

Abstract
The structure of the catalytically inactive mutant (C215S) of the human protein-tyrosine phosphatase 1B (PTP1B) has been solved to high resolution in two complexes. In the first, crystals were grown in the presence of bis-(para-phosphophenyl) methane (BPPM), a synthetic high-affinity low-molecular weight nonpeptidic substrate (Km = 16 μM), and the structure was refined to an R-factor of 18.2% at 1.9 Å resolution. In the second, crystals were grown in a saturating concentration of phosphotyrosine (pTyr), and the structure was refined to an R-factor of 18.1% at 1.85 Å. Difference Fourier maps showed that BPPM binds PTP1B in two mutually exclusive modes, one in which it occupies the canonical pTyr-binding site (the active site), and another in which a phosphophenyl moiety interacts with a set of residues not previously observed to bind aryl phosphates. The identification of a second pTyr molecule at the same site in the PTP1B/C215S–pTyr complex confirms that these residues constitute a low-affinity noncatalytic aryl phosphate-binding site. Identification of a second aryl phosphate binding site adjacent to the active site provides a paradigm for the design of tight-binding, highly specific PTP1B inhibitors that can span both the active site and the adjacent noncatalytic site. This design can be achieved by tethering together two small ligands that are individually targeted to the active site and the proximal noncatalytic site.
Protein-tyrosine phosphatases (PTPases), working in concert with protein-tyrosine kinases, regulate a vast array of cellular events, including passage through the cell cycle, proliferation and differentiation, metabolism, cytoskeletal organization, neuronal development, and the immune response (1, 2). PTPases constitute a large family of enzymes that parallel protein-tyrosine kinases in their structural diversity and complexity. A recent estimate suggests that as many as 500 PTPase genes may be encoded within the human genome (2). In vivo, PTPases catalyze the removal of the phosphoryl group from phosphotyrosine (pTyr) residue(s) in protein substrates. However, the Km value for free pTyr is three to four orders of magnitude higher than the best protein/peptide substrates (3, 4). Several groups have demonstrated that PTPases display a range of kcat/Km values for pTyr-containing peptides. This sequence specificity has been exploited in the design of potent and selective PTPase inhibitors by incorporating a nonhydrolyzable pTyr analog into optimal peptide templates (5). However, owing to proteolytic susceptibility and weak partitioning across the plasma membrane, peptide-based compounds are not highly desirable for the development of medicinally effective drugs.
As an initial step toward the development of selective, low-molecular weight nonpeptidic PTPase inhibitors, we investigated the active site substrate specificity of PTP1, the rat structural homologue of human protein-tyrosine phosphatase 1B (PTP1B). PTP1B (3) is the prototypical intracellular PTPase and is found in a wide variety of human tissues (6). PTP1B is overexpressed in human breast cancer (7) and has been implicated as a negative regulator of insulin-stimulated pathways (8). The catalytic domain of PTP1 consists of residues 1–321 (9) and is 97% identical to the corresponding 321 residues of the human PTP1B. The kinetic properties of the two enzymes are indistinguishable. Although nonpeptidic species tend to be much poorer substrates than peptide-based compounds (9, 10), we have recently shown that appropriately functionalized aromatic phosphates exhibit Km values in the low μM range and are hydrolyzed by PTP1 as efficiently as the best peptide substrates reported for this enzyme (11). Furthermore, the Km values for several of these substrates correlate well with their affinity for PTP1.
To understand the structural basis of substrate recognition by PTP1B, and to lay the groundwork for future design of specific compounds that can interfere with PTP1B-related processes, we must first characterize those regions that form the substrate binding sites. We describe here the 1.9 Å crystal structure of the active site Cys-215 to Ser mutant PTP1B complexed with bis-(para-phosphophenyl) methane (BPPM, Fig. 1a), which is one of the highest-affinity low-molecular weight nonpeptidic substrates identified for PTP1 [kcat = 6.9 s−1, Km = 16 μM (11)]. We also report the 1.85 Å crystal structure of PTP1B/C215S complexed with pTyr [Fig. 1b; kcat = 44 s−1, Km = 4.9 mM (4)]. We have found that both BPPM and pTyr bind, as expected, at the pTyr-binding site (the active site), and we provide a rationale for the higher affinity of BPPM relative to pTyr. Unexpectedly, these structures reveal a second site, adjacent to the active site, that binds BPPM and pTyr as well. These results have significant implications for PTP1B inhibitor design, as it should be possible to develop compounds that can simultaneously occupy both sites to gain higher affinity and selectivity.
Figure 1.
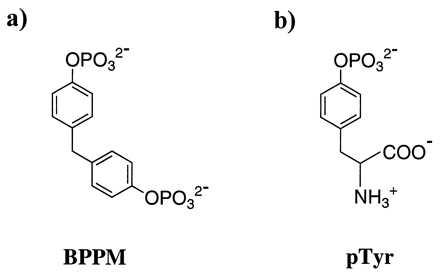
Structures of PTP1B substrates BPPM (a) and pTyr (b).
EXPERIMENTAL PROCEDURES
Construction of the C215S Mutant of PTP1B.
The cDNA encoding the catalytic domain of human PTP1B (amino acids 1–321) was obtained using PCR from a human fetal brain cDNA library (Stratagene). The PCR primers used were 5′-AGCTGGATCCATATGGAGATGGAAAAGGAGTT (encoding both a BamHI and a NdeI site), and 3′-ACGCGAATTCTTAATTGTGTGGCTCCAGGATTCG (encoding an EcoRI site). The PCR product was digested with BamHI and EcoRI and subcloned into a pUC118 vector. The PTP1B coding sequence was confirmed by DNA sequencing. Site-directed mutagenesis was carried out by using the Muta-Gene in vitro mutagenesis kit from Bio-Rad. The oligonucleotide primer used for C215S was 5′-TGGTGCACTCCAGTGCAGG-3′, where the underlined base indicates the change from the naturally occurring nucleotide. The mutation was confirmed by DNA sequencing.
Subcloning, Expression, and Purification of PTP1B/C215S.
The coding region for the PTP1B/C215S mutant was cut from pUC118-PTP1B/C215S with NdeI and EcoRI and ligated to the corresponding sites of plasmid pT7-7. The resulting pT7-7(PTP1B/C215S) was used to transform Escherichia coli BL21(DE3). An overnight culture of the transformed BL21(DE3) cells was diluted 1:100 into 1 liter of 2× YT medium containing 100 μg/ml ampicillin. The culture was grown at 37°C until the absorbance at 600 nm reached 0.6, at which point the cells were induced with 0.4 mM isopropyl β-d-thiogalactoside for 6 h. The cells were harvested by centrifugation and resuspended in 30 ml of ice-cold buffer A [100 mM 2-(4-morpholino)-ethane sulfonic acid, pH 6.5/1 mM EDTA/1 mM DTT] and lysed by two passes through a French press at 1,300 psi. All of the following steps were then carried out at 4°C. The lysate was centrifuged at 15,000 rpm (DuPont SS-34 rotor) for 30 min. The supernatant was incubated with 50 ml of CM-Sephadex C50 equilibrated with buffer A and shaken gently for 40 min. The resin was washed three times with the same volume of buffer A, loaded onto a column, and washed again with 10 bed volumes of buffer A. PTP1B/C215S was eluted from the column by a linear gradient from 0 to 0.5 M NaCl in 200 ml of buffer A.
Cocrystallization of PTP1B/C215S with Substrates.
Crystals were grown by hanging-drop vapor diffusion at 4°C by using conditions modified from Barford et al. (12, 13). A stock was made of PTP1B/C215S at 10 mg/ml in buffer B (10 mM Tris⋅HCl, pH 7.5/25 mM NaCl/0.2 mM EDTA/3.0 mM DTT) with either 3.4 mM BPPM (11) or 53 mM pTyr. For crystal growth, a 5-μl drop of this stock solution was mixed with an equal volume of precipitating solution [0.1 mM Hepes, pH 7.0–7.5/0.2 M magnesium acetate/12%-14% (wt/vol) polyethylene glycol 8000 (Fluka)] and equilibrated against 1 ml of the precipitating solution.
Data Collection and Processing.
All data were collected at beamline X9B of the National Synchrotron Light Source at Brookhaven National Laboratories (Upton, NY). The beamline utilizes radiation from a bending magnet that collects substantial horizontal acceptance with a sagittally focused Si[111] monochromator combined with vertical focusing by a nickel-surfaced aluminum substrate mirror. The accessible wavelength range is 1.1–2.0 Å. The flux is 1.8–3.1 × 1011 photons/s per 100 mAmp and the beam can be focused to 0.3 mm horizontally and 0.2 mm vertically. A vertically mounted conventional rotary table is used as the rotation stage, and an LT-2 low-temperature system mounted nearly coaxially with the rotation axis was employed for cryogenic data collection.
Single crystals (≈0.3 × 0.3 × 0.3 mm3) were successively transferred at 4°C in increments of 5% glycerol concentration to a final cryoprotectant solution that contained crystallization buffer, 25% (vol/vol) glycerol, and either 1.5 mM BPPM or 30 mM pTyr. The crystal was then transferred to a loop and flash-frozen in a stream of nitrogen gas at ≈140 K. A wavelength of 1.20 Å was used to collect 1.0° frames with typical exposure times of 10–20 s. Data were recorded on Fuji image plates and reduced and merged with the hkl package (14).
Structure Solution of the PTP1B Complexes.
For the BPPM complex, the initial model used for phase determination was the unliganded PTP1B structure (13) with the loop residues 179–187 deleted and Cys-215 replaced by Ala. Refinement with x-plor (15) was alternated with rounds of manual rebuilding with the program o (16), resulting in a model comprising residues 2–298 (out of 1–321), 233 ordered water molecules, and two overlapping BPPM molecules. Electron density for two overlapping BPPM molecules (Fig. 2a) accommodated models for both ligands with starting occupancies of 0.5, which refined to values of 0.54 and 0.46. Simulated annealing omit maps (17) and the free R value (18) were used to verify accurate model building, and the quality of the model at all stages of refinement was assessed using the programs procheck (19) and what if (20). In the final stages of refinement, the reflections that had been sequestered for calculating the free R-factor were then merged. All data from 22.0 to 1.9 Å were used in refinement employing an isotropic bulk solvent model (21), yielding a final crystallographic R-factor of 18.1% with good geometry (Table 1).
Figure 2.
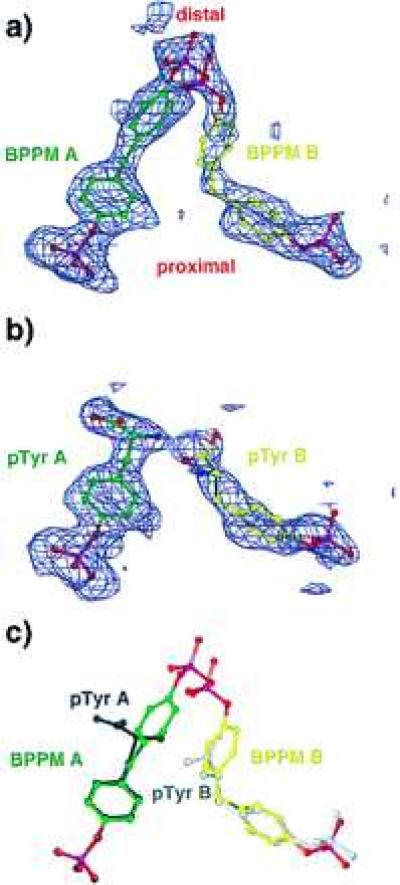
(a and b) Simulated annealing omit maps showing unbiased electron density for bound BPPM (a) and phoshphotyrosine (b) molecules. The density shown is an Fo − Fc map contoured at 2.0 σ, with the refined models superimposed. Molecules bound to the canonical pTyr binding site are colored green, and ligands bound to the second site are colored yellow. (c) Superposition of BPPM and pTyr ligands. pTyr A is drawn in black, and pTyr B is gray. [Diagrams were generated with program o (16)].
Table 1.
Crystal, data collection, and refinement statistics
| BPPM complex | 53 mM pTyr complex | |
|---|---|---|
| Crystal parameters | ||
| Space group | P3121 | P3121 |
| Cell constants | a = b = 88.37 Å | a = b = 87.91 Å |
| c = 104.53 Å | c = 103.82 Å | |
| α = ß = 90°, γ = 120° | α = ß = 90°, γ = 120° | |
| Data collection | ||
| Resolution range, Å | 22.0–1.9 (1.97–1.90)* | 25.0–1.85 (1.85–1.92) |
| Measurements | 138,889 | 185,058 |
| Unique reflections I > 0σ | 31,197 (2,558) | 39,868 (3,754) |
| Completeness, % | 82.1 (69.2) | 98.8 (93.9) |
| Rmerge,† % | 5.2 (26.7) | 4.2 (23.0) |
| Mean I/σ of merged data | 24.7 (2.9) | 30.7 (3.0) |
| Wavelength, Å | 1.20 Å | 1.20 Å |
| Refinement statistics | ||
| Resolution used, Å | 22.0–1.9 | 25.0–1.85 |
| Reflections used | 31,197 (2,558) | 39,868 (3,754) |
| Crystallographic R-factor,‡ % | 18.2 (27.0) | 18.1 (27.1) |
| rms bond length deviation,§ Å | 0.009 | 0.009 |
| rms bond angle deviation,§ ° | 1.85 | 1.85 |
| Number of atoms | ||
| Protein | 2,426 | 2,438¶ |
| Water | 233 | 263 |
| Mg2+ | 1 | 1 |
| BPPM A/pTyr A | 23 | 17 |
| BPPM B/pTyr B | 23 | 17 |
| All atoms | 2,706 | 2,736¶ |
| Average temp. factors, Å2 | ||
| Protein | 25.3 | 25.5¶ |
| Water | 32.6 | 36.1 |
| Mg2+ | 38.7 | 30.0 |
| BPPM A/pTyr A | 11.8 | 27.8 |
| BPPM B/pTyr B | 20.8 | 33.6 |
| All atoms | 25.8 | 26.5¶ |
| Occupancy | ||
| BPPM A/pTyr A | 0.54 | 1.00 |
| BPPM B/pTyr B | 0.46 | 0.51 |
In both structures a Mg2+ ion that is coordinated to six waters, four of which form hydrogen bonds to protein atoms (in residues Glu-129, Glu-130, and residue His-54 from a symmetry-related molecule) was built into octahedral density that could not be otherwise accounted for.
Numbers in parentheses refer to the shell of highest-resolution data.
Rmerge = Σhkl Σn |〈I(hkl)〉 − In(hkl)|/Σhkl Σn In(hkl) where In(hkl) and 〈I(hkl)〉 are the nth and mean measurements of the intensity of reflection hkl.
R-factor = Σhkl |Fo − Fc|/Σhkl Fo where Fo and Fc are the observed and calculated structure factor amplitudes for reflection hkl.
Stereochemical criteria are those of Engh and Huber (37).
Included are the additional three atoms from the alternate conformation of Asp-48.
The refined coordinates of the complex with BPPM were used as a starting model for the PTP1B complex with 53 mM pTyr. Refinement proceeded in a fashion essentially identical to that of the BPPM complex, except that the resolution range used was 25.0–1.85 Å, residues 2–299 were visualized, and 263 ordered water molecules were observed. Electron density for two pTyr molecules (Fig. 2b) was observed, where the density for the pTyr in the noncatalytic site (pTyr B) appeared to overlap the density for one of two conformations of Asp-48, so the occupancies of pTyr B and the two conformations of Asp-48 were refined. The final model had an R-factor of 18.2% with good geometry (Table 1).
Least-squares superposition (22) of the 5 mM (low) pTyr complex (23) onto either the BPPM complex or the 53 mM (high) pTyr complex yielded rms backbone deviations of 0.25 Å and 0.19 Å, respectively. As observed in the low-pTyr complex, binding of either BPPM or pTyr to PTP1B/C215S induces a conformational change of the surface loop 179–187 to bring Asp-181 and Phe-182 into the active site.
RESULTS
Binding Mode of the BPPM Molecules.
Two BPPM molecules of approximately half occupancy were each visualized as strong features in simulated annealing omit maps calculated with coefficients Fo − Fc and 2Fo − Fc (Fig. 2a). One molecule occupies the canonical pTyr-binding site (active site) and will be designated as ligand A, whereas the second molecule, which binds to a different site in the vicinity of the active site, will be designated as ligand B. The terms “proximal” and “distal” will be taken to refer to the phosphophenyl moieties of BPPM that are bound or surface-exposed, respectively (Fig. 2).
Electron density for BPPM A and B allowed unambiguous fitting of the entirety of both molecules (Fig. 2a). The distal phosphate densities of ligands A and B display substantial overlap. Consequently, the two BPPM molecules cannot be simultaneously bound to the same PTP1B molecule. The ligand molecules refined to occupancies of 0.54 for ligand A and 0.46 for ligand B (Table 1).
The proximal half of BPPM molecule A superimposes on the pTyr seen in the active site in both the low-pTyr (23) and high-pTyr structures (Fig. 2c), engaging in similar contacts: electrostatic interactions with Arg-221, hydrogen bonds with the main-chain nitrogens of Ser-216–Arg-221, van der Waals contacts with several aliphatic side chains, and aromatic–aromatic interactions with Tyr-46 and Phe-182 (Figs. 3a and 4a). In addition, a conserved water (Wat28 in the BPPM complex, Wat505 in the low-pTyr structure) forms hydrogen bonds between the proximal phenolic oxygen of BPPM A, Phe-182 N, and Gln266 Nɛ2. The distal half of BPPM A also has significant interactions with the protein (Figs. 3a and 4a): the distal ring makes a typical aromatic-aromatic contact with Phe-182 (24, 25), and an oxygen of the distal phosphate makes a water-mediated hydrogen bond with Gln-262.
Figure 3.
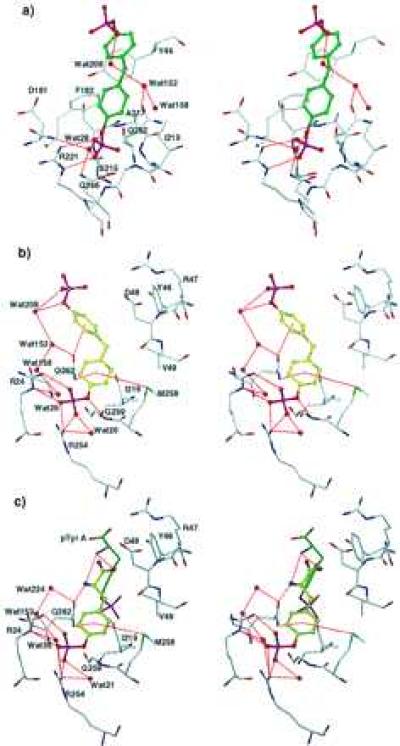
Stereo representations of the binding modes of BPPM A (a), BPPM B (b), and pTyr B (c). Contacts represented by dashed lines are distances less than 3.6 Å, except for certain interactions with aromatic rings. Interactions between the amide nitrogens of residues 216–221 and the phosphate groups of ligand A are too numerous to represent. [Diagrams were generated with the program o (16)].
Figure 4.
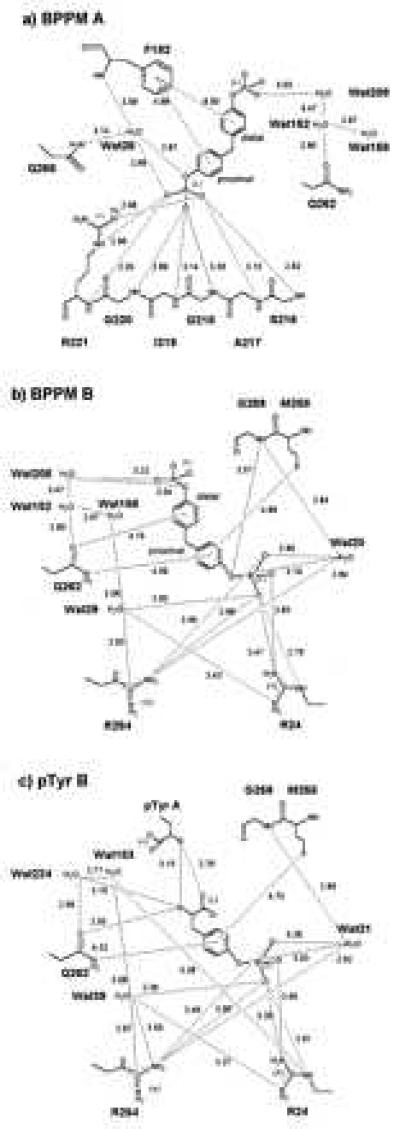
Schematic representations of the interactions between PTP1B/C215S and BPPM A (a), BPPM B (b), and pTyr B (c). A distance cutoff of 3.6 Å was used, except for certain interactions with aromatic rings.
The proximal half of BPPM molecule B highlights a new set of residues capable of binding aryl phosphates (Figs. 3b and 4b). The guanidinium groups of both Arg-24 and Arg-254 are both responsible for ionic interactions with oxygen atoms of the proximal phosphate. An additional determinant of binding may be Wat20, which is conserved in the low-pTyr and high-pTyr complexes. Wat20 has a low B factor of 18 Å2, multiple protein ligands, and it is within hydrogen-bonding distance of both a terminal and a phenolic oxygen in ligand B, suggesting that it is stably bound in solution and has a role in binding BPPM. The phenolic oxygen may also be stabilized by a long (3.6 Å) interaction with the amide nitrogen of Gly-259. The proximal aromatic ring of ligand B is held in place by a number of weakly polar interactions with Gln-262 and Met-258 [Fig. 4b (25)] and several van der Waals contacts with the aliphatic portions of the side chains of Ile-219 and Met-258, and the Cα of Gly-259, effectively burying most of its hydrophobic surface. The bridging methylene group makes van der Waals contacts with the carbonyl oxygen of Asp-48 and the side chain of Val-49. The distal ring of BPPM B may have an equatorial oxygen–aromatic interaction with Gln-262 Oɛ1, and it also makes a van der Waals contact with the Cβ of Asp-48 (Fig. 3b). The distal phosphate of BPPM molecule B, like its counterpart in ligand A, interacts with Wat208, which mediates hydrogen bonds with a terminal and a phenolic phosphate oxygen.
Binding Mode of pTyr Molecules.
Two pTyr molecules are clearly visibly bound to PTP1B. Both pTyr molecules superimpose closely onto the proximal halves of the two BPPM molecules (Fig. 2c). Electron density and refinement statistics indicate that pTyr B has a lower occupancy and larger thermal factors than pTyr A, suggesting that pTyr B is less tightly bound and more disordered. The pTyr A molecule bound at the active site has a binding mode identical to that described by Jia et al. (23), whereas the aryl phosphate of pTyr B makes the same contacts seen for the proximal aryl phosphate of BPPM B. The α-amino and carboxylate functionalities of pTyr also participate in polar interactions not available to BPPM B, as shown in Fig. 4c.
Buried Nonpolar Surface Area.
Upon binding PTP1B, BPPM A and B bury 222.5 Å2 (77%) and 200.4 Å2 (74%) of their nonpolar surface area [calculated by the method of Lee and Richards (26) as implemented in x-plor (15), by using a 1.4-Å probe], respectively. The analogous calculation on pTyr A and B shows that 177.0 Å2 (86%) and 143.2 Å2 (75%) of their nonpolar surface area are buried upon binding. In all cases, the greatest fraction of buried surface area is contributed by the proximal aromatic ring.
DISCUSSION
The in vivo phosphorylation of tyrosine residues within proteins is a reversible and dynamic process governed by the opposing action of protein tyrosine kinases (PTKs), which catalyze protein tyrosine phosphorylation, and PTPases, which are responsible for dephosphorylation (1, 2). PTPases comprise a large family of enzymes and can function to both antagonize and potentiate the action of PTKs. Hundreds of protein kinases and protein phosphatases and their substrates are integrated within an elaborate signal transducing network. The defective or inappropriate operation of this network is at the root of such widespread diseases such as cancers and diabetes. Indeed, like PTKs, overexpression or altered activity of the PTPases can contribute to the symptoms and progression of various diseases. For example, PTPases have been identified as both tumor suppressors (27) and oncogenes (28). A number of PTPases, including PTP1B, have been implicated as negative regulators of insulin receptor regulated processes (8). The importance of PTPases in cellular physiology is further emphasized by the fact that they are often targets for microbial or viral intervention. For instance, the pathogenic bacteria Yersinia encodes a PTPase essential for its virulence (29), and vaccinia virus encodes a dual specificity phosphatase in the PTPase super family (30).
Given the large number of PTPases identified so far and the emerging role played by PTPases in disease, it is not surprising that interest in the development of inhibitors for these enzymes has intensified in recent years (5). Unfortunately, PTPases represent a significant challenge in terms of inhibitor design. Until recently, these enzymes were known to only efficiently hydrolyze protein and peptide substrates, species that are not readily converted into inhibitory agents. In addition, nonpeptidic substrates, such as p-nitrophenylphosphate, are comparatively inefficient PTPase substrates and exhibit little or no selectivety for individual members of the PTPase family. The latter observation is not surprising given the fact that the majority of the invariant amino acid residues conserved in PTPases are located in and around the enzyme active site (13, 31–33). Nevertheless, we recently showed that the Km for the low-molecular weight species BPPM is nearly three orders of magnitude lower than that displayed by phosphotyrosine in the PTP1-catalyzed reaction (11). We now report the crystal structures of PTP1B complexed with BPPM as well as with phosphotyrosine. Quite unexpectedly, both crystal structures reveal the presence of a second aryl phosphate-binding site positioned adjacent to the active site. This second site lies within a region that is nonconserved among PTPases. As a consequence, this unanticipated observation has important implications in terms of the design of highly selective PTPase inhibitory agents.
Identification of a Novel Aryl Phosphate-Binding Site.
The existence of a second site (“site B”) was suggested by an unexpected binding mode observed in the complex with the high-affinity substrate BPPM, and confirmed by the structure of PTP1B/C215S in 53 mM pTyr. The most important residues in site B appear to be Arg-24 and Arg-254, which coordinate the phosphate. Additional favorable interactions include water-mediated hydrogen bonds, weakly polar interactions with Met-258 and Gln-262, and van der Waals contacts with Ile-219, Asp-48, and Val-49 (Figs. 3c and 4c). Hydrophobic driving forces may also play a role, because 143 Å2 (75%) of nonpolar surface area of pTyr are buried within binding site B. In the catalytic site (“site A”), most of the ring atoms of a bound ligand are not exposed to the external aqueous milieu, whereas each atom of the buried proximal ring of BPPM or pTyr in site B has several Å2 of solvent-exposed surface. The fact that the phosphophenol moiety in site B has a greater solvent-exposed area than its counterpart in site A suggests that the former does not form a hydrophobic binding pocket possessing the same degree of complementarity observed in the latter. Indeed, in spite of the fact that the solution concentration of pTyr was 53 mM, the occupancy of pTyr B refined to only approximately one-half. Thus, it is clear from both this structure and the fact that pTyr B was not observed at all in a 5 mM pTyr complex (23) that the affinity of site B for pTyr is lower than that of site A.
Binding Mode of BPPM.
PTP1B/C215S possesses two sites capable of binding the low-molecular weight nonpeptide substrate BPPM. The two BPPM molecules appear to bind in mutually exclusive modes, and electron density, B-factors, and occupancies suggest that BPPM A binds somewhat more tightly than BPPM B (Fig. 2a and Table 1). However, in contrast to the results outlined above for pTyr, it is evident that the difference in the affinities of the two sites for BPPM is not very large. The concentration of BPPM in the crystallization solution is sufficient to saturate both sites, and as a consequence BPPM should partition between sites A and B in proportion to its affinity for each site. The similar occupancy of BPPM in the two sites indicates that BPPM binds to both sites with similar affinity. PTP1B has an ≈300-fold greater preference for BPPM over pTyr, which may be explained by protein–ligand interactions unavailable to pTyr at either site. In addition, we note that it is unlikely that the presence of amino and carboxyl groups in pTyr adversely influences the Km of pTyr (4.9 mM), because the Km for phenyl phosphate itself is 2.5 mM. Finally, the crystallographic finding that BPPM can bind PTP1B in two mutually exclusive modes also provides structural support for the hypothesis that BPPM might be engaged in a combination of productive and nonproductive binding modes (11).
The high affinity of PTP1B for BPPM can be explained by additional interactions that are unavailable to pTyr. BPPM molecule A is not only bound by those interactions responsible for pTyr coordination (Figs. 3a and 4a; ref. 23), but may also experience favorable aromatic–aromatic interactions between its distal ring and Phe-182, as well as water-mediated hydrogen bonds with its distal phosphate (Fig. 4a). The burial of an additional 68 Å2 of hydrophobic surface area by the distal ring of BPPM A may provide an additional entropic driving force on the order of −1.0 kcal/mol (34). BPPM molecule B is also bound by a combination of electrostatic interactions (Arg-24, Arg-254), van der Waals contacts, weakly polar interactions, and water-mediated hydrogen bonds (Fig. 4b), and its distal ring also may also contribute −1.0 kcal/mol by burying 69 Å2 of nonpolar surface.
Implications for Inhibitor Design.
The results from the structural determinations of PTP1B/C215S-BPPM and PTP1B/C215S–pTyr complexes demonstrate the utility of studying the interactions between PTPases and small molecule ligands. PTPases utilize pTyr-containing proteins as their in vivo substrates; and the associated enzyme–substrate complexes possess multiple functional-group binding sites that confer a high complementary fit between substrate and enzyme. In contrast, and as exemplified by BPPM and pTyr, small molecule substrates are able to associate with PTP1B in more than one binding mode. The identification of these alternate binding modes immediately suggests a new strategy for the design of inhibitors with enhanced affinity and specificity. Indeed, prior studies devoted to PTPase inhibitors have generally been forced to ignore the issue of specificity because the majority of the invariant amino acid residues conserved in PTPases are located in and around the enzyme active site (13, 31–33). In short, PTPase active sites are likely to recognize pTyr in a similar way, exhibit the same catalytic mechanisms, and bind pTyr mimetics (i.e., inhibitors) equally well. However, on the basis of the results described herein, it should be possible to design and synthesize nonhydrolyzable aryl phosphate analogs that are able to simultaneously occupy both sites A and B. The rationale for this approach to achieve higher affinity is based on the principle of additivity of free energy of binding and is illustrated in Fig. 5. Indeed, the utility of this strategy to obtain high affinity ligands for proteins has been successfully demonstrated by Fesik and colleagues (35, 36). By using NMR techniques, a weak inhibitor (acetohydroxamic acid, Kd = 17 mM) that is targeted to the catalytic site and a modest inhibitor [3-(cynomethyl)-4′-hydroxybiphenyl, Kd = 0.02 mM]) that binds to the S1′ site of stromelysin have been identified. A high affinity (15 nM) inhibitor was then produced by tethering together the two weakly bound ligands with an appropriate linker. The interaction of an inhibitor with two independent sites (e.g., site A and site B) on PTP1B would be expected to confer exquisite specificity, if other PTPases do not possess an identical second site interaction.
Figure 5.
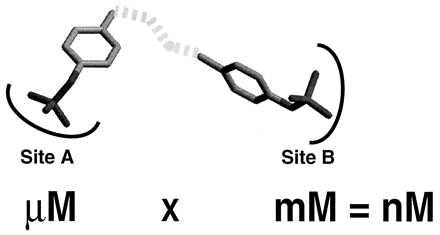
A strategy for creating selective and high-affinity PTP1B inhibitors. Based on the principle of additivity of free energy of binding, high-affinity ligands can be designed by linking two functional groups (each with modest affinity to the target protein) identified experimentally. The added specificity arises from the fact that the tethered ligand has to bind both sites simultaneously.
Are similar spatial arrangements of amino acid side chains and solvent molecules that form site B present in other PTPases? Arg-254 and Gln-262, which comprise part of the binding pocket for site B, are invariant amongst all PTPases (31). Amino acid sequence alignment suggests that an Arg residue equivalent to Arg-24 in site B of PTP1B may also exist in T-cell PTPase, PTP–PEST, and the Yersinia PTPase, but not in PTPα, PTPɛ, leukocyte common antigen-related (LAR), PTPγ, CD45, SH2 domain-containing PTPases, and PTP-meg (data not shown). Nevertheless, it appears that, with the exception of Arg-254 and Gln-262, residues that form the second site in PTP1B (e.g., Gln-21, Met-258, and Gly-259) are less conserved among PTPases than residues that form the active site. Further structural and biochemical studies on PTPases other than PTP1B are required to resolve this issue. Interestingly, residues Val-49, Ile-219, and Gln-262 that are part of site B are also found to make van der Waals contacts with the side chain of Leu at the P+1 position of the peptide substrate DADEpYL-NH2 (23). Because these side chains make van der Waals contacts and polar interactions with the aromatic ring and the bridging methylene unit in BPPM (or the Cβ in pTyr), appropriately designed compounds that are specifically targeted to the second site may be effective in disrupting the binding of pTyr-containing peptide–protein substrates to PTP1B. This finding may represent another approach for PTPase inhibitor design that targets the extended binding sites rather than the immediate catalytic site.
In summary, we report the high-resolution structures of PTP1B/C215S complexed with BPPM, a tight-binding substrate, and with pTyr. These structures reveal the details of key interactions between amino acid residues in PTP1B and a novel class of bis-phenyl substrates. We also describe a second aryl phosphate-binding mode in PTP1B that has significant implications for PTP1B inhibitor design. These studies provide a solid molecular basis for further structure-based rational design of PTPase inhibitors. Selective and potent PTPase inhibitors may not only serve as useful probes to help define the physiological functions of PTPases, but may ultimately constitute a unique family of therapeutic agents for the treatment of cancers and diabetes.
Acknowledgments
We thank Dr. M. R. Chance (Albert Einstein College of Medicine, National Synchrotron Light Source supported by DOE) for the opportunity to develop x-ray crystallography at beamline X9B, supported by RR01633. The assistance of Dr. R. Fischetti (Argonne National Laboratories) was indispensable in the early stages of development of the the data collection apparatus. The assistance of Dr. A. A. Fedorov, S. Goldsmith, and N. M. Mahoney in data collection is greatly appreciated. We would also like to thank Dr. J. C. Hanson and Dr. C. Ogata (National Synchrotron Light Source) for the generous use of their equipment during data collection. This work was supported in part by a Pilot Research Project Award from the Howard Hughes Medical Institute–Research Resources Program for Medical Schools, and National Institutes of Health Grant CA69202 (Z.-Y.Z.), National Institutes of Health Grant GM50121, an award from the W. M. Keck Foundation (S.C.A.), and Medical Scientist Training Program Grant T32 GM07288 (Y.A.P.) from the National Institutes of Health.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: PTPase, protein-tyrosine phosphatase; BPPM, bis-(para-phosphophenyl) methane; pTyr, phosphotyrosine; PTP1B, protein-tyrosine phosphatase 1B.
Data deposition: Coordinates have been deposited at the Brookhaven Protein Data Bank (accession codes 1AAX for BPPM complex and 1PTY for pTyr complex).
References
- 1.Hunter T. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 2.Tonks N K, Neel B G. Cell. 1996;87:365–368. doi: 10.1016/s0092-8674(00)81357-4. [DOI] [PubMed] [Google Scholar]
- 3.Tonks N K, Diltz C D, Fischer E H. J Biol Chem. 1988;263:6731–6737. [PubMed] [Google Scholar]
- 4.Zhang Z-Y, Maclean D, McNamara D J, Dobrusin E M, Sawyer T K, Dixon J E. Biochemistry. 1994;33:2285–2290. doi: 10.1021/bi00174a040. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z-Y. Curr Top Cell Regul. 1997;35:21–68. doi: 10.1016/s0070-2137(97)80002-7. [DOI] [PubMed] [Google Scholar]
- 6.Chernoff J, Schievella A R, Jost C A, Erikson R L, Neel B G. Proc Natl Acad Sci USA. 1990;87:2735–2739. doi: 10.1073/pnas.87.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiener J R, Kerns B-J M, Harvey E L, Conaway M R, Iglehart J D, Berchuck A, Bast R C., Jr J Natl Cancer Inst. 1994;86:372–378. doi: 10.1093/jnci/86.5.372. [DOI] [PubMed] [Google Scholar]
- 8.Kenner K A, Anyanwu E, Olefsky J M, Kusari J. J Biol Chem. 1996;271:19810–19816. doi: 10.1074/jbc.271.33.19810. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Z-Y. J Biol Chem. 1995;270:11199–11204. doi: 10.1074/jbc.270.19.11199. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Z-Y. J Biol Chem. 1995;270:16052–16055. [PubMed] [Google Scholar]
- 11.Montserat J, Chen L, Lawrence D S, Zhang Z-Y. J Biol Chem. 1996;271:7868–7872. doi: 10.1074/jbc.271.13.7868. [DOI] [PubMed] [Google Scholar]
- 12.Barford D, Keller J C, Flint A J, Tonks N K. J Mol Biol. 1994;239:726–730. doi: 10.1006/jmbi.1994.1409. [DOI] [PubMed] [Google Scholar]
- 13.Barford D, Flint A J, Tonks N K. Science. 1994;263:1397–1404. [PubMed] [Google Scholar]
- 14.Otwinowski Z. In: Proceedings of the CCP4 Study Weekend. Wolf W, Evans P R, Leslie A G W, editors. Warrington, U.K.: SERC Daresbury Lab.; 1991. pp. 80–86. [Google Scholar]
- 15.Brünger A T. x-plor Manual. New Haven, CT: Yale Univ.; 1993. , Version 3.1. [Google Scholar]
- 16.Jones T A, Zhou J-Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 17.Hodel A, Kim S-H, Brünger A T. Acta Crystallogr A. 1992;48:851–858. [Google Scholar]
- 18.Brünger A T. Nature (London) 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 19.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 20.Vriend G. J Mol Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 21.Jiang J S, Brünger A T. J Mol Biol. 1994;243:100–115. doi: 10.1006/jmbi.1994.1633. [DOI] [PubMed] [Google Scholar]
- 22.Martin A C R. pro fit: Protein Least Squares Fitting. Ashtead, Surrey, U.K.: SciTech Software; 1995. , Version 1.6. [Google Scholar]
- 23.Jia Z, Barford D, Flint A J, Tonks N K. Science. 1995;268:1754–1758. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]
- 24.Burley S K, Petsko G A. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 25.Burley S K, Petsko G A. Adv Prot Chem. 1988;39:125–189. doi: 10.1016/s0065-3233(08)60376-9. [DOI] [PubMed] [Google Scholar]
- 26.Lee B, Richards F M. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang S I, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner S H, Giovanella B C, Ittmann M, Tycko B, hibshoosh H, Wigler M H, Parsons R. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 28.Galaktionov K, Lee A K, Eckstein J, Draetta G, Meckler J, Loda M, Beach D. Science. 1995;269:1575–1577. doi: 10.1126/science.7667636. [DOI] [PubMed] [Google Scholar]
- 29.Bliska J B, Guan K L, Dixon J E, Falkow S. Proc Natl Acad Sci USA. 1991;88:1187–1191. doi: 10.1073/pnas.88.4.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guan K L, Broyles S, Dixon J E. Nature (London) 1991;350:359–361. doi: 10.1038/350359a0. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z-Y, Wang Y, Dixon J E. Proc Natl Acad Sci USA. 1994;91:1624–1627. doi: 10.1073/pnas.91.5.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stuckey J A, Schubert H L, Fauman E, Zhang Z-Y, Dixon J E, Saper M A. Nature (London) 1994;370:571–575. doi: 10.1038/370571a0. [DOI] [PubMed] [Google Scholar]
- 33.Bilwes A M, den Hertog J, Hunter T, Noel J P. Nature (London) 1996;382:555–559. doi: 10.1038/382555a0. [DOI] [PubMed] [Google Scholar]
- 34.Eisenberg D, McLachlan A D. Nature (London) 1986;319:199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- 35.Shuker S B, Hajduk P J, Meadows R P, Fesik S W. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 36.Hajduk P J, Sheppard G, Nettesheim D G, Olejniczak E T, Shuker S B, Meadows R P, Steinman D H, carrera G M, Marcotte P A, Severin J, Walter K, Smith H, Gubbins E, Simmer R, Holzman T F, Morgan D W, Davidsen S K, Summers J B, Fesik S W. J Am Chem Soc. 1997;119:5818–5827. [Google Scholar]
- 37.Engh R A, Huber R. Acta Crystallogr A. 1991;47:392–400. [Google Scholar]