

Abstract
Congenital human cytomegalovirus infections are the major infectious cause of birth defects in the United States. How this virus crosses the placenta and causes fetal disease is poorly understood. Guinea pig cytomegalovirus (GPCMV) is a related virus that provides an important model for studying cytomegaloviral congenital transmission and pathogenesis. In order to facilitate genetic analysis of GPCMV, the 232 kb GPCMV genome was cloned as an infectious bacterial artificial chromosome (BAC). The BAC vector sequences were flanked by LoxP sites to allow efficient excision using Cre recombinase. All initial clones contained spontaneous deletions of viral sequences and reconstituted mutant viruses with impaired growth kinetics in vitro. The deletions in one BAC were repaired using E. coli genetics. The resulting repaired BAC reconstituted a virus with in vitro replication kinetics identical to the wild type parental virus; moreover, its genome was indistinguishable from that of the wild type parental virus by restriction pattern analysis using multiple restriction enzymes. These results suggest that the repaired BAC is an authentic representation of the complete GPCMV genome. It should provide a valuable tool for evaluating the impact of genetic modifications on the safety and efficacy of live attenuated vaccines and for identifying genes important for congenital transmission and fetal disease.
Keywords: GPCMV, cytomegalovirus, congenital infection, BAC
1. Introduction
Human cytomegalovirus (HCMV) is a herpesvirus in the beta-herpesvirinae subfamily. HCMV is ubiquitous and generally non-pathogenic in the general population but is frequently pathogenic in the immunocompromised host. In adults, HCMV causes disease primarily in AIDS and transplant patients. The developing fetus, however, is also immune compromised, and unfortunately, HCMV can be transmitted transplacentally from mother to fetus. Indeed, congenital HCMV infections comprise the major infectious cause of birth defects in the United States, resulting in an estimated 8000 seriously affected newborns each year and perhaps as many as 8000 additional cases of late-onset hearing loss in children born with HCMV infections but asymptomatic at birth (Fowler and Boppana, 2006; Fowler et al., 1992).
Like other herpesviruses, HCMV has a large (235-kb) linear double-stranded DNA genome that encodes for a large number of genes (Chee et al., 1990). While the functions of many of these genes are known, the majority remain poorly characterized. Moreover, because HCMV does not replicate in any species other than humans, the importance of specific viral genes in congenital transmission and fetal pathogenesis cannot be investigated by targeted mutagenesis. While related cytomegaloviruses of rat and mouse provide important models for viral latency, immune control, and pathogenesis, these CMVs have only limited capacity to cross the placenta and infect the fetus, and have had limited usefulness as models of congenital infection (Loh et al., 2006; Woolf et al., 2007).
The guinea pig is unique as the only small animal model for congenital cytomegalovirus infection. Guinea pig cytomegalovirus (GPCMV) shares many biological similarities to HCMV and in experimental settings can cross the placenta to cause fetal infection and disease (Griffith et al., 1985; Kumar and Nankervis, 1978; Schleiss, 2002). The guinea pig model therefore provides an important tool for testing vaccines or other intervention strategies aimed at preventing congenital cytomegalovirus infection or for elucidating the roles of specific viral factors in congenital transmission and pathogenesis (Bravo et al., 2006; Chatterjee et al., 2001; Schleiss et al., 2006a; Schleiss et al., 2005; Schleiss et al., 2004; Schleiss et al., 2007; Schleiss et al., 2006b).
Bacterial artificial chromosome (BAC) clones of herpesvirus genomes have proven extremely valuable for mutagenesis, as mutations at even the single nucleotide level can be constructed, characterized, and clonally isolated in E. coli using powerful E. coli genetic tools without regard for the impact that such mutations might have on virus viability. Once constructed, mutant viral genomes in the form of BAC DNA can be transfected into appropriate permissive mammalian cells. BACs containing wild type sequences or non-lethal mutations reconstitute viruses that spread within the transfected cell cultures. In some instances complementing cell lines can be used to reconstitute viruses from BACs containing lethal mutations. BAC-based mutagenesis of both human and animal cytomegaloviruses has provided a powerful tool for elucidation of viral gene functions both in vitro and in vivo (Borst et al., 1999; Chang and Barry, 2003; Cicin-Sain et al., 2003; Hahn et al., 2003; McGregor and Schleiss, 2001; Rue et al., 2004; Yu et al., 2003; Yu et al., 2002).
McGregor and Schleiss (2001) reported previously on the construction of an infectious BAC clone of the GPCMV genome. However, in this first generation BAC the 9-kb BAC ori cassette (E. coli BAC origin of replication and marker genes) was not engineered to be excised. Hence, GPCMV derived from this BAC was not authentic, as it contained a large insertion of non-viral sequence, and was potentially unstable. In the present study, new generation GPCMV BAC was constructed in which the BAC vector sequences are flanked by LoxP sites such that co-transfection of BAC DNA with plasmid DNA encoding Cre recombinase results in excision of the BAC ori sequences, leaving only a single 34-bp LoxP site in the viral genome. All initial BAC clones contained substantial deletions on one or both sides of the BAC vector insertion and mutant viruses reconstituted from these clones replicated with impaired kinetics relative to the wild type parental virus. The deletions in one BAC clone were then repaired using several allelic exchange reactions in E. coli. The repaired BAC was shown to reconstitute a virus that lacks detectable deletions relative to the wild type parental virus, and importantly, replicates in vitro with fully wild type kinetics. This new GPCMV BAC clone should provide an important tool for genetic analysis of GPCMV. It will be useful for evaluating the impact on safety and efficacy of rationally designed genetic modifications to live attenuated vaccines and may facilitate identification and analysis of viral genes that are important for congenital transmission and fetal disease.
2. Methods
2.1 Virus and cell culture
GPCMV strain 22122 (ATTC VR682) and BAC-derived viruses were propagated in guinea pig lung fibroblast (GLF) cells (ATCC CCL 158) in minimum essential medium (Gibco-BRL) supplemented with 10% fetal calf serum (FCS, HyClone Laboratories), 10,000 IU/L penicillin, 10 mg/L streptomycin (Gibco-BRL).
2.2 Plasmid Construction
2.2.1 Construction of BAC ori recombination vector pACYC-NLKGG
The BAC cassette pKGG (McGregor and Schleiss, 2001) contains BAC ori sequences from pKSO, a screenable marker expressing enhanced green fluorescent protein (EGFP), a selectable marker encoding E. coli xanthine-guanine phophoribosyltransferase (GPT) (Greaves et al., 1995), and a unique PacI restriction site. The 4.6-kb HindIII N fragment of the GPCMV genome was previously cloned in pBluescript SK(+) to create plasmid pKTS398 (McGregor and Schleiss, 2001) (Fig. 1). Four synthetic oligonucleotides (Invitrogen) were used to create a BglII-LoxP-PacI-LoxP-BgII construct: LoxP1f (5′-AGATCTATAACTTCGTATAGCATACATTATACGAAGTTATTAATT), LoxP1r (5′-pAATAACTTCGTATAATGTATGCTATACGAAGTTATAGATCTA), LoxP2f (5′pAATAACTTCGTATAGCATACATTATACGAAGTTATAGATCTA), and LoxP2r (5′AGATCTATAACTTCGTATAATGTATGCTATACGAAGTTATTAATT). LoxP1f and LoxP1r were annealed to form double-stranded oligonucleotide LoxP1, while LoxP2f and LoxP2r were annealed to form double-stranded oligonucleotide LoxP2. The PacI cohesive ends of LoxP1 and LoxP2 were annealed and ligated to generate a LoxP1–2 fragment that was T/A cloned into pCR®XL TOPO TA (Invitrogen) to create plasmid pCRXL-LPL. The BglII-LoxP-PacI-LoxP-BglII cassette was released from pCRXL-LPL by BglII digestion and ligated into the unique BglII site in pKTS398 to generate clone pKTS398-LPL (Fig. 1). To minimize the potential for instability from dual origins upon subsequent insertion of BAC ori the modified HindIII N insert was excised from pKTS398-LPL by HindIII digestion and ligated into HindIII-digested pACYC177 to create pACYC-NLPL (Fig. 1). Finally, pKGG was linearized by PacI digestion and inserted into the unique PacI site of pACYC-NLPL to generate pACYC-NLKGG (Fig. 1). Orientation of the BAC ori insertion relative to HindIII N was determined by sequencing using primers N3LF (5′ CGTGTCGTATCGTAAATCATC 3′) and N3LR (5′ CACCGACAATTCTAGTCTGGTC 3′).
Fig. 1. Construction of the BAC ori recombination vector.

A HindIII map of the GPCMV genome (top) shows the locations of relevant HindIII restriction fragments. The HindIII N fragment cloned in pKTS398 is expanded below and shows the BglII site at which the BglII-LoxP-PacI-LoxP-BglII fragment from pCRXL-LPL was inserted. This modified HindIII N fragment was then transferred into the HindIII site of pACYC177 to make pACYC-NLPL. The final step shows insertion of BAC ori and marker genes as PacI-linearized pKGG into the unique PacI site of pACYC-NLPL to make the recombination vector pACYC-NLKGG.
2.2.2 Construction of allelic exchange vectors
The allelic exchange vector pGS284 was a gift from Greg Smith (Smith and Enquist, 1999) and was modified by SphI digestion and religation to remove a unique NotI site, resulting in plasmid pGS284ΔNotI. Virion DNA from GPCMV strain 22122 was restricted with SacI and a 14.7-kb fragment was purified by agarose gel electrophoresis, extracted from the agarose using the QIAquick Gel Extraction Kit (Qiagen), and ligated into the unique SacI site of pGS284ΔNotI to create plasmid p15k-ND-pGS284. A marker gene cassette encoding kanamycin-resistance and lacZα (kanr/lacZ) was excised from pYD-Tn1721 (Yu et al., 2002) (kindly provided by Dong Yu) as a 1.8-kb NotI fragment and ligated into a unique NotI site within the 14.7-kb SacI fragment in p15k-ND-pGS284 to create plasmid p15k-ND-kanr/lacZ-pGS284. A 4.0-kb NsiI fragment was similarly gel purified from NsiI-digested GPCMV strain 22122 virion DNA and ligated into the NsiI site of pGS284 to create plasmid p4k-RN-pGS248.
2.3 DNA preparation
Virion DNA was prepared from culture supernatants of virus-infected cells when the cells reached full cytopathic effect (CPE). Virions were pelleted from supernatants at 25,000 rpm for 30 min and viral DNA was isolated as previously described (McVoy et al., 1997). Midi-prep BAC DNAs were prepared from 100 ml overnight E. coli cultures grown in LB with 20 μg/ml chloramphenicol using Nucleobond BAC DNA purification kits (ClonTech) and redisolved in 100 μl H2O. Mini-prep BAC DNAs were prepared from 5 ml of overnight E. coli cultures using alkaline lysis Solutions I, II and III from a mini-prep kit (Qiagen); however, the matrix purification step was omitted and BAC DNAs were instead precipitated directly from lysate supernatants with ethanol and redisolved in 10 μl H2O.
2.4 Recombinant virus construction
Plasmid pACYC177NLKGG was digested with HindIII and the 13.4-kb fragment containing HindIII N with the BAC ori insertion was purified by agarose electrophoresis and extracted from the agarose using the QIAquick Gel Extraction Kit (Qiagen). One μg of this DNA was mixed with one μg of GPCMV strain 22122 virion DNA and transfected into subconfluent GLF cells in 100-mm dishes using 60 μl Effectene Transfection Reagent (Qiagen). After the appearance of EGFP+ foci (10–20 days), 200 μM mycophenolic acid (Gibco/BRL) and 25 μM xanthine (Sigma) were applied to the medium to enrich for GFP+ recombinant viruses. Incubation was continued another 5–7 days until extensive CPE was observed. The culture supernatant was then transferred to a 75 cm2 flask of confluent GLF cells and again incubated with medium containing 200 μM mycophenolic acid and 25 μM xanthine for 5–7 days until extensive CPE was observed. A significant degree of enrichment was confirmed by a high percentage of CPE+/EGFP+ cells.
2.5 Isolation and initial screening of BAC clones
Supernatants from the enriched cultures were used to infect a 75 cm2 flask of confluent GLFs at an MOI of 1–5. Infected-cell DNA was isolated 2 days post infection (dpi) by the method of Hirt (Yu et al., 2002) and transformed into competent GeneHog strain E. coli (Invitrogen) by electroporation at 1.8 kv in a 0.1 cm cuvette using a Gene Pulser electroporator (Bio-Rad). Transformed bacteria were selected by plating on LB containing 20 μg/ml chloramphenicol. BAC clones were screened by restriction pattern analysis of midi-prep DNAs using EcoRI, HindIII, XbaI, and SacI.
2.6 PCR screening of BAC clones
BACs were screened for the presence of sequences within the 2.3-kb EcoRI fragment using primers F10207 (5′-GTCACGATACACCATCAGACACGGG) and R10722 (5′-CGAGAGGAGGAGGAGAGGAAGAAAG) or for the presence of sequences within HindIII R using primers H3R1 (5′-CTGTGTTTCGGGTGAGGTGAAG) and H3R2 (5′-TAGGGAGGCGTGTGTGTGCG). Ten μl of mini-prep DNA was amplified using Taq DNA polymerase (Takara) according to the manufacturer’s protocol. The cycler was programmed for 4 min at 94°C followed by 35 cycles of 1 min at 94°C, 1 min at 60°C, and 1 min at 72°C. PCR products were analyzed by electrophoretic separation on 1% agarose gels and visualization with ethidium bromide staining and UV light.
2.7 Sequencing
Sanger dideoxy sequencing was performed by the Nucleic Acids Research Facilities of Virginia Commonwealth University using either midi-prep BAC or virion DNA templates. Deletions were identified relative to the complete sequence of the GPCMV genome (Schleiss et al., manuscript in preparation).
2.8 Allelic exchange
Allelic exchange was conducted essentially as described previously (Hahn et al., 2003). Briefly, the deletion in HindIII D was repaired by transforming BAC pN13 into E. coli strain GS500 (kindly provided by Greg Smith) (Smith and Enquist, 1999). The allelic exchange vector p15k-ND-kanr/lacZ-pGS284 was transferred into these cells by mating and cointegrates were selected on plates containing 20 μg/ml ampicillin, 10 μg/ml kanamycin, and 20 μg/ml chloramphenicol. Colonies containing BACs that had lost sacB by resolution were then negatively selected on plates containing 10 μg/ml kanamycin, 20 μg/ml chloramphenicol, 5% sucrose, 100 μg/ml X-gal, and 40 μg/ml IPTG. Blue colonies were characterized by restriction pattern analysis and DNA sequencing to confirm restoration of sequences in HindIII D. To remove the residual kanr/lacZ insertion from the resulting BAC, p15k-ND-pGS284 was introduced by mating and cointegrates were selected as before. Resolution products were selected using plates containing 20 μg/ml chloramphenicol, 5% sucrose, 100 μg/ml X-gal, and 40 μg/ml IPTG. White colonies were then screened for kanamycin-sensitivity and characterized by restriction pattern analysis to confirm removal of kanr/lacZ.
The deletion in HindIII R was repaired by transfer of p4k-NR-pGS284, selection for cointegrates (without kanamycin), and selection for resolution products as described above. Candidate clones were screened by PCR using primers H3R1 and H3R2 as described in 2.6. Positive clones were confirmed for restoration of HindIII R sequences by restriction pattern analysis.
2.9. Virus reconstitution from BAC DNA and isolation of EGFP-negative viruses
Midi-prep BAC DNA (0.2–0.4 μg) was co-transfected with 0.2 μg pCre plasmid DNA (constructed by Wolfram Brune and kindly provided by Gabriele Hahn) into subconfluent GLF cells in 6-well plates using 20 μl Effectene (Qiagen). Cells were incubated at 37°C for 10–14 days until extensive CPE was observed. Typically, BAC ori excision was ~50% efficient, giving rise to both EGFP+ and EGFP-negative CPE+ cells. BAC ori-excised (EGFP-negative) viruses were isolated by limiting-dilution in 96-well plates containing confluent GLF cells and selection of CPE+ wells that exhibited no detectable EGFP expression.
2.10 Viral growth curves
Frozen viral stocks were titrated carefully by limiting-dilution in 96-well plate cultures (see below). Confluent GLF cells in 75 cm2 flasks were infected with carefully matched viral inocula at an MOI of 3 for single-step and 0.01 for multi-step growth curves. The cultures were washed 3 h post infection to remove unattached virus. Samples of culture media were titrated daily for 5 days for single-step and every 3 or 4 days for 14 days for multi-step curves. Titrations were performed by preparing 10-fold serial dilutions (10−1 to 10−6) of clarified supernatants and using each dilution to inoculate 32 replicate wells (100 μl/well) in 96-well plates containing confluent GLFs. After incubation at 37°C for two weeks, wells were scored as CPE+ or CPE-. Titers were calculated by assigning x as the number of positive wells out of 32 from the greatest dilutions that contained positive wells. The Poisson distribution was then used to correct for the probability of multiple virus hits per well using the equation:
3. Results
3.1. Recombinant virus construction and characterization of initial BAC clones
To construct BAC clones of the GPCMV genome it was first necessary to derive recombinant GPCMVs in which a BAC ori cassette was inserted into the GPCMV genome by homologous recombination. A BglII site within the HindIII N region near the left end of the GPCMV genome was used for insertion of the BAC ori (Fig. 1) as this location had been successfully utilized for construction of the first generation GPCMV BAC (McGregor and Schleiss, 2001). However, in this case the BAC ori was to be flanked with LoxP sites such that Cre-mediated excision would delete all non-viral sequences with the exception of one residual 34-bp LoxP site. Hence, recombination vector pACYC-NLKGG was constructed in which BAC ori sequences with adjacent GPT and EGFP marker cassettes were flanked by LoxP sites and inserted into the BglII site within a plasmid clone of HindIII N (Fig. 1). This HindIII fragment was then co-transfected into GLF cells with GPCMV DNA extracted from virions of wild type parental reference strain 22122. The resulting virus stock was enriched for recombinant viruses by passage in the presence of mycophenolic acid and xanthine, which selects for GPT+ viruses (Greaves et al., 1995), while EGFP fluorescence served as a visual marker to monitor the extent of enrichment.
To derive BAC clones, DNA was extracted from cells infected with the enriched but still mixed virus population and transformed into E. coli GeneHog strain cells. Colonies containing BACs were selected on chloramphenicol plates. The authenticity of BAC clones was first assessed by restriction pattern analysis of BACs derived from 24 colonies selected at random. Eleven of the 24 BACs clearly lacked large regions of the viral genome and were not considered further. The remaining 13 clones had patterns very similar to the parental virus but several restriction fragments were notably different. Results for one representative BAC clone, pN13, are shown in Figure 2. Some of these differences were expected. For example, insertion of the BAC ori cassette was anticipated to increase the size of HindIII N from 4.6 kb to 13.6 kb. Moreover, because BACs are circular, restriction fragments from the ends of viral genomes were expected to be fused in BACs in the form of junction fragments that result from genome circularization (McVoy et al., 1997). Thus, all BACs were predicted to lack HindIII R (2.6 kb), EcoRI Y (2.6 kb), and XbaI N (5.0 kb), which derive from the left end of the genome, as well as HindIII O (4.0 kb) and HindIII M (5.0 kb), which derive from two alternate forms of the genome’s right end (Fig. 1) (Gao and Isom, 1984). Instead of terminal fragments BACs were expected to contain one of two possible junction fragments, HindIII OR (6.6 kb) or MR (7.6 kb) (McVoy et al., 1997).
Fig. 2. Restriction pattern analyses of BAC clones and viral DNAs.

Virion DNAs from wild type virus (v22122) or virus reconstituted from BAC pN13R10 (vN13R10-LoxP) were compared to DNAs from BACs pN13 and pN13R10 by restriction with the indicated enzymes, separation on agarose gels, and staining of DNA fragments with ethidium bromide. Numbers to the left of each gel indicate the sizes (in kb) of 1 kb ladder markers (M). Arrows indicate the positions and sizes (in kb) of relevant restriction fragments. Letters in parentheses indicate letter designations for restriction fragments that are referred to in the text.
As expected, the terminal fragments and HindIII N were absent from BAC clones (Fig. 2). However, several additional pattern anomalies remained that could not be explained. All but one BAC (pN2) contained neither a 6.6-kb OR nor 7.6-kb MR junction fragment but instead contained novel fragments of ~5.9 kb (for example, see pN13, Fig. 2A, lane 2). Moreover, all BACs were missing several restriction fragments that derive from interior regions of the viral genome: 1.4- and 2.3-kb EcoRI fragments (Fig. 2B, lane 2) and 8.6-, 7.6-, 5.2-, 4.0-, and 3.1-kb XbaI restriction fragments (Fig. 2C, lane 2). All pattern anomalies were ascribed ultimately to either (i) deletions within viral sequences causing the genuine loss of restriction fragments; or (ii) failure of XbaI to cleave at certain restriction sites in E. coli-derived BAC DNA due to methylation, causing an apparent (but erroneous) loss of restriction fragments. Anomalies of the latter type were resolved upon reconstitution of virus from BAC DNA, which removed the E. coli methylation (see section 3.3). In order to explain the remaining anomalies the deletions in several representative BACs were characterized in detail, as described below.
3.1.1 All BACs contain deletions in HindIII D
Five of the missing fragments (1.4- and 2.3-kb EcoRI, 3.1-, 4.0-, and 7.6-kb XbaI) were clustered together within HindIII D, which lies adjacent to and to the right of HindIII N (Fig. 3). This suggested that BACs may have deletions that span a common region. Several BAC clones (pN2, pN3, pN13) were characterized by additional restriction mapping (not shown) followed by direct sequencing to identify the deletion break points precisely. In each BAC substantial deletions were found within HindIII D. The leftward break points coincided precisely with the HindIII site defining the boundary between HindIII N and HindIII D, whereas the rightward break points differed significantly between BACs and resulted in deletions that varied in size from 7.9 kb in pN3 to 17.9 kb in pN2 (Fig 3). These deletions accounted for the absence of the 1.4- and 2.3-kb EcoRI fragments and the 3.1-, 4.0-, and 7.6-kb XbaI fragments from BAC restriction digests (Fig 3).
Fig. 3. Mapping and repair of deletions in BAC clones.
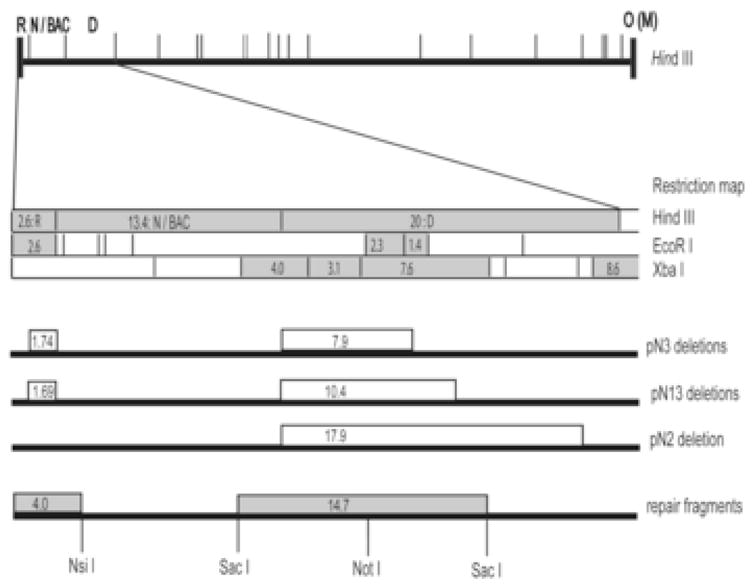
A HindIII map of the BAC ori-containing GPCMV genome (top) shows the locations of HindIII fragments near the left end of the genome. The region expanded below shows HindIII, EcoRI, and XbaI restriction maps the region spanning HindIII R-D. Restriction fragments that were missing or altered in BAC clones are highlighted in gray. Open boxes on lines below the restriction maps indicate regions deleted in BACs pN2, pN3, and pN13. Gray boxes on the bottom line indicate the positions of NsiI and SacI fragments that were used to repair the two deletions in pN13 as well as the location of the NotI site that was used for insertion of a kanr/lacZ marker cassette to facilitate allelic exchange. Numbers inside boxes indicate restriction fragment sizes in kb.
3.1.2 Some BACs contain a second deletion in HindIII R
The absence of an OR or MR junction fragment from pN3 and pN13 suggested that these BACs contain additional mutations in regions that comprise the ends of the viral genome. To determine which ends were affected BAC DNAs were used to reconstitute infectious viruses. Linear virion DNAs were prepared from BAC-derived viruses and from the wild type parental virus and their restriction patterns were compared. The BAC-derived viral DNAs exhibited normal terminal fragments from the right end of the genome (HindIII O, 4.0 kb; and HindIII M, 5.0 kb); however, only the virus derived from BAC pN2 exhibited a normal 2.6-kb HindIII R fragment from the left-end of the genome (not shown). Viruses derived from both pN3 and pN13 lacked the 2.6-kb R fragment and instead contained a novel ~1-kb fragment (not shown), suggesting that these clones have additional deletions within HindIII R. Direct sequencing of this region confirmed that both pN3 and pN13 have similar but not identical deletions. The rightward break points coincided precisely with the HindIII site that defines the boundary between HindIII R and HindIII N, while the leftward break points were slightly different and resulted in a 1.69-kb deletion in pN13 and a 1.74-kb deletion in pN3 (Fig. 3). These results explain the lack of HindIII OR (6.6 kb) or MR (7.6 kb) junctions from pN3 and pN13 BAC DNAs, as the ~1.7-kb deletions in HindIII R would reduce the size of the junctions by an equal amount, thus shifting a 7.6-kb MR fragment down to ~5.9 kb, as was observed in pN13 (Fig. 2A, lane 2) and pN3 (not shown).
3.1.3 Screening a large number of colonies failed to identify a BAC that lacks deletions
The above findings suggested that insertion of the 9-kb BAC ori cassette into the viral genome was poorly tolerated and resulted in large spontaneous deletions of viral sequences from regions flanking the restriction fragment that was used for recombination. Of the 13 BAC clones that were characterized, all contained deletions in HindIII D that removed the 2.3-kb EcoRI fragment. In hopes of finding a rare BAC clone that lacked deletions a PCR assay was devised to detect sequences specifically within the 2.3-kb EcoRI fragment and used to screen 59 additional BAC-containing colonies. Although one positive clone was identified it was not infectious and restriction analysis revealed that it lacked large regions from elsewhere in the viral genome (not shown).
3.2 Repair of BAC pN13 to derive pN13R10, a fully authentic BAC clone of the GPCMV genome
As the search for a complete BAC clone was not fruitful, an alternative solution was undertaken to repair the deletions in one of the BAC clones characterized above. While pN2 had only a single deletion, restoration of so large a missing sequence (17.9 kb) posed a significant challenge. Instead it was decided to repair the two smaller deletions in clone pN13. The 10.4-kb deletion in HindIII D was repaired in a two step process. First, a 14.7-kb SacI fragment that spans the deletion in HindIII D (Fig. 3) was derived from parental virus virion DNA and cloned into the allelic exchange vector pGS284. This fragment contained a unique and centrally located NotI site into which we inserted an E. coli marker cassette encoding kanr/lacZ. Following allelic exchange between this vector and BAC pN13, screening for blue colonies on kanamycin/X-gal plates allowed us to obtain a BAC clone in which the missing sequences were restored but were disrupted by the kanr/lacZ marker cassette. Removal of the cassette was then achieved by a second allelic exchange reaction using the pGS284 vector that contained only the wild type 14.7-kb SacI fragment and screening for white colonies and loss of kanamycin-resistance. Direct sequencing of the resulting BAC verified the authenticity of sequences flanking both ends of the inserted 14.7-kb SacI fragment, and restriction pattern analysis confirmed that the 1.4- and 2.3-kb EcoRI and 7.6-kb XbaI fragments had been restored (not shown).
To repair the smaller deletion a 4.0-kb NsiI fragment from parental virus virion DNA that spans the HindIII R deletion (Fig. 3) was cloned into pGS284. As there was no convenient means of inserting the marker cassette, allelic exchange was performed without markers and three candidate clones were identified by screening 103 colonies with a PCR reaction designed to detect the HindIII R sequences that are missing from pN13. The three BACs were characterized further by restriction pattern analysis and one candidate clone designated pN13R10 was selected for further evaluation.
3.3 Restriction analysis of BAC pN13R10 and pN13R10-derived virus
BAC pN13R10 was co-transfected into GLF cells with pCre. From the resulting virus mix an EGFP-negative virus (lacking the BAC ori) was isolated by limiting-dilution and designated vN13R10-LoxP. The restriction patterns of pN13 and pN13R10 BAC DNAs and virion DNAs derived from vN13R10-LoxP and the parental virus strain 22122 were compared using different restriction enzyme digestions: HindIII, EcoRI, XbaI, and SacI/SphI. All of the fragments that were absent from pN13 due to the two deletions were restored in pN13R10. For example, the 5.9-kb HindIII junction fragment in pN13 (Fig. 2A, lane 2) was replaced in pN13R10 by the correctly-sized 7.6-kb HindIII MR fragment (Fig. 2A, lane 3), and within HindIII D, the 2.3- and 1.4-kb EcoRI fragments (Fig. 2B, lane 3), the 7.6-kb XbaI fragment (Fig. 2C, lane 3), and the 14.7-kb SacI fragment (Fig. 2D, lane 3) were also restored.
When linear vN13R10-LoxP virion DNA was examined, HindIII N was restored to its correct 4.6-kb size, indicating proper excision of BAC ori (Fig. 2A). This was confirmed further by direct sequencing, which revealed precise excision of BAC ori leaving a single 34-bp LoxP site within HindIII N. Virion DNA from vN13R10-LoxP also exhibited all of the expected terminal fragments (HindIII R, M, and O, EcoRI Y, and XbaI N) and lacked the 7.6-kb HindIII MR junction observed in circular pN13R10 BAC DNA (Fig. 2A–C). Finally, vN13R10-LoxP virion DNA contained the 8.6-, 5.2-, 4.0-, and 3.1-kb XbaI fragments that were not detected in pN13R10 BAC DNA (Fig. 2C). These XbaI fragments were not detected in E. coli-derived BAC DNA because XbaI cleavage of certain restriction sites was blocked by methylation. This gave rise to larger fragments that were fused with their neighbors. In the case of the 8.6- and 5.2-kb XbaI fragments these fusions were too large to discern; however, fusion of the 4.0- and 3.1-kb XbaI fragments was apparent as a 7.1-kb fragment in pN13R10 DNA that was not present in vN13R10-LoxP DNA (Fig. 2C).
Importantly, no restriction fragment differences were observed between virion DNAs from the parental virus and vN13R10-LoxP using the four different restriction digests (Fig. 2). The SacI/SphI double-digestions were particularly revealing as this combination of enzymes produced a broad range of well-separated fragments. These results indicate that within this level of analysis, BAC pN13R10 represents a complete and authentic clone of the GPCMV genome.
3.4 In vitro growth properties of BAC-derived viruses
An important test of the authenticity of any BAC-derived virus is its replication kinetics in vitro. BAC-derived viruses were therefore compared to the parental strain using both single step (high MOI) and multi-step (low MOI) growth curves. Viruses derived from BACs that contained deletions (pN2, pN3, and pN13) all exhibited similar growth delays and approximately 10-fold reductions in peak titers. A representative growth curve for pN13-derived virus vN13-LoxP is shown in Fig. 4A. Growth curves for pN2- and pN3-derived viruses are not shown but were very similar. In contrast, vN13R10-LoxP replicated with kinetics and titers essentially identical to the parental strain 22122 under both single and multi-step conditions (Fig. 4B and 4C).
Fig. 4. Growth curves of wild type and BAC-derived viruses.
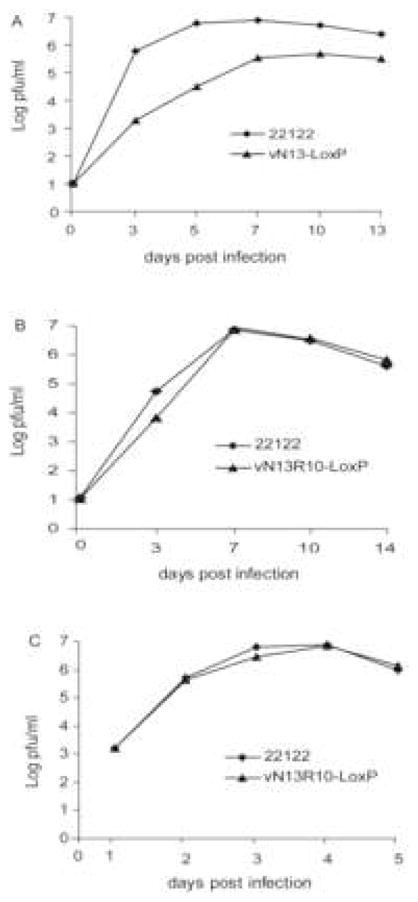
GPL cells were infected with the indicated viruses at MOIs of 0.01 (A and B) or 3 (C). Viral titers in the culture supernatants were determined on the days post infection indicated.
4. Discussion
BAC clones of herpesvirus genomes provide powerful tools for mutagenesis of viral sequences as genetic changes can be created irrespective of their impact on virus replication. In a previous report McGregor and Schleiss described construction of a BAC clone of the GPCMV genome (McGregor and Schleiss, 2001). This clone, however, lacked a means to excise the BAC ori and hence was unable to reconstitute a fully authentic virus. A second generation GPCMV BAC was constructed in which the BAC ori can be excised using Cre recombinase. In theory, the reconstituted virus should be identical to the parental virus with the exception of a single residual 34-bp LoxP site.
The BAC clones that were obtained initially all contained significant deletions and gave rise to mutant viruses that replicated with delayed kinetics and reduced efficiencies relative to the parental strain. These deletions must have arisen during the process of recombination between the plasmid DNA fragment containing the BAC ori and the parental viral DNA, as the deletional break points coincided with the HindIII sites at each end of the plasmid DNA fragment. Subsequent selection against propagation of viruses with over length genomes (i.e., those containing the BAC ori but lacking compensatory deletions) probably occurred at the time of viral DNA packaging, as this process presumably imposes a limit to the length of DNA that can be packaged efficiently into viral capsids. The selection must have been quite stringent as it was not possible to identify BAC clones lacking deletions despite screening a substantial number of colonies. Moreover, retrospective analysis of the first generation GPCMV BAC (McGregor and Schleiss, 2001) revealed that it too had a deletion to the right of the BAC ori that had escaped detection previously (not shown).
Through a series of three allelic exchange reactions in E. coli plasmid-cloned sequences derived from parental virus DNA were used to repair two deletions in one of these initial BACs (pN13). The repaired BAC, pN13R10, reconstitutes a virus that by restriction pattern and limited sequence analyses appears to be complete and authentic. In vitro, virus derived from pN13R10 replicates with kinetics and efficiencies identical to the wild type parental virus. How virulent the BAC-derived vN13R10-LoxP virus is in vivo is currently under investigation. Preliminary studies suggest that it can disseminate, cause viremia, and transmit to the fetus. However, as with other experimental models of pathogenesis, the virulence of GPCMV stocks can vary significantly. Highly virulent stocks are obtained by serial transfer of salivary gland extracts from one animal to the next (SG virus) (Hartley et al., 1957), whereas passage of virus in tissue culture (TC virus) results in attenuation such that higher doses of inocula are needed to achieve equivalent pathogenic effects (Schleiss and Lacayo, 2005). BAC pN13R10 was derived from a TC virus stock. Thus, vN13R10-LoxP is likely to resemble TC virus in vivo. However, this may not preclude the use of pN13R10 for in vivo genetic studies as, under appropriate experimental conditions, TC virus can disseminate, transmit in utero, and cause fetal damage (Kumar and Nankervis, 1978; Schleiss et al., 2006a). Moreover, as an attenuated virus, N13R10 should serve to model the live attenuated approach to vaccination. The ability to manipulate its genome through E. coli genetics will facilitate studies to evaluate the impact of rationally designed genetic modifications on vaccine safety and efficacy.
An attempt was made to derive BAC clones from an SG virus stock of GPCMV using essentially the same approach described here. Surprisingly, despite what appeared to be EGFP+ viruses in the cultures used to prepare DNA for E. coli transformation, all BAC clones that were recovered were non-infectious and the majority lacked large regions of the viral genome (Cui and McVoy, unpublished data). Why SG virus behaved so differently from TC virus in this respect is unknown. Perhaps SG virus contains sequences that are toxic to E. coli or is unable to undergo the spontaneous deletions that compensated for the BAC ori insertion in TC virus recombinants. Nevertheless, identification of the genetic determinants that render SG virus more virulent than TC virus remains an important area for future investigations. Indeed, it may be possible to identify sequences unique to SG virus and then modify pN13R10 to match SG in order to derive a BAC clone that reconstitutes a fully pathogenic virus. A thus-modified pN13R10 would provide an important tool for evaluating the importance of specific viral genes or subtle genetic modifications thereof for congenital infection and fetal disease.
Acknowledgments
The authors are grateful to Gabriele Hahn and Wolfram Brune for providing pCre, Dong Yu for providing pYD-Tn1721 and very helpful advice on BAC cloning, and Greg Smith for providing pGS284, E. coli strain GS500, and very helpful advice on allelic exchange. This work was supported by a grant R01HD044864 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Borst EM, Hahn G, Koszinowski UH, Messerle M. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in escherichia coli: a new approach for construction of HCMV mutants. J Virol. 1999;73(10):8320–9. doi: 10.1128/jvi.73.10.8320-8329.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo FJ, Cardin RD, Bernstein DI. Effect of maternal treatment with cyclic HPMPC in the guinea pig model of congenital cytomegalovirus infection. J Infect Dis. 2006;193(4):591–7. doi: 10.1086/499603. [DOI] [PubMed] [Google Scholar]
- Chang WL, Barry PA. Cloning of the full-length rhesus cytomegalovirus genome as an infectious and self-excisable bacterial artificial chromosome for analysis of viral pathogenesis. J Virol. 2003;77(9):5073–83. doi: 10.1128/JVI.77.9.5073-5083.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Harrison CJ, Britt WJ, Bewtra C. Modification of maternal and congenital cytomegalovirus infection by anti-glycoprotein b antibody transfer in guinea pigs. J Infect Dis. 2001;183(11):1547–53. doi: 10.1086/320714. [DOI] [PubMed] [Google Scholar]
- Chee MS, Bankier AT, Beck S, Sohni R, Brown CM, Cerny R, Horsnell T, Hutchinson CA, Kouzarides T, Marignetti JA, Preddie E, Satchwell SC, Tomlinson P, Weston K, Barrell BG. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. In: McDougall JK, editor. Cytomegaloviruses. Current topics in microbiology and immunology. Vol. 154. Springer-Verlag; New York, N.Y: 1990. pp. 125–169. [DOI] [PubMed] [Google Scholar]
- Cicin-Sain L, Brune W, Bubic I, Jonjic S, Koszinowski UH. Vaccination of mice with bacteria carrying a cloned herpesvirus genome reconstituted in vivo. J Virol. 2003;77(15):8249–55. doi: 10.1128/JVI.77.15.8249-8255.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler KB, Boppana SB. Congenital cytomegalovirus (CMV) infection and hearing deficit. J Clin Virol. 2006;35(2):226–31. doi: 10.1016/j.jcv.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Fowler KB, Stagno S, Pass RF, Britt WJ, Boll TJ, Alford CA. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med. 1992;326(10):663–7. doi: 10.1056/NEJM199203053261003. [DOI] [PubMed] [Google Scholar]
- Gao M, Isom HC. Characterization of the guinea pig cytomegalovirus genome by molecular cloning and physical mapping. J Virol. 1984;52(2):436–47. doi: 10.1128/jvi.52.2.436-447.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves RF, Brown JM, Vieira J, Mocarski ES. Selectable insertion and deletion mutagenesis of the human cytomegalovirus genome using the Escherichia coli guanosine phosphoribosyl transferase (gpt) gene. J Gen Virol. 1995;76(Pt 9):2151–60. doi: 10.1099/0022-1317-76-9-2151. [DOI] [PubMed] [Google Scholar]
- Griffith BP, McCormick SR, Fong CK, Lavallee JT, Lucia HL, Goff E. The placenta as a site of cytomegalovirus infection in guinea pigs. J Virol. 1985;55(2):402–9. doi: 10.1128/jvi.55.2.402-409.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn G, Jarosch M, Wang JB, Berbes C, McVoy MA. Tn7-mediated introduction of DNA sequences into bacmid-cloned cytomegalovirus genomes for rapid recombinant virus construction. J Virol Methods. 2003;107(2):185–94. doi: 10.1016/s0166-0934(02)00232-x. [DOI] [PubMed] [Google Scholar]
- Hartley JW, Rowe WP, Huebner RJ. Serial propagation of the guinea pig salivary gland virus in tissue culture. Proc Soc Exp Biol Med. 1957;96(2):281–5. doi: 10.3181/00379727-96-23455. [DOI] [PubMed] [Google Scholar]
- Kumar ML, Nankervis GA. Experimental congenital infection with cytomegalovirus: a guinea pig model. J Infect Dis. 1978;138(5):650–4. doi: 10.1093/infdis/138.5.650. [DOI] [PubMed] [Google Scholar]
- Loh HS, Mohd-Lila MA, Abdul-Rahman SO, Kiew LJ. Pathogenesis and vertical transmission of a transplacental rat cytomegalovirus. Virol J. 2006;3:42. doi: 10.1186/1743-422X-3-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor A, Schleiss MR. Molecular cloning of the guinea pig cytomegalovirus (GPCMV) genome as an infectious bacterial artificial chromosome (BAC) in Escherichia coli. Mol Genet Metab. 2001;72(1):15–26. doi: 10.1006/mgme.2000.3102. [DOI] [PubMed] [Google Scholar]
- McVoy MA, Nixon DE, Adler SP. Circularization and cleavage of guinea pig cytomegalovirus genomes. J Virol. 1997;71(6):4209–17. doi: 10.1128/jvi.71.6.4209-4217.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rue CA, Jarvis MA, Knoche AJ, Meyers HL, DeFilippis VR, Hansen SG, Wagner M, Fruh K, Anders DG, Wong SW, Barry PA, Nelson JA. A cyclooxygenase-2 homologue encoded by rhesus cytomegalovirus is a determinant for endothelial cell tropism. J Virol. 2004;78(22):12529–36. doi: 10.1128/JVI.78.22.12529-12536.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleiss MR. Animal models of congenital cytomegalovirus infection: an overview of progress in the characterization of guinea pig cytomegalovirus (GPCMV) J Clin Virol. 2002;25(Suppl 2):S37–49. doi: 10.1016/s1386-6532(02)00100-2. [DOI] [PubMed] [Google Scholar]
- Schleiss MR, Anderson JL, McGregor A. Cyclic cidofovir (cHPMPC) prevents congenital cytomegalovirus infection in a guinea pig model. Virol J. 2006a;3:9. doi: 10.1186/1743-422X-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleiss MR, Bernstein DI, McVoy MA, Stroup G, Bravo F, Creasy B, McGregor A, Henninger K, Hallenberger S. The non-nucleoside antiviral, BAY 38–4766, protects against cytomegalovirus (CMV) disease and mortality in immunocompromised guinea pigs. Antiviral Res. 2005;65(1):35–43. doi: 10.1016/j.antiviral.2004.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleiss MR, Bourne N, Stroup G, Bravo FJ, Jensen NJ, Bernstein DI. Protection against congenital cytomegalovirus infection and disease in guinea pigs, conferred by a purified recombinant glycoprotein B vaccine. J Infect Dis. 2004;189(8):1374–81. doi: 10.1086/382751. [DOI] [PubMed] [Google Scholar]
- Schleiss MR, Lacayo J. The guinea pig model of congenital cytomegalovirus infection. In: Reddehase MJ, editor. Cytomegaloviruses: Pathogenesis, Molecular Biology and Infection Control. Horizon Scientific Press Ltd; Wymondham, Norfolk, UK: 2005. [Google Scholar]
- Schleiss MR, Lacayo JC, Belkaid Y, McGregor A, Stroup G, Rayner J, Alterson K, Chulay JD, Smith JF. Preconceptual administration of an alphavirus replicon UL83 (pp65 homolog) vaccine induces humoral and cellular immunity and improves pregnancy outcome in the guinea pig model of congenital cytomegalovirus infection. J Infect Dis. 2007;195(6):789–98. doi: 10.1086/511982. [DOI] [PubMed] [Google Scholar]
- Schleiss MR, Stroup G, Pogorzelski K, McGregor A. Protection against congenital cytomegalovirus (CMV) disease, conferred by a replication-disabled, bacterial artificial chromosome (BAC)-based DNA vaccine. Vaccine. 2006b;24(37–39):6175–86. doi: 10.1016/j.vaccine.2006.06.077. [DOI] [PubMed] [Google Scholar]
- Smith GA, Enquist LW. Construction and transposon mutagenesis in Escherichia coli of a full- length infectious clone of pseudorabies virus, an alphaherpesvirus. J Virol. 1999;73(8):6405–14. doi: 10.1128/jvi.73.8.6405-6414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf NK, Jaquish DV, Koehrn FJ. Transplacental murine cytomegalovirus infection in the brain of SCID mice. Virol J. 2007;4:26. doi: 10.1186/1743-422X-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Silva MC, Shenk T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A. 2003;100(21):12396–401. doi: 10.1073/pnas.1635160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Smith GA, Enquist LW, Shenk T. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol. 2002;76(5):2316–28. doi: 10.1128/jvi.76.5.2316-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]