

Abstract
The in vivo morphology and phenotype of circulating cells that spontaneously contribute to new vessel formation in adults remain unclear. Here, we use high-resolution imaging and flow cytometry to characterize the morphology and phenotype of a distinct population of circulating mononuclear cells contributing to spontaneous new vessel formation after hyperoxia acute lung injury (HALI). We identify a subpopulation of myeloid (CD11b/Mac1+) haematopoietic cells co-expressing vascular endothelial growth factor receptor 2 (VEGFR2) and platelet derived growth factor receptor beta (PDGFRβ). Moreover, we show that these CD11b+VEGFR2+PDGFRβ+ circulating precursor cells (CPCs) contribute structurally to the luminal surface of capillaries re-forming 2 weeks post-HALI. This indicates that these myeloid CPCs may function, at least transiently, as putative vascular precursors, and has important implications for capillary growth and repair in injury and in pathologies of the lung and other organs.
Keywords: hyperoxia, lung capillary injury/restoration, circulating precursor cell
Introduction
The distal vasculature in the adult lung—distributed through approximately 300,000,000 alveoli—is a major site of injury and remodelling in acute lung injury (ALI). Once destroyed by injury or disease the spontaneous restoration of small vessel and capillary networks, leading to better lung function, has proved limited. Although major advances have been made in the treatment and management of patients with ALI, vascular loss remains a significant clinical problem. Moreover, supplemental oxygen levels (>50%), typically administered to improve oxygen delivery to the brain and other vital organs and to peripheral tissues, can exacerbate ventilator-induced lung injury in ALI [1, 2]. Similarly, in patients developing radiation pneumonitis, chronic obstructive lung disease, or pulmonary hypertension injury to the distal lung leading to alveolar disruption and vascular loss is often extensive and progressive.
New vessel formation during vascular repair in adult organs, previously thought to be limited to sprouting angiogenesis by endothelial cells (ECs) from nearby vessels, may involve circulating ‘vasculogenic’ cells which have been shown to home to sites of tissue injury and contribute to neovascularization [3]. Blood circulating precursor cells (CPCs) originating from the bone marrow may give rise to endothelial as well as perivascular cells during new vessel formation in tumours or Matrigel plugs [4–7]. Although such cells present an attractive target for use in cell-based therapies, it remains currently unclear if CPCs can affect spontaneous vascular growth in lungs.
We have shown in an in vivo rodent model that pulmonary capillary networks injured in hyperoxia acute lung injury (HALI) spontaneously re-form post-HALI—in a robust, vascular endothelial growth factor (VEGF)-dependent manner [8]. Based on our finding that a population of haematopoietic cells interacts with lung endothelium post-HALI, we hypothesized that they are CPCs that contribute to capillary repair. The goal of this study was to identify the phenotype of these cells by combining high-resolution immunogold labelling with flow cytometric analyses, and determine the role of these CPCs in capillary restoration post-HALI. We show that a population of CD11b+VEGFR2+PDGFRβ+ CPCs interacts with the endothelium of spontaneously re-forming pulmonary capillaries.
Materials and methods
Animals and hyperoxia acute lung injury model
We used adult male Sprague-Dawley rats (Charles River, Wilmington, MA, USA), after obtaining approval for this protocol from the Subcommittee on Research Animal Care of the Massachusetts General Hospital, Boston, USA. The exposure chamber and the post-HALI neovascularization model used in this study have been described in detail [8, 9]. In brief, after weaning from 87% O2 to 21% O2 over 1 week, air breathing over 4 weeks triggers spontaneous capillary repair: as ECs re-establish patent capillary networks, these markedly increase 2 weeks post-HALI and by 4 weeks approach a normal distribution. We collected lung tissue and blood from adult male Sprague-Dawley rats that breathed air (n= 19), high oxygen (87%) at normobaric pressure for 4 weeks (HALI, n= 13), or high oxygen for 4 weeks (followed by ∼10% less oxygen each day for 7 days, week 5), and then air for 1, 2, 3 or 4 weeks (post-HALI, n= 11, 13, 8 and 10, respectively). The distribution of capillaries in the normal lung provided the baseline from which to assess capillary loss after high oxygen and restoration in air.
Immunocytochemistry
Tissue preparation was performed as previously described [8]. All Unicryl sections were pre-treated with 1% bovine serum albumin (BSA) in PBS (5 min at RT), incubated (overnight at 4°C) with (i) mouse anti-rat monoclonal antibody to CD11b (Calbiochem, La Jolla, CA, USA); (ii) rabbit anti-rat polyclonal antibodies to VEGFR2 or PDGFRβ (Calbiochem); or (iii) a rabbit polyclonal antibody to von Willebrand factor (vWF, DAKO, Carpinteria, CA, USA), rinsed in PBS, and incubated (60 min at RT) in protein A-gold (pA-AU) (10-nm diameter, Auroprobe AG10, Amersham, UK) diluted 1:50 in PBS prior to double-staining. The electron-dense gold particles map antigenic sites for expressed proteins. As a negative control, we omitted treatment with the primary antibody and incubated sections in 0.5% BSA in PBS alone: no reactive sites were detected in these control sections.
Morphologic analyses of CPCs
Using a macro designed to work with NIH Scion (1.62) software (public domain), digital images of capillaries and CPCs were collected and analysed by TEM via a high sensitivity CCD-300-RC camera (MTI) attached to the 35-mm port of the microscope (Advanced Microscopy Techniques Corp., Danvers, MA, USA). We routinely acquired images at ×3456–29,623 magnification. For reference, additional 1–2 mm thick epon-araldite and Unicryl sections were mounted on glass slides, stained with 0.1% toluidine blue: digital images from these sections were collected by brightfield microscopy using a Nikon system (COOLSCOPE CS1, MicroVideo Instruments Inc, Avon, MA, USA), to confirm the sequence of capillary injury and repair (663 representative images).
Analyses of CPC phenotype
Data for CPC morphology, phenotype and interactions with capillary endothelium during capillary repair were based on high-resolution images of CPCs in HALI (week 4), post-HALI (weeks 6–9) and normal lung (779 CPCs in 1191 capillaries, 2853 images). These images were routinely collected to obtain an overview of capillary ECs and CPCs, to identify apposing and adherent regions of cell membranes and visualize gold-labelled antigenic sites.
Phenotypic analyses and kinetic measurements of CPCs by flow cytometry
Blood was collected by cardiac puncture with a 26G needle from rats at 2 weeks post-HALI and from rats breathing air. Blood cells were stained after their separation from plasma and RBC lysis using ACK lysis buffer (Invitrogen, Carlsbad, CA, USA). We used mouse anti-rat CD31-PE, CD11b-PE and CD45-PE-Cy5 (all from BD Pharmingen, San Jose, CA, USA), and rabbit anti-rat VEGFR2 and PDGFRβ antibodies (Calbiochem). Immunostaining for VEGFR2 and PDGFRβ on CPC’s surface was performed using a secondary goat anti-rabbit IgG. CPCs were measured on a BD-FACSCalibur system using Cellquest software. The percentage of CPCs was calculated as the fraction of VEGFR2+ cells among mononuclear cells in peripheral blood. Additionally, VEGFR2+CD11b+ cells were gated and sorted by fluorescence-activated cell sorting. Fifty thousand sorted cells were fixed (in 2% paraformaldehyde in PBS) and centrifuged onto microdissection slides: the cytospin preparations were air-dried, stained with Giemsa and analysed by brightfield microscopy. For phenotypic analyses, we labelled a rabbit anti-rat VEGFR2 antibodies with Alexa Fluor® 488 and a rabbit anti-rat PDGFRβ antibody with Alexa Fluor® 647 using Invitrogen labelling kits. Peripheral blood cells were immunostained for CD31, CD11b, CD45, VEGFR2 and PDGFRβ and the samples were analysed on an LSR-II cytometer (BD Biosciences). For compensation, we used unstained blood cells and single colour controls for each channel.
Quantitative analysis of CPCs
To estimate the number of capillary CPCs within the lung, we examined a further sample of >200 capillaries at each study time-point (1077 CPCs in 1837 capillaries, 3833 images). Starting at an opening at the top of the grid, we systematically collected successive images of all patent/residual capillary units and their associated CPCs in consecutive fields across the (200 mesh) grid face. We collected images from two grids per lung and analysed those fields containing only alveolar-capillary membrane structures, i.e. an occasional field was excluded if a conducting airway, or vessel, was also present. CPC number per lung was estimated from the average number of cells per microscopic field, volume of tissue per field and total lung volume (total cells in the lung = VL/HC× CS/AS× 200 / NR×[104cm]3/cm3, where VL= total lung volume, HC= average height of cell, CS= number of cells counted in the grid, AS= surface area of grid [πr2], NR= number of grid openings with capillary tissue). A subset of these data further demonstrated the proportion of CPCs adherent to capillary endothelium.
Results
Capillary segments injured in HALI spontaneously repair post-HALI
We studied the CPCs at time-points that correspond to phases of capillary injury in HALI and active repair post-HALI (Fig. 1a). At these time-points, the alveolar-capillary membrane was characterized by endothelial loss/avascular zones in HALI, and by restored capillary networks post-HALI reminiscent of the open (lace-like) networks in normal lungs (Fig. 1b–d). EC injury, frequent in both patent and residual capillary structures in HALI, was infrequent post-HALI, suggesting initial sloughing of injured ECs and later expansion of endothelial surfaces by local or by circulating precursors.
Figure 1.
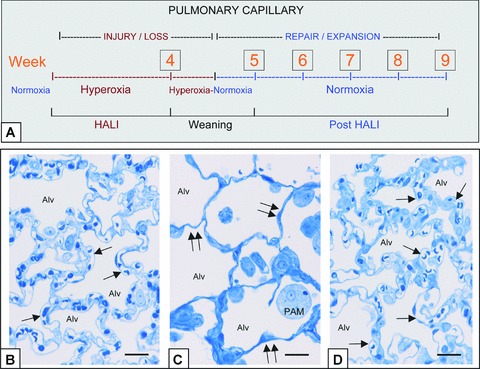
In vivo model of capillary remodelling in HALI and post-HALI. Schema of in vivo model (a) and representative brightfield images illustrating patent capillaries in normal lung (b, arrows), their loss in HALI (c, arrows, week 4) and the restoration of patent capillaries early in the post-HALI phase following spontaneous repair (d, arrow, week 6). In the model, rodents breathe 87% oxygen for 4 weeks (HALI) or 87% oxygen for 4 weeks, followed by weaning to air for 1 week (week 5) and air breathing for up to 4 weeks (post-HALI, weeks 6–9). Initially in HALI (week 1), ECs of small vessels and capillaries in the alveolar-capillary membrane are severely injured. As these cells (and epithelial cells) adapt to the high oxygen tension, the membrane remodels; however there is extensive capillary loss (HALI, weeks 2–4). Because of the greatly reduced capillary bed present at this time, the oxygen tension is lowered (∼less 10%) daily during the transition to breathing air (week 5) to prevent asphyxia, dyspnoea and cyanosis—the lower oxygen tension of air (i.e. relative hypoxia) triggering a burst of spontaneous vascular repair post-HALI (weeks 6 and 7). Spontaneous capillary repair and expansion continue to restore the membrane post-HALI until, as patent capillary networks approach their normal distribution, the response wanes (week 9) [8]. Two-μm-thick resin sections stained with toluidine blue. Bars = 25 μm (b–d).
CPCs have a distinct morphology and are numerous in normal lung, in HALI and post-HALI
We identified CPCs as a morphologically distinct mononuclear cell population in the normal lung, in HALI and in post-HALI (Fig. 2)—monocytic cells of approximately 9 μm (maximum) diameter, with a high nucleus to cystosol ratio, and a cytsol characterized by abundant free ribosomes and large mitochondria. The cells were distinct from capillary ECs and from mature intravascular haematopoietic cells circulating through the lung (e.g. granulocytes, eosinophils, mast cells and monocytes, see Fig. S1). CPCs rarely interacted with capillary walls. In normal lung, CPCs rarely interacted with capillary walls (9% of CPCs), i.e., the vast majority were freely circulating. In HALI, CPCs were trapped in narrowed capillaries. Early in the post-HALI phase (weeks 6 and 7), 35% of the CPCs interacted with ECs while later the number of interacting cells fell to only 6% (weeks 8 and 9). But not at later time-points (6% at weeks 8 and 9). CPCs formed a significant intravascular population (i.e. estimated at approximately 5–8 million cells/lung) at all of the time-points evaluated in this study.
Figure 2.
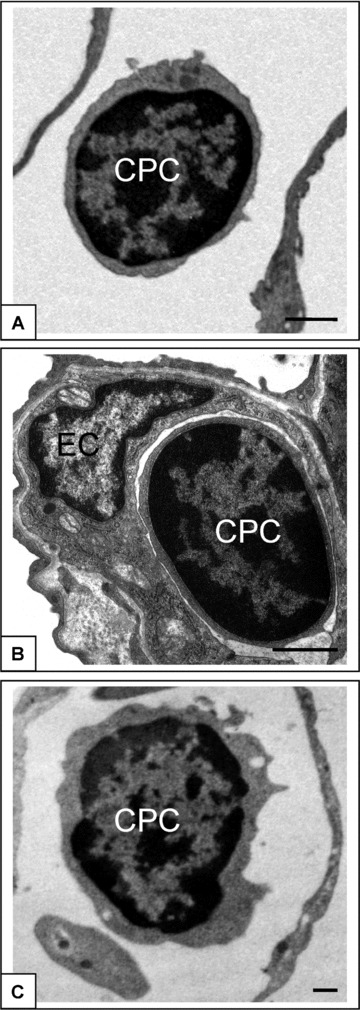
CPC in capillaries of normal lung, in HALI and post-HALI. CPCs were identified by their similar morphology as freely circulating cells in patent capillaries of normal lung (a), in the narrowed capillary segments in HALI (b), and again as freely circulating cells in patent capillary segments post-HALI (c, week 9). Characterized by a high ratio of nucleus to cytoplasm and dense arrangement of heterochromatin CPCs were clearly identified from adjacent capillary ECs (see b). Eighty-nm-thick epon resin sections stained with uranyl acetate and lead citrate. Bars = 1 μm (a–c).
A subset of blood CPCs expresses CD11b, VEGFR2 and PDGFR on cell surface
By flow cytometric analysis, in post-HALI (week 8) we found in peripheral blood that a subset of mononuclear cells (approximately 0.5%) co-express the endothelial-selective marker VEGFR2 and the mesenchymal-selective marker PDGFRβ (Fig. 3a–f). The scattering properties of these cells, consistent with the morphology of CPCs, were similar to those of lymphocytes. Of interest, all VEGFR2+PDGFRβ+ cells expressed CD45 and CD31, and some expressed the myeloid-cell associated integrin CD11b (Mac1) on their surface (Fig. 3e). Cytospin preparations of VEGFR2+ cells revealed the same monocytic morphology as CPCs in tissue sections. VEGFR2+CD45+ CPCs represented 1–2% of the circulating mononuclear cells, at all of the time-points evaluated in this study.
Figure 3.
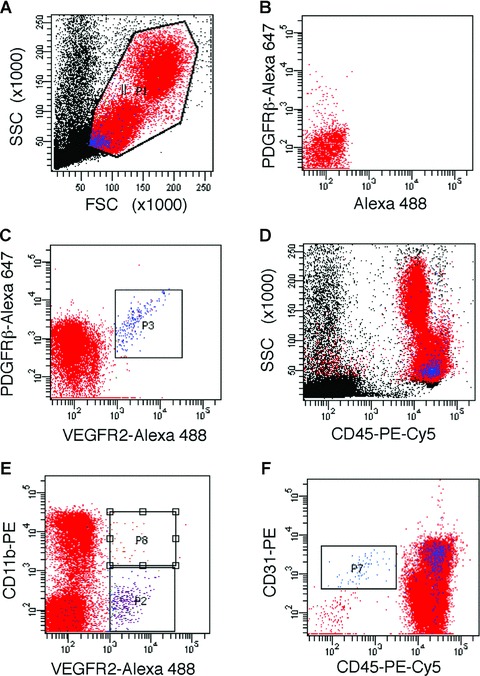
Phenotypic characterization of CPCs. LSR-II flow cytometric analyses of (gated, a) mononuclear cells in peripheral blood, identified a distinct population of CPCs that co-express VEGFR2 and PDGFRβ (c); single-colour controls were used for compensation (e.g. see staining for PDGFRβ-Alexa Fluor 647 in b). These CPCs (in blue) are positive for CD45 (d) and show scattering properties typical of small cells with a high nucleus to cytoplasm ratio (a, d). A subset of CPCs is positive for CD11b (e), whereas all VEGFR2+PDGFRβ+ CPCs are positive for CD31 (f); CPCs are distinct from cells with a phenotype consistent with ‘circulating ECs’, i.e. CD31+CD45− (see gated population in f).
CPCs detected in lung capillaries are CD11b+VEGFR2+PDGFRβ+
Next, by high-resolution imaging and immunogold labelling of lung tissue, we detected antigenic sites for VEGFR2 on all intravascular CPCs (Fig. 4a). Further analysis showed that these CPCs expressed PDGFRβ (Fig. 4b). Moreover, analysis of CPCs in serial sections demonstrated co-expression of VEGFR2 and PDGFRβ (Fig. 4c and d). Furthermore, these CPCs stained positively for CD11b with similar cytoplasmic patterns of staining as compared to other myeloid-lineage cells (e.g. eosinophils and PMNs) (Fig. 4e). Finally, these CPCs expressed the endothelial marker vWF (Fig. 4f).
Figure 4.
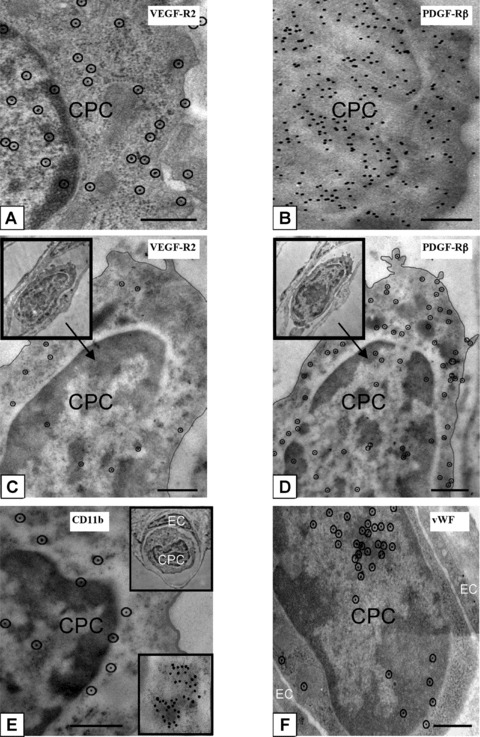
Phenotypic characterization of CPCs in lungs post-HALI. Antigenic sites, visualized with 10-nm protein A-gold by high-resolution microscopy, demonstrated that CPCs were VEGFR2+ (a) and PDGFRβ+ (b) post-HALI (weeks 6 and 7). Representative images of the same CPC profile in adjacent sections of lung tissue (c and d, and insets) demonstrated co-expression of VEGFR2 and PDGFRβ antigenic sites post-HALI (week 7). Other representative images of additional antigenic sites expressed by CPCs post-HALI (week 7): CD11b (e and inset) and for vWF (f). Typically, the sites for CD11b were uniformly distributed over CPCs but also appeared as clusters (e, see bottom inset, illustrating an example of a cluster of ∼40 CD11b+ labelled sites). Ten-nm gold-labelled antigenic sites (a, c–f) are circled for clarity to distinguish these from 7 to 8 nm ribosomes on cell profiles. Ninety-nm-thick Unicryl resin sections stained with uranyl acetate and lead citrate. Bars = 0.5 μm (a–f).
CPCs participate in lung capillary restoration post-HALI
To study the interaction between CPCs and the endothelial of re-forming capillaries, we reconstructed (from sets of ∼20–25 serial high-resolution digital images) single CPCs and adjacent ECs from ∼90 capillary segments forming during the height of the regenerative response post-HALI (after 1–2 weeks). Attaching first focally to the endothelial apical plasmalemmal membrane via multiple small cytoplasmic extensions (Fig. S2), CPCs aligned with endothelial surfaces and, in regions, became adherent, i.e. the two cells were no longer delineated by intact plasmalemmal membranes (Fig. 5a–d). They adhered to ECs with normal morphology and not to injured (oncotic, necrotic, apoptotic) ECs. CPCs and ECs were judged adherent by loss of integrity of the two plasmalemmal membranes (Fig. 5b–d). This is in contrast to inflammatory haematopoietic cells transiting endothelium cells via diapedesis, where each cell type clearly retains an intact plasmalemmal membrane. We did not observe inflammatory cell infiltration or adherence between CPCs in the lungs during HALI or post-HALI.
Figure 5.
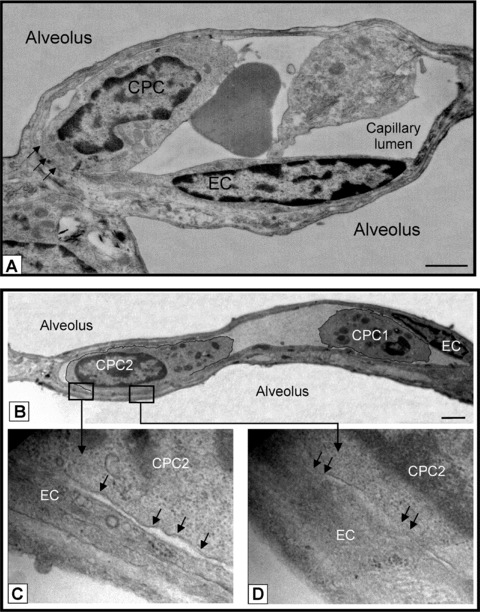
CPCs contact and adhere to endothelium post HALI. (a) Representative image of CPC aligning and adhering to a capillary EC (see arrows) post-HALI (week 7). At this time, approximately 35% of CPCs form contacts with lung capillary ECs. (b) Representative image of two CPCs in a lung capillary post-HALI (week 6): CPC1 appears free in the lumen and CPC2 is in the process of adhering to the adjacent EC (see boxed areas): higher magnification shows regional separation of two distinct cell membranes (lower left image, arrows), and regional loss of membrane delineation between the two cells (lower right image, arrows). Eighty-nm-thick epon resin sections stained with uranyl acetate and lead citrate. Bars = 1 μm (a) and 2 μm (b).
The insertion of CPC processes between ECs indicated their retention within the capillary luminal surface (Fig. 6a and b). At this transitional stage, the CPCs were still clearly identified from ECs by morphology. All the cells forming a contiguous capillary surface, however, displayed characteristic endothelial morphology. Nevertheless, the cells forming the capillary surfaces expressed focal accumulations of CD11b antigenic sites, typical of CPCs (Fig. 6c). In a non-biased sample of ECs in the early post-HALI phase, approximately 32% of them (33/103 nucleated ECs) were CD11b+.
Figure 6.
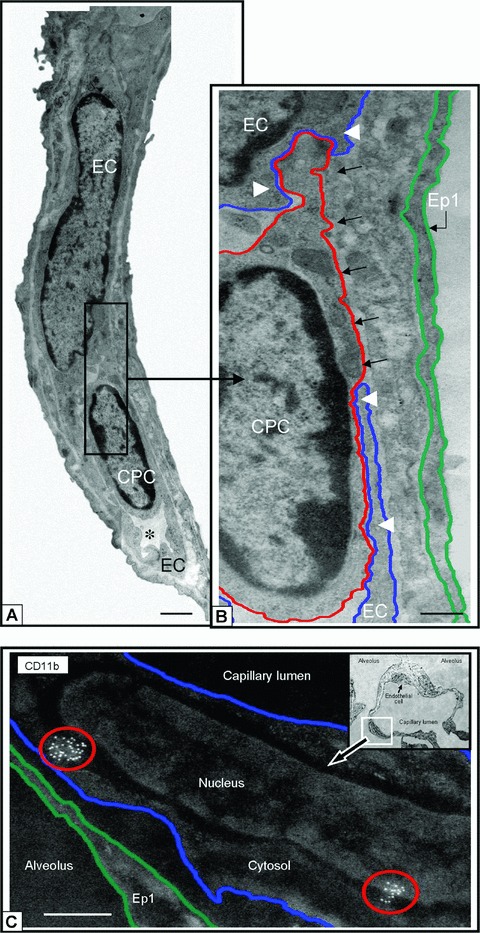
CD11b+ CPCs and cells integrate into capillary surfaces post HALI. Illustrations of CD11b+ CPC (a) and capillary EC in a residual capillary structure post-HALI (week 6). An asterisk marks the restricted capillary lumen at this time. A region of the CPC cytosol is inserted between the processes of two capillary ECs (b, black arrows). Note the location of the CPC within the capillary lumen in relation to the processes of adjacent ECs (white arrowheads), to (an un-delineated) perivascular cell process, and to the underlying epithelial (Ep1) cell process. The blue line delineates the plasmalemmal membrane of the capillary EC, the red line outlines the CPC, and the green line demarcates the adjacent epithelial type 1 (Ep1). (c) Representative image of lung capillary (see boxed area of inset) and higher magnification of the luminal surface illustrating cell clusters of antigenic sites for CD11b (circled in red) typical of CPCs expressing this protein post-HALI (week 7). Image is inverted to highlight the location of this cell with antigenic sites. The other EC indicated in the same capillary (see inset, small arrow) does not express CD11b. Eighty-nm-thick epon resin section (a, b), and 90-nm-thick Unicryl resin section (c), stained with uranyl acetate and lead citrate. Bars = 1 μm (a), 0.5 μm (b, c).
Rare CPCs were present post-HALI in the interstitium of the alveolar-capillary membrane (after transit from the capillary lumen and across endothelium), where they aligned as CD11b+ perivascular cells (Fig. S3). They were identified by their morphology from interstitial fibroblasts, which are the major source of PDGFRβ+ perivascular cells developing around small vessels in the alveolar-capillary membrane in this model [10, 11].
Discussion
We have characterized by morphology and phenotype a novel population of CPCs in normal lung, in HALI and post-HALI; and demonstrated the interaction of these cells with capillary endothelium re-forming after injury. Other studies have failed to detect any contribution by bona fide endothelial CPCs to the extensive vascular growth and remodelling that occurs in a model of regenerative lung growth [12]. Cells identified as vascular progenitors have been identified in the pulmonary arteries of patients with chronic obstructive pulmonary disease, however, where they contribute to capillary repair and pulmonary vascular remodelling [13]. In line with this, and our present findings, studies in parabiotic mice (GFP+ mice and wild mice with a co-joined circulation) demonstrated that GFP+ cells derived from the circulation are retained in pulmonary tissue following injury [14]. In addition, Balasubramaniam and colleagues reported that circulating bone marrow-derived (BMD) cells are necessary to maintain vascular density in the developing and adult mouse lung challenged by hyperoxia [15]. Several groups have demonstrated a direct interaction between CPCs—including VEGFR2+ or PDGFRβ+ myeloid cells—and blood vessels in tumours [16, 17]. In addition, myeloid growth factor overexpression has been shown to protect lung capillaries during HALI [18].
Here, we characterize a previously undescribed CPC population of VEGFR2+PDGFRβ+ cells, expressing haematopoietic markers, and morphologically distinct from mature haematopoietic cells. Our data highlight a role for this CPC population in the luminal and abluminal surfaces of capillaries re-forming in the adult lung post-HALI. Only cells with CPC morphology were identified interacting with capillary walls, and these cells expressed detectable levels of CD11b, VEGFR2 and PDGFRβ. Furthermore, at this time of increased capillary repair, the presence of few monocyte/monocytic or PMN cell infiltrates in the lung’s interstitium indicated the absence of an overt inflammatory response.
Our data support the adhesion of myeloid VEGFR2+PDGFRβ+ CPCs to capillary ECs early in the post-HALI phase, and indicate that these cells integrate into the capillary surface. Because the CPCs studied by us were not genetically marked, their progeny could not be defined morphologically at later time-points. Thus, in this model we cannot discern whether the CPCs eventually ‘differentiate’ into ECs with a ‘mature’ (i.e. non-haematopoietic) phenotype. Nor can we detect if the vascular CPCs are replaced by local-derived, proliferating lung capillary endothelial or perivascular cells. Future studies should determine if CPCs are only transiently incorporated into the vessels or, conversely, if they ‘differentiate’ into mature capillary endothelial and/or perivascular cells. The CPCs identified in the lung tissue studies (by intracellular detection of the two proteins) express both VEGFR2 and PDGFRβ at some level, and only cells with these markers interact with endothelium. Thus, VEGFR2 and PDGFRβ expression could not be used to discriminate between CPCs aligning as endothelial or perivascular cells. Moreover, ECs may express PDGFRβ[19] and conversely, perivascular cells may express VEGFR2 [20].
A greater understanding of these cells, including the molecular mechanisms mediating the vascular responses reported here, might improve therapeutic strategies to address vascular loss following injury or disease in the lung and other organs. Future work should determine if CPCs could be used/engineered to promote capillary restoration in the developing or adult lung. Moreover, it will be critical to establish if VEGFR2+PDGFRβ+ CPCs have any relevance for other types of physiological and pathological capillary formation in adults, such as occurs in tumours, wounds and ischaemic tissues.
Acknowledgments
The work of the authors is supported by grants from the NIH (R01-HL070866, R01-HL089252, R01-CA115767, P01-CA80124, R01-CA85140, R01-CA126642, K08-HL071938 and a Federal Share/NCI Proton Beam Program Income Grant). We thank Dr LL Munn for help with quantitative measurements and C Koppel for technical support.
Supporting Information
Fig. S1 CPC morphology is distinct from that of maturehematopoietic cells circulating in lung capillaries. Representativeimage of CPC for comparison with a circulating PMN, eosinophil,mast cell and monocyte. Each cell type is clearly identified by itscharacteristic arrangement of nuclear heterochromatin andcytoplasmic organelles. A dense arrangement of free ribosomes andpaucity of organelles other than mitochrondria characterize thecytosol of the CPC (a and b); in addition to a highlylobulated nucleus, an electron-dense cytosol with numerous granulescharacterize the PMN (c and d); a lobulated nucleusand granules with a crystalline structure characterize theeosinophil (e); the monocyte is characterized by a typicalhorseshoe-shaped nucleus and a sparse arrangement of nuclearheterochomatin to euchromatin (f). 80 nm-thick epon resinsections stained with uranyl acetate and lead citrate. Bars = 1 mm(a-f).
Fig. S2 CPCs contact endothelium post-HALI. Representative image of CPC forming small contact sites (arrows) with the process of an adjacent endothelial cell post-HALI (week 6). Note the large mitochondria (MT) and dense arrangement of free ribsomes in the CPC cytosol. 80 nm-thick epon resin section stained with uranyl acetate and lead citrate: Bar = 0.5 mm.
Fig. S3 CPCs align as capillary perivascular cellspost-HALI. Region of the alveolar capillary membrane (post-HALI,week 7), with multiple capillary structures (evident byerythrocytes in their lumen) and with CPCs that have crossed acapillary wall into the interstitium (a, see boxed area). Athigher magnification, these cells are seen to retain the typicalmorphology of CPCs within the capillary lumen: note the extendedprocess of the cell aligning as a perivascular cell along theabluminal surface of a capillary wall (b, arrows). Highmagnification illustrating 10 nm protein-A gold labeled CD11bantigenic sites (inset) typically expressed by capillaryperivascular cells in the model (post-HALI, week 6). Red delineatesthe plasmalemmal membrane of CPCs; blue delineates that ofcapillary endothelial cell processes. 80 nm-thick epon resinsections stained with uranyl acetate and lead citrate. Bars = 10 mm(a), 6 mm (b), 0.1 mm (c).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
References
- 1.Dos Santos CC. Hyperoxic acute lung injury and ventilator-induced/associated lung injury: new insights into intracellular signaling pathways. Crit Care. 2007;11:126. doi: 10.1186/cc5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li LF, Liao SK, Ko YS, et al. Hyperoxia increases ventilator-induced lung injury via mitogen-activated protein kinases: a prospective, controlled animal experiment. Crit Care. 2007;11:R25. doi: 10.1186/cc5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med. 2003;9:702–12. doi: 10.1038/nm0603-702. [DOI] [PubMed] [Google Scholar]
- 4.Du R, Lu KV, Petritsch C, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–20. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rafii S, Lyden D, Benezra R, et al. Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy. Nat Rev Cancer. 2002;2:826–35. doi: 10.1038/nrc925. [DOI] [PubMed] [Google Scholar]
- 6.Rajantie I, Ilmonen M, Alminaite A, et al. Adult bone marrow-derived cells recruited during angiogenesis comprise precursors for periendothelial vascular mural cells. Blood. 2004;104:2084–6. doi: 10.1182/blood-2004-01-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anghelina M, Krishnan P, Moldovan L, et al. Monocytes/macrophages cooperate with progenitor cells during neovascularization and tissue repair: conversion of cell columns into fibrovascular bundles. Am J Pathol. 2006;168:529–41. doi: 10.2353/ajpath.2006.050255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones R, Capen D, Cohen KS, et al. A protocol for a lung neovascularization model in rodents. Nat Protoc. 2008;3:378–87. doi: 10.1038/nprot.2007.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones R, Capen D, Cohen KS, et al. A protocol for phenotypic detection and characterization of vascular cells of different origins in a lung neovascularization model in rodents. Nat Protoc. 2008;3:388–97. doi: 10.1038/nprot.2007.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones R. Ultrastructural analysis of contractile cell development in lung microvessels in hyperoxic pulmonary hypertension. Fibroblasts and intermediate cells selectively reorganize nonmuscular segments. Am J Pathol. 1992;141:1491–505. [PMC free article] [PubMed] [Google Scholar]
- 11.Jones R, Capen D, Jacobson M. PDGF and microvessel wall remodeling in adult lung: imaging PDGF-Rbeta and PDGF-BB molecules in progenitor smooth muscle cells developing in pulmonary hypertension. Ultrastruct Pathol. 2006;30:267–81. doi: 10.1080/01913120600820336. [DOI] [PubMed] [Google Scholar]
- 12.Ziegelhoeffer T, Fernandez B, Kostin S, et al. Bone marrow-derived cells do not incorporate into the adult growing vasculature. Circ Res. 2004;94:230–8. doi: 10.1161/01.RES.0000110419.50982.1C. [DOI] [PubMed] [Google Scholar]
- 13.Peinado VI, Ramirez J, Roca J, et al. Identification of vascular progenitor cells in pulmonary arteries of patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2006;34:257–63. doi: 10.1165/rcmb.2005-0255OC. [DOI] [PubMed] [Google Scholar]
- 14.Abe S, Boyer C, Liu X, et al. Cells derived from the circulation contribute to the repair of lung injury. Am J Respir Crit Care Med. 2004;170:1158–63. doi: 10.1164/rccm.200307-908OC. [DOI] [PubMed] [Google Scholar]
- 15.Balasubramaniam V, Mervis CF, Maxey AM, et al. Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: implications for the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1073–84. doi: 10.1152/ajplung.00347.2006. [DOI] [PubMed] [Google Scholar]
- 16.Conejo-Garcia JR, Benencia F, Courreges MC, et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med. 2004;10:950–8. doi: 10.1038/nm1097. [DOI] [PubMed] [Google Scholar]
- 17.Song S, Ewald AJ, Stallcup W, et al. PDGFRbeta+ perivascular progenitor cells in tumours regulate pericyte differentiation and vascular survival. Nat Cell Biol. 2005;7:870–9. doi: 10.1038/ncb1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paine R, 3rd, Wilcoxen SE, Morris SB, et al. Transgenic overexpression of granulocyte macrophage-colony stimulating factor in the lung prevents hyperoxic lung injury. Am J Pathol. 2003;163:2397–406. doi: 10.1016/S0002-9440(10)63594-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones R, Capen D, Jacobson M, et al. PDGF and microvessel wall remodeling in adult rat lung: imaging PDGF-AA and PDGF-Ralpha molecules in progenitor smooth muscle cells developing in experimental pulmonary hypertension. Cell Tissue Res. 2006;326:759–69. doi: 10.1007/s00441-006-0177-5. [DOI] [PubMed] [Google Scholar]
- 20.Greenberg JI, Shields DJ, Barillas SG, et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature. 2008;456:809–13. doi: 10.1038/nature07424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item
Supporting info item