

Abstract
The adenylyl and guanylyl cyclases catalyze the formation of 3′,5′-cyclic adenosine or guanosine monophosphate from the corresponding nucleoside 5′-triphosphate. The guanylyl cyclases, the mammalian adenylyl cyclases, and their microbial homologues function as pairs of homologous catalytic domains. The crystal structure of the rat type II adenylyl cyclase C2 catalytic domain was used to model by homology a mammalian adenylyl cyclase C1-C2 domain pair, a homodimeric adenylyl cyclase of Dictyostelium discoideum, a heterodimeric soluble guanylyl cyclase, and a homodimeric membrane guanylyl cyclase. Mg2+ATP or Mg2+GTP were docked into the active sites based on known stereochemical constraints on their conformation. The models are consistent with the activities of seven active-site mutants. Asp-310 and Glu-432 of type I adenylyl cyclase coordinate a Mg2+ ion. The D310S and D310A mutants have 10-fold reduced Vmax and altered [Mg2+] dependence. The NTP purine moieties bind in mostly hydrophobic pockets. Specificity is conferred by a Lys and an Asp in adenylyl cyclase, and a Glu, an Arg, and a Cys in guanylyl cyclase. The models predict that an Asp from one domain is a general base in the reaction, and that the transition state is stabilized by a conserved Asn-Arg pair on the other domain.
Adenylyl cyclases (ACs) catalyze the formation of the prototypical second messenger, cAMP, from ATP. In mammals, AC isoforms types I–IX (AC1–AC9) are among the key effectors of hormones that bind seven-transmembrane receptors (1–4). In fungi and bacteria, the ACs function in development and in metabolic regulation, respectively (3). The guanylyl cyclases (GCs) catalyze the formation of cGMP from GTP. Soluble GC (sGC) is the key effector of nitric oxide, and membrane GCs serve in atrial natriuretic peptide (ANP) signaling and in phototransduction (5). The central importance of these enzymes in so many biological processes has led to intense interest in their catalytic mechanism.
The enzymatic activities of the eukaryotic ACs, other homologous ACs, and the GCs are conferred by homologous catalytic domains (refs. 1–8; Fig. 1). Mammalian ACs contain two copies, designated C1 and C2, which are ≈35% identical to each other (1–4, 9). Recombinant C1 and C2 form an active heterodimeric enzyme when mixed and stimulated by forskolin (7, 8). The isolated C1 homodimer is inactive, whereas the C2 homodimer has trace activity (7, 8, 10). The class III bacterial and most of the fungal and parasite ACs contain one catalytic domain per polypeptide chain (3). Transmembrane GCs also contain a single cyclase catalytic domain per polypeptide chain and function as homooligomers (11–13). sGCs are heterodimers of the α and β subunits (14), which contain one catalytic domain each. Many cyclases clearly function as domain pairs, and we postulate that this property is general to the superfamily (Fig. 1).
Figure 1.
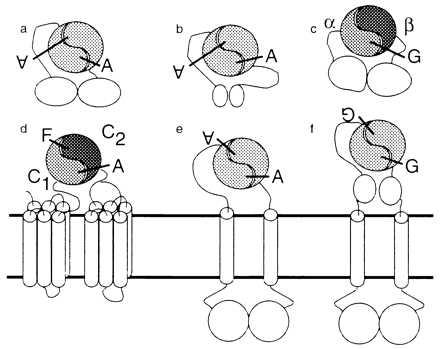
Topologies (3) of yeast AC (a), bacterial class III AC (b), sGC (c), mammalian and Drosophila ACs and ACA (d), ACG and parasite ACs (e), and ANP receptor and photoreceptor GCs (f). A, ATP; G, GTP; and F, forskolin. Mammalian ACs and their homologues are known as “class III” (3) to distinguish them from nonhomologous class I and II bacterial ACs.
The ACs and GCs catalyze stereochemically analogous reactions, which proceed with inversion of configuration, presumably by direct in-line attack of the 3′ hydroxyl on the α-phosphate (15–18). The (Sp)- but not the (Rp)-diastereomer of NTPαS is a substrate, and the reaction requires Mg2+ or Mn2+ (Me2+). Me2+GTP binds to photoreceptor GC as a β,γ chelate with the metal ion coordinated to the pro-R oxygen of the β phosphate (18). The stereochemical data substantially define the conformation of the bound Mg2+ nucleotide complex.
The crystal structure of the homodimer of the C2 cyclase catalytic domain revealed a wreath-like dimer (19) and an active site located in a deep cleft together with the forskolin binding site. It was not possible to obtain the C2 homodimer structure in complex with ATP, however. This left key questions open about the enzyme mechanism, which we try to address here.
The C2 homodimer appears to be a useful template for understanding the larger family of dimeric enzymes because the dimer interface residues are well conserved (Fig. 2). A model of the AC1 C1/AC2 C2 heterodimer was used to predict a region on the AC1 C1 domain that binds Gsα (20). The modeling has now been extended to four major cyclase groups. In view of the stereochemical constraints on Me2+ NTP binding, unambiguous solutions were found for the docking of substrate. The models are consistent with sequence conservation and mutational data. We confirmed the predicted role of a conserved Asp in Mg2+ binding by characterizing the AC1 mutant D310A.
Figure 2.
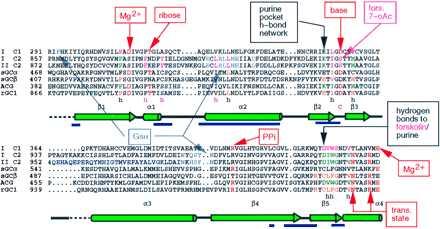
Sequence alignment colored by residue function. Based on modeling, catalytic, Me2+, pyrophosphate ligands are red; forskolin, magenta; adenine, dark green; and guanine, gold. The Gsα binding site is gray (20), and Gβγ is blue (21). Dimer interface: blue underscores; h, hydrophobic pocket. The C-terminal 60 aa are omitted.
METHODS
Homology Modeling.
Thirty other sequences were aligned with AC2 C2 (22) using clustalw (23). This alignment differs from our previous one (19), in that the α1 helix is present in all domains. Homology models of AC1 (9), ACG (24), ret-GC1 (25), and bovine sGC (26, 27) were constructed by using look 2.0 (Molecular Applications Group) for side-chain rotamer placement and energy minimization. The rat AC2 C2 domain homodimer (ref. 19; 1AB8) was used as the template.
Substrate Docking and Molecular Dynamics.
To model the reactive conformation of the ATP, the coordinates of cAMP were minimized without a 3′ O—P bond restraint after adding one oxygen to the phosphate. The pyrophosphate moiety of ATP was incorporated and energy-minimized with x-plor 3.8 (28) by using the parameters of Parkinson et al. (29). Docking was carried out using O (30) by placing trial Mg2+ ions coordinated by the acidic pairs (AC1 310/354 or 310/432). ATP was docked onto AC1 as a β,γ chelate through the (Rp) oxygen of the β-phosphate. Four permutations were considered of two potential Mg2+ sites and two possible Δ-screwsense β,γ chelates related by a 180° rotation about the axis between the metal ion and the β,γ ester oxygen. Three out of four models were eliminated because the adenosine moiety either had few or no contacts with the enzyme (two cases) or had steric overlap with protein atoms (one case). Hydrogen bond donor/acceptor similarities between forskolin and adenosine were used to manually improve the docked conformation. The resulting structure was minimized to convergence to a gradient of less than 1.0 kcal/Å by using x-plor 3.8 and the parameters of Engh and Huber (31) and Parkinson et al. (29). ATP or GTP was docked into the other homology models as above. Each of the models was subjected to 4,000 steps of molecular dynamics at T = 300 K and minimized subject to restraints based on identified interactions (Table 1). Distance restraints were implemented as smoothed square-well potentials (28). Charges were removed from all net charged groups, and a dielectric constant ɛ = 20 was used. No disulfide bond restraints were used in refinement.
Table 1.
Interatomic distance restraints
| AC1 | ACG | sGC | rGC1 | Atom | Tgt | Min | Max | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| NTP | 1 | o3′ | Asn | 1007 | 520B | 548B | 1004B | nd2 | 2.8 | 0.3 | 0.5 |
| NTP | 1 | o3′ | Asp | 354 | 445A | 531A | 929A | od1 | 2.8 | 0.3 | 0.5 |
| NTP | 1 | o3′ | Asp | 354 | 445A | 531A | 929A | od2 | 2.8 | 0.3 | 0.5 |
| ATP | 1 | n6 | Asp | 1000 | 513B | od1 | 2.8 | 0.2 | 1.2 | ||
| ATP | 1 | n6 | Asp | 1000 | 513B | od2 | 2.8 | 0.2 | 1.2 | ||
| GTP | 1 | o6 | Cys | 541B | 997B | sg | 2.8 | 0.2 | 1.2 | ||
| NTP | 1 | o2b | Mg | 1 | 1 | 1 | 1 | mg | 2.3 | 0.1 | 0.1 |
| NTP | 1 | o3g | Mg | 1 | 1 | 1 | 1 | mg | 2.3 | 0.1 | 0.1 |
| NTP | 1 | pa | NTP | 1 | 1 | 1 | 1 | o3′ | 3.5 | 0.1 | 0.1 |
| NTP | 1 | o1a | NTP | 1 | 1 | 1 | 1 | o3′ | 3.9 | 0.5 | 0.2 |
| NTP | 1 | o2a | NTP | 1 | 1 | 1 | 1 | o3′ | 3.9 | 0.5 | 0.2 |
| NTP | 1 | o1a | Arg | 1011 | 524B | 552B | 1008B | nh1 | 3.0 | 0.5 | 0.7 |
| NTP | 1 | o1a | Arg | 1011 | 524B | 552B | 1008B | nh2 | 3.0 | 0.5 | 0.7 |
| NTP | 1 | o2b | Arg | 398 | 491A | 575A | 976A | nh1 | 3.0 | 0.5 | 1.0 |
| NTP | 1 | o2b | Arg | 398 | 491A | 575A | 976A | nh2 | 3.0 | 0.5 | 1.0 |
| Mg | 1 | mg | Arg | 398 | 491A | 575A | 976A | nh1 | 4.0 | 0.5 | 4.0 |
| Mg | 1 | mg | Arg | 398 | 491A | 575A | 976A | nh2 | 4.0 | 0.5 | 4.0 |
| Mg | 1 | mg | Glu | 432 | 526A | 609A | 1010A | oe1 | 2.4 | 0.1 | 0.5 |
| Mg | 1 | mg | Asp | 310 | 401A | 487A | 885A | od1 | 2.4 | 0.1 | 0.5 |
| Mg | 1 | mg | Glu | 432 | 526A | 609A | 1010A | oe2 | 2.4 | 0.1 | 1.1 |
| Mg | 1 | mg | Asp | 310 | 401A | 487A | 885A | od2 | 2.4 | 0.1 | 1.1 |
| NTP | 2 | o3′ | Asn | 520A | 1004A | nd2 | 2.8 | 0.3 | 0.5 | ||
| NTP | 2 | o3′ | Asp | 445B | 929B | od1 | 2.8 | 0.3 | 0.5 | ||
| NTP | 2 | o3′ | Asp | 445B | 929B | od2 | 2.8 | 0.3 | 0.5 | ||
| ATP | 2 | n6 | Asp | 513A | od1 | 2.8 | 0.2 | 1.2 | |||
| ATP | 2 | n6 | Asp | 513A | od2 | 2.8 | 0.2 | 1.2 | |||
| GTP | 2 | o6 | Cys | 997A | sg | 2.8 | 0.2 | 1.2 | |||
| NTP | 2 | o2b | Mg | 2 | 2 | mg | 2.3 | 0.1 | 0.1 | ||
| NTP | 2 | o3g | Mg | 2 | 2 | mg | 2.3 | 0.1 | 0.1 | ||
| NTP | 2 | pa | NTP | 2 | 2 | o3′ | 3.5 | 0.1 | 0.1 | ||
| NTP | 2 | o1a | NTP | 2 | 2 | o3′ | 3.9 | 0.5 | 0.2 | ||
| NTP | 2 | o2a | NTP | 2 | 2 | o3′ | 3.9 | 0.5 | 0.2 | ||
| NTP | 2 | o1a | Arg | 524A | 1008A | nh1 | 3.0 | 0.5 | 0.7 | ||
| NTP | 2 | o1a | Arg | 524A | 1008A | nh2 | 3.0 | 0.5 | 0.7 | ||
| NTP | 2 | o2b | Arg | 491B | 976B | nh1 | 3.0 | 0.5 | 1.0 | ||
| NTP | 2 | o2b | Arg | 491B | 976B | nh2 | 3.0 | 0.5 | 1.0 | ||
| Mg | 2 | mg | Arg | 491B | 976B | nh1 | 4.0 | 0.5 | 4.0 | ||
| Mg | 2 | mg | Arg | 491B | 976B | nh2 | 4.0 | 0.5 | 4.0 | ||
| Mg | 2 | mg | Glu | 526B | 1010B | oe1 | 2.4 | 0.1 | 0.5 | ||
| Mg | 2 | mg | Asp | 401B | 885B | od1 | 2.4 | 0.1 | 0.5 | ||
| Mg | 2 | mg | Glu | 526B | 1010B | oe2 | 2.4 | 0.1 | 1.1 | ||
| Mg | 2 | mg | Asp | 401B | 885B | od2 | 2.4 | 0.1 | 1.1 | ||
| For | 1 | oAc | Ser | 927 | og | 2.8 | 0.3 | 0.5 | |||
| For | 1 | o1 | Asp | 419 | od1 | 2.8 | 0.3 | 0.5 | |||
| For | 1 | o1 | Val | 420 | o | 2.8 | 0.3 | 0.5 | |||
| For | 1 | o11 | Ser | 422 | n | 2.8 | 0.3 | 0.5 |
Tgt, min, and max are target distance d, and allowed deviations from it, d− and d+. The restraint potential is E (kcal/mol) = 50Δ2, where Δ = R − d + d+ for R > d + d+, Δ = 0 for d − d− < R < d + d+; Δ = d − d− − R for R < d − d−, where R is the interatomic distance.
Stereochemical Quality.
The Ramachandran plots for each of the final models were within the acceptable range for low resolution structures (32), with 0–5 outliers per dimer. All other Procheck stereochemical criteria are acceptable (32). Average packing scores for each dimer range from −1.9 to −1.5, as compared with −0.7 for the C2 homodimer crystal structure. These are within the range for correctly threaded structures (33). Individual outliers were inspected manually and did not reveal any uncompensated buried charges.
Site-Directed Mutagenesis.
Site-directed mutagenesis was carried out by the PCR from the template pProEX-HAH6-IC1 (8) with primers 5′-AGCGGATAACAATTTCACACAGG-3′, 5′-CCTCTTTGCATCCATCGTGGGC-3′ (D310S), and 5′-CCTCTTTGCAGCATCGTGGGC-3′ (D310A) by using Taq DNA polymerase (Promega). Secondary reactions were carried on pProEX-HAH6-IC1 with the primer 5′-TGTGGAATTGTGAGCGGATAAC-3′ and the 0.5-kb products of the first PCR. PCR products were purified from agarose gels with Geneclean (Bio 101). The 0.7-kb products were digested with EcoRI and HindIII, ligated into pProEX-HAH6-IC1 digested with the same enzymes, and sequenced.
Enzyme Kinetics.
Mutant proteins were expressed in BL21(DE3) and purified (8). AC activity was assayed in a 100-μl volume for 10 min at 30°C with 1 μg each of AC1 C1 domain and AC2 C2 domain (8) in a buffer of 20 mM Tris (pH 7.8), 1 mM EDTA, 0.5 mM ATP, 0.1 mM 3-isobutyl-1-methylxanthine, 2 mM DTT, and 0 or 100 μM forskolin while [MgCl2] or [MnCl2] was varied, or with 20 mM MgCl2 while [ATP] was varied. cAMP production was measured with a cAMP assay kit (Amersham; ref. 34). Kinetic parameters were obtained by nonlinear fitting to the Michaelis–Menten equation.
RESULTS
Forskolin Binding Site.
The two forskolin binding sites in the C2 homodimer crystal structure were compared with the homology models. In all of the domains except C2, an Asp (354 in AC1 numbering) replaces a Ser (AC2 S942), which donates a hydrogen bond to the 7-acetyl group of forskolin (19, 20). The S→D replacement destroys a hydrogen bond to forskolin and creates an unfavorable 2.5-Å O—O close contact. This suggests that forskolin does not bind to sites containing the Asp substitution, consistent with a single forskolin binding site on mammalian AC containing the β2-β3 turn of the C2 domain and β5 of the C1 domain.
Nucleotide and Metal Ion Binding Site.
A triad of acidic residues is present on all of the domains except C2 and the sGC β subunit. In AC1, D354, D310, and E432 (Fig. 3a) are arrayed in an arc from near one end of the cleft to the center. D354 is 4.5 Å and 8.0 Å away from D310 and E432, respectively, and D310 and E432 are 3.9 Å apart. The heterodimeric cyclases contain one of these clusters, whereas the homodimers contain two. The β,γ biphosphate moiety of ATP docked to AC1 is bridged by Mg2+ to D310 and E432 (Figs. 3a, 4a, and 5). R398 from C1 β4 approaches the β-phosphate. The α-phosphate interacts with R1011. The ribose interacts with the side-chains of T315, D354, and N1007. The adenosine moiety is in the syn conformation.
Figure 3.
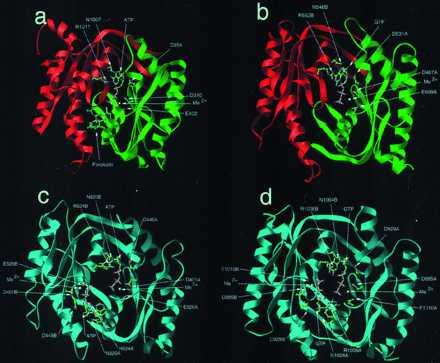
Ribbon diagram (35) of AC1 (a), ACG (b), sGC (c), and retGC-1 (d). Heterodimers are green (C1/α) and red (C2/β), and homodimers are cyan.
Figure 4.
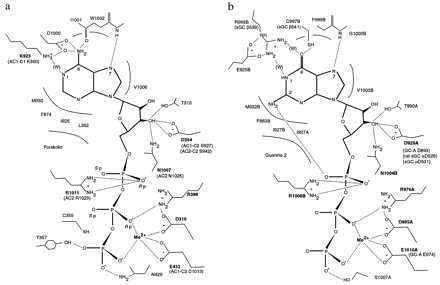
Schematic of the ATP site of AC1 (a), and one of the two GTP sites on retGC-1 (b). Mutational data are available for boldface residues. Homologous positions discussed in the text are shown in parentheses. sGC numbering is for bovine, except where marked.
Figure 5.
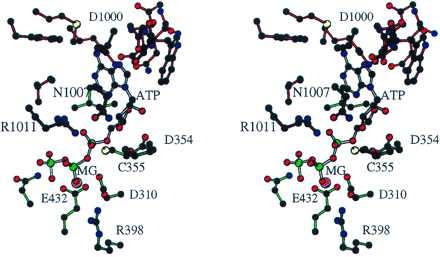
Stereoview (36) of nucleotide site in AC1. Atoms are black (carbon), red (oxygen), blue (nitrogen), yellow (sulfur), green (phosphorous), and magenta (magnesium). Protein bonds are colored as in Fig. 3; NTP bonds are white.
GTP binds to a single site on the sGC dimer (Figs. 3b and 4b). The α subunit binds the Mg2+ and triphosphate moieties, whereas the β subunit binds the guanosine moiety. Residues interacting with the ribose, triphosphate, and metal ion are conserved between ACs and GCs, but interactions with the purine ring differ. The guanine O6 accepts a weakly polar hydrogen bond from Cβ541 (Fig. 4b), which replaces AC1 D1000. The O6 may interact, perhaps via a water molecule, with the side-chain of Rβ539.
The ATP-bound ACG homodimer differs from the mammalian ACs in that it contains two active sites (Fig. 3c). The γ-phosphates of the two ATP molecules approach within 6 Å of each other. The GTP-bound homodimeric photoreceptor retGC-1 has two GTP binding sites (Fig. 3d). The retGC-1-bound GTP molecules approach within 3 Å of each another in the model, with an apparent interaction between the guanine 2-amino group of one and the γ-phosphate of the other. This may reflect the presence of the guanine 2-amino or may be an artifact of the modeling.
Kinetics of D310A and D310S.
The D310A and D310S mutants of AC1 C1 domain were expressed at normal levels and purified to homogeneity. The mutant proteins therefore are presumed to be normally folded. The Vmax for each is 9% that of wild type at 20 mM MgCl2 in the absence of forskolin stimulation. Mutant activities are increased less than 2-fold in the presence of MnCl2, whereas wild type is stimulated almost 25-fold (Fig. 6). The activity of D310S increases about 2-fold in the presence of 100 μM forskolin, as compared with 900-fold activation of wild type (not shown). Both mutants have maximum activity at [Me2+] = 20–25 mM, as compared with 2 to 8 mM for wild type (Fig. 6). Both mutants and wild type exhibit nonlinear kinetics with respect to [Mg2+], as does intact AC (37, 38).
Figure 6.
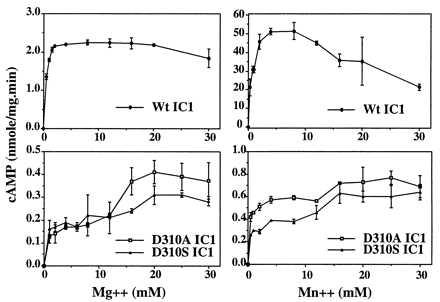
Enzyme activity as a function of [MgCl2] or [MnCl2] with error bars from duplicate measurements.
DISCUSSION
Mutational Analysis.
The wealth of existing mutational data on ACs and GCs provides stringent tests of the models. The AC1 mutant D354A is inactive (39), as are its counterparts D893A and αD529A in rat GC-A (ref. 11; α531 in bovine) and sGC (40), respectively (Figs. 2 and 4). Inactivation is not a result of global misfolding. N1025A and R1029A in AC2 reduce activity by 30- to 50-fold (41). These three residues cluster around the reactive 3′ hydroxyl and α-phosphate in a manner consistent with a plausible catalytic mechanism (Fig. 7).
Figure 7.
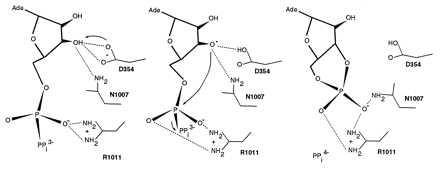
Hypothetical reaction mechanism, using AC1 as an example.
Two mutants increase Km (ATP) in AC more than 2-fold: K923A and E432A in AC1 (39). The C2 domain K923 is in the adenosine binding pocket and is predicted to contact N1 or to stabilize the conformation of the adenine 6-amino group ligand D1000. Its C1 domain counterpart, K350A, has normal activity. The asymmetric effects of mutating the two Lys residues on Km (ATP) are consistent with their nonequivalent roles in the structure.
E432A is predicted to impair Me2+ binding, consistent with its hypersensitivity to Mn2+ activation and the increase in Km(ATP) at fixed [Mn2+] (39). The AC1 C2 counterpart of E432A, D1013A, has normal activity. This agrees with the prediction that the catalytic Me2+ site is on C1. The pyrophosphate contact mutant R398A increases Km(ATP) 2-fold and the IC50 for P-site inhibition 24-fold (39).
D310A does not abolish activity as does D354A (39); hence, D310 is unlikely to be an essential catalytic base. The reduction in Vmax is greater than for the other predicted Me2+ ligand, E432. This is consistent with the proximity of D310 to the 3′ OH of the NTP. D310 may be a linchpin for positioning the ADP moiety in the active site. Alternatively, D310 could bind a second metal ion together with D354. The lack of Mn2+ activation suggests a fundamental alteration in the Me2+ response because this phenotype has not been reported previously. The lack of forskolin stimulation suggests the reaction catalyzed by D310S does not proceed preferentially through the normal activated conformation. D310A and D310S have markedly different [Mg2+] dose-response curves from wild type. The increase in activity is biphasic, reaching a maximum at 20 mM MgCl2 compared with 2–8 mM for wild type. The actual increase in the [Mg2+] optimum might have been larger if not for the inhibitory response to high [Mg2+] (37, 38).
Forskolin and Nucleotide Binding Sites.
The catalytic core of mammalian AC binds one molecule each of forskolin and ATP (42). Knowledge of the binding determinants allows us to predict the NTP stoichiometries of other cyclases. The domains fall into six groups (Fig. 5): mammalian AC and ACA C1 and C2; soluble GC α and β; homodimeric AC; and homodimeric GC. C1 domains of mammalian ACs and Dictyostelium discoideum ACA contain the catalytic Asp, Me2+ ligands, and part of the forskolin and adenine binding sites. C2 domains of mammalian ACs and ACA contain the catalytic Asn and Arg and the remainder of the adenine and forskolin (except ACA and AC9) binding sites. The model is consistent with the binding of two molecules of forskolin in the C2 homodimer crystal.
Catalytic domains from membrane GCs, microbial ACs, and ACG contain all the determinants for activity and substrate binding. Therefore, they are predicted to have two catalytic sites per homodimer. This agrees with the cooperativity with respect to GTP seen in the catalytic fragment of GC-A (13). Each α4 helix contributes the catalytic Asn and Arg to one active site and a Me2+-binding Glu to the other (Fig. 3d). This could partly explain why mutation of this E974 abolishes positive cooperativity in the soluble form of GC-A (43). The AC and GC families can be divided into two fundamentally different subfamilies. Mammalian ACs and sGC have a single active site, whereas ACG and other homodimeric ACs, as well as the membrane GCs, have two.
The forskolin binding site appears to have evolved by “cannibalizing” one of the two active sites of an ancestral homodimer. The three rings of forskolin and adenosine have similar combinations of hydrophobic and hydrogen bonding groups. The forskolin O1 hydroxyl donates, but does not accept, hydrogen bonds from the enzyme, as does the adenine 6-amino group. The adenine N7 and forskolin O11 keto are both hydrogen bond acceptors.
Metal Binding Site.
Thiophosphate analogue studies (18) and modeling locate one catalytic metal ion per active site. There is kinetic evidence for two Me2+ binding sites, however (37, 38, 44). The thiophosphate studies do not rule out a second catalytic metal ion bound near D310, T315, D354 (AC1), and the NTP α-phosphate, nor are there structural grounds to exclude this possibility. Uranyl ion and ordered solvent sites on the C2 homodimer (19) suggest possible allosteric Me2+ binding sites outside the catalytic cleft.
Like AC, DNA polymerase I catalyzes the Mg2+-dependent attack of a hydroxyl group on the α-phosphate of a (deoxy)NTP. DNA polymerase I and AC both contain within larger folds a βαββαβ motif, a ubiquitous supersecondary structure (45, 46). dCTP bound to DNA polymerase I (1KFD; ref. 47) overlaid with the βαββαβ motif of AC1 is 7.4 Å from ATP docked to AC1 (Pγ-Pγ). The difference in binding modes can be explained by the absence of intermolecular interactions that are conserved in the two enzymes. The functionally relevant structural similarity is confined to the two active-site Asps.
Domain Closure.
The central cleft is 19.8 Å across in the C2 crystal structure (Cα-Cα of R1029A and D1031B). In contrast, it is 14.0–16.1 Å in the homodimer models and 16.2–16.8 Å in the heterodimer models after restrained molecular dynamics. The catalytically competent conformation of AC is proposed to be more closed than the C2 crystal structure by 3–6 Å. The ability of the enzyme the adopt the proper closed conformation may be an important point of regulation.
Substrate Specificity.
The adenine 6-amino group interacts with AC1 D1000, which would discriminate against the guanine O6. The GC counterpart of the Asp is Cys (997 in retGC-1), which could form a weak hydrogen bond to the 6-group of either adenine or guanine. This is consistent with the absence of GC activity in ACs but the presence of AC activity in GCs at 10% of the normal GC activity (48). A conserved Lys (AC1 923) and a Glu/Arg pair (retGC-1 925 and 995) occupy analogous parts of the binding site (Fig. 4). RetGC-1 E925 does not contact the purine but appears to position R995. R995 interacts directly or indirectly with the N1 or O6 of the guanine. Mutations occur in the dimer interface outside the pocket that have low activity and little NTP selectivity, suggesting that altering the dimer interfaces disrupts the pocket (49).
Catalytic Mechanism.
The proximity of the AC1 D354 to the ribose 3′ hydroxyl suggests it is optimally positioned to serve as a catalytic base (Fig. 7). The essential Asn (AC1 N1007) is directly across from the Asp and within hydrogen bond distance of the 3′ hydroxyl, suggesting a possible role in stabilizing an oxyanion-like nucleophile. The essential Arg (AC1 R1011) is positioned to stabilize a pentavalent transition state at the α-phosphate. The Asn/Arg pair is analogous to the Gln/Arg pair in GTPases (50–52), as suggested by Tang and coworkers (41). The build-up of positive charge in the vicinity of the 3′ hydroxyl might promote binding of inhibitors possessing a negative charge at this position. This might explain in part why 3′ phosphorylated adenosine derivatives (P-site inhibitors) bind so tightly to AC (53) and why their binding is dramatically impaired by the AC2 N1025A and R1029A mutants (41).
Several startling insights have emerged from the modeling. First, heterodimeric cyclases have one active site, whereas homodimeric cyclases have two sites within a single cleft. Second, the forskolin binding site in mammalian ACs appears to have evolved from an adenosine binding site. Third, the catalytic center consists of three residues, an Asp from one domain and an Asn and Arg from the other. There are considerable uncertainties in model-built structures such as these. Nevertheless, the modeling accounts for a large body of existing experimental data and successfully predicted D310 to have a key role in the active site of AC1.
Acknowledgments
We thank many colleagues in the cyclase field for discussions and D. R. Duan for comments on the manuscript. This work was supported in part by National Institutes of Health Grant GM33138 to A.E.R.
ABBREVIATIONS
- AC
adenylyl cyclase
- GC
guanylyl cyclase
- sGC
soluble guanylyl cyclase
- AC1
type I adenylyl cyclase
- AC2
type II adenylyl cyclase
- ACG
germination-specific adenylyl cyclase
- retGC-1
photoreceptor guanylyl cyclase-1
Footnotes
Data deposition: Model coordinates have been deposited in the Protein Data Bank at Brookhaven National Laboratory, Upton, NY 11973 (accession nos. 1awk–1awn).
References
- 1.Sunahara R K, Dessauer C W, Gilman A G. Annu Rev Pharmacol Toxicol. 1996;36:461–480. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- 2.Smit M J, Iyengar R. Adv Second Messenger and Phosphoprotein Res. 1997;32:1–21. doi: 10.1016/s1040-7952(98)80003-7. [DOI] [PubMed] [Google Scholar]
- 3.Tang W-J, Yan S, Drum C. Adv Second Messenger and Phosphoprotein Res. 1997;32:137–151. doi: 10.1016/s1040-7952(98)80009-8. [DOI] [PubMed] [Google Scholar]
- 4.Hanoune J, Pouille Y, Tzavara E, Shen T, Lipskaya L, Mayamoto N, Suzuki Y, Defer N. Mol Cell Endocrinol. 1997;128:179–194. doi: 10.1016/s0303-7207(97)04013-6. [DOI] [PubMed] [Google Scholar]
- 5.Drewett J G, Garbers D L. Endocrine Rev. 1994;15:135–162. doi: 10.1210/edrv-15-2-135. [DOI] [PubMed] [Google Scholar]
- 6.Tang W-J, Gilman A G. Science. 1995;268:1769–1772. doi: 10.1126/science.7792604. [DOI] [PubMed] [Google Scholar]
- 7.Whisnant R E, Gilman A G, Dessauer C W. Proc Natl Acad Sci USA. 1996;93:6621–6625. doi: 10.1073/pnas.93.13.6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan S-Z, Hahn D, Huang Z-H, Tang W-J. J Biol Chem. 1996;271:10941–10945. doi: 10.1074/jbc.271.18.10941. [DOI] [PubMed] [Google Scholar]
- 9.Krupinski J, Coussen F, Bakalyar H, Tang W-J, Feinstein P G, Orth K, Slaughter C, Reed R R, Gilman A G. Science. 1989;244:1558–1564. doi: 10.1126/science.2472670. [DOI] [PubMed] [Google Scholar]
- 10.Zhang G, Liu Y, Qin J, Vo B, Tang W-J, Ruoho A E, Hurley J H. Protein Sci. 1997;6:903–908. doi: 10.1002/pro.5560060417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson D K, Garbers D L. J Biol Chem. 1995;270:425–430. doi: 10.1074/jbc.270.1.425. [DOI] [PubMed] [Google Scholar]
- 12.Wilson E M, Chinkers M. Biochemistry. 1995;34:4696–4701. doi: 10.1021/bi00014a025. [DOI] [PubMed] [Google Scholar]
- 13.Yang R-B, Garbers D L. J Biol Chem. 1997;272:13738–13742. doi: 10.1074/jbc.272.21.13738. [DOI] [PubMed] [Google Scholar]
- 14.Wedel B, Harteneck C, Foerster J, Friebe A, Schultz G, Koesling D. J Biol Chem. 1995;270:24871–24875. doi: 10.1074/jbc.270.42.24871. [DOI] [PubMed] [Google Scholar]
- 15.Gerlt J A, Coderre J A, Wolin M S. J Biol Chem. 1980;255:331–334. [PubMed] [Google Scholar]
- 16.Eckstein F, Romaniuk P J, Heideman W, Storm D R. J Biol Chem. 1981;256:9118–9120. [PubMed] [Google Scholar]
- 17.Senter P D, Eckstein F, Mulsch A, Bohme E. J Biol Chem. 1983;258:6741–6745. [PubMed] [Google Scholar]
- 18.Koch K-W, Eckstein F, Stryer L. J Biol Chem. 1990;265:9659–9663. [PubMed] [Google Scholar]
- 19.Zhang G, Liu Y, Ruoho A E, Hurley J H. Nature (London) 1997;386:247–253. doi: 10.1038/386247a0. [DOI] [PubMed] [Google Scholar]
- 20.Yan S-Z, Huang Z-H, Rao V D, Hurley J H, Tang W-J. J Biol Chem. 1997;272:18849–18854. doi: 10.1074/jbc.272.30.18849. [DOI] [PubMed] [Google Scholar]
- 21.Chen J, Devivo M, Dingus J, Harry A, Li J, Sui J, Carty D J, Blank J L, Exton J H, Stoffel R H, Inglese J, Lefkowitz R J, Logothetis D E, Hildebrandt J D, Iyengar R. Science. 1995;268:1166–1169. doi: 10.1126/science.7761832. [DOI] [PubMed] [Google Scholar]
- 22.Feinstein P G, Schrader K A, Bakalyar H A, Tang W J, Krupinski J, Gilman A G, Reed R R. Proc Natl Acad Sci USA. 1991;88:10173–10177. doi: 10.1073/pnas.88.22.10173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pitt G S, Milona N, Borleis J, Lin K C, Reed R R, Devreotes P N. Cell. 1992;69:305–315. doi: 10.1016/0092-8674(92)90411-5. [DOI] [PubMed] [Google Scholar]
- 25.Shyjan A W, de Sauvage F J, Gillett N A, Goeddel D V, Lowe D G. Neuron. 1992;9:727–737. doi: 10.1016/0896-6273(92)90035-c. [DOI] [PubMed] [Google Scholar]
- 26.Koesling D, Herz J, Gausepohl H, Niroomand F, Hinsch K D, Mulsch A, Bohme E, Schultz G, Frank R. FEBS Lett. 1988;239:29–34. doi: 10.1016/0014-5793(88)80539-8. [DOI] [PubMed] [Google Scholar]
- 27.Koesling D, Harteneck C, Humbert P, Bosserhoff A, Frank R, Schultz G, Bohme E. FEBS Lett. 1990;266:128–132. doi: 10.1016/0014-5793(90)81523-q. [DOI] [PubMed] [Google Scholar]
- 28.Brunger A T. x-plor. Yale University, New Haven, CT: Dept. of Molecular Biophysics and Biochemistry; 1996. , Version 3.8. [Google Scholar]
- 29.Parkinson G, Vojtechovsky J, Clowney L, Brunger A T, Berman H M. Acta Cryst D. 1996;52:57–64. doi: 10.1107/S0907444995011115. [DOI] [PubMed] [Google Scholar]
- 30.Jones T A, Zou J Y, Cowan S W, Kjelgard M W. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 31.Engh R, Huber R. Acta Crystallogr A. 1991;47:392–400. [Google Scholar]
- 32.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 33.Hooft R W W, Vriend G, Sander C, Abola E E. Nature (London) 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 34.Voss T, Wallner E. Anal Biochem. 1992;207:40–43. doi: 10.1016/0003-2697(92)90496-t. [DOI] [PubMed] [Google Scholar]
- 35.Carson M. J Mol Graphics. 1987;5:103–106. [Google Scholar]
- 36.Kraulis P. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 37.Somkuti S G, Hildebrandt J D, Herberg J T, Iyengar R. J Biol Chem. 1982;257:6387–6393. [PubMed] [Google Scholar]
- 38.Pieroni J P, Harry A, Chen J, Jacobowitz O, Magnusson R P, Iyengar R. J Biol Chem. 1995;270:21368–21373. doi: 10.1074/jbc.270.36.21368. [DOI] [PubMed] [Google Scholar]
- 39.Tang W-J, Stanzel M, Gilman A G. Biochemistry. 1995;34:14563–14572. doi: 10.1021/bi00044a035. [DOI] [PubMed] [Google Scholar]
- 40.Yuen P S, Doolittle L K, Garbers D L. J Biol Chem. 1994;269:791–793. [PubMed] [Google Scholar]
- 41.Yan S-Z, Huang Z-H, Shaw R S, Tang W J. J Biol Chem. 1997;272:12342–12349. doi: 10.1074/jbc.272.19.12342. [DOI] [PubMed] [Google Scholar]
- 42.Dessauer C W, Scully T T, Gilman A G. J Biol Chem. 1997;272:22272–22277. doi: 10.1074/jbc.272.35.22272. [DOI] [PubMed] [Google Scholar]
- 43.Wedel B J, Foster D C, Miller D E, Garbers D L. Proc Natl Acad Sci USA. 1997;94:459–462. doi: 10.1073/pnas.94.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garbers D L, Johnson R A. J Biol Chem. 1975;250:8449–8456. [PubMed] [Google Scholar]
- 45.Artymiuk P J, Poirrett A R, Rice D W, Willett P A. Nature (London) 1997;388:33–34. doi: 10.1038/40310. [DOI] [PubMed] [Google Scholar]
- 46.Bryant S H, Madej T, Janin J, Liu Y, Ruoho A E, Zhang G, Hurley J H. Nature (London) 1997;388:34. [Google Scholar]
- 47.Beese L S, Friedman J M, Steitz T A. Biochemistry. 1993;32:14095–14101. doi: 10.1021/bi00214a004. [DOI] [PubMed] [Google Scholar]
- 48.Mittal C K, Murad F. J Biol Chem. 1977;252:3136–3140. [PubMed] [Google Scholar]
- 49.Beuve A, Danchin A. J Mol Biol. 1992;225:933–938. doi: 10.1016/0022-2836(92)90093-y. [DOI] [PubMed] [Google Scholar]
- 50.Sondek J, Lambright D G, Noel J P, Hamm H E, Sigler P B. Nature (London) 1994;372:276–279. doi: 10.1038/372276a0. [DOI] [PubMed] [Google Scholar]
- 51.Coleman D E, Berghuis A M, Lee E, Linder M E, Gilman A G, Sprang S R. Science. 1994;265:1405–1412. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- 52.Scheffzek K, Ahmadian M R, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, Wittinghofer A. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 53.Desaubry L, Shoshani I, Johnson R A. J Biol Chem. 1996;271:14028–14034. doi: 10.1074/jbc.271.24.14028. [DOI] [PubMed] [Google Scholar]