherweghem JL: Toxic nephropathies. In: Atlas of Diseases of the Kidney, edited by Schrier RW. Philadelphia, Current Medicine, 1999, pp 10.1–10.19 [Google Scholar]
- 40.Mazzali M, Hughes J, Kim YG, Jefferson JA, Kang DH, Gordon KL, Lan HY, Kivlighn S, Johnson RJ: Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 38: 1101–1106, 2001 [DOI] [PubMed] [Google Scholar]
- 41.Kumagai H, Katoh S, Hirosawa K, Kimura M, Hishida A, Ikegaya N: Renal tubulointerstitial injury in weanling rats with hyperhomocysteinemia. Kidney Int 62: 1219–1228, 2002 [DOI] [PubMed] [Google Scholar]
- 42.Harfe BD, McManus MT, Mansfield JH, Hornstein E, Tabin CJ: The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci U S A 102: 10898–10903, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pentz ES, Lopez ML, Kim HS, Carretero O, Smithies O, Gomez RA: Ren1d and Ren2 cooperate to preserve homeostasis: Evidence from mice expressing GFP in place of Ren1d. Physiol Genomics 6: 45–55, 2001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.