

Abstract
The primary limitation of solution state NMR with larger, highly dynamic, or paramagnetic systems originates from signal losses due to fast transverse relaxation. This is related to the high gyromagnetic ratio γ of protons, which are usually detected. Thus, it is attractive to consider detection of nuclei with lower γ, such as 13C, for extending the size limits of NMR. Here, we present an approach for complete assignment of Cα and N resonances in fast relaxing proteins using a Cα detected 3D CANCA experiment for perdeuterated proteins. The CANCA experiment correlates alpha carbons with the sequentially adjacent and succeeding nitrogen and alpha carbons. This enables elongation of the chain of assigned residues simply by navigating along both nitrogen and carbon dimensions using a “stairway” assignment procedure. The simultaneous use of both Cα and N sequential connectivities makes the experiment more robust than conventional 3D experiments, which rely solely on a single 13C indirect dimension for sequential information. The 3D CANCA experiment, which is very useful for mainchain assignments of higher molecular weight proteins at high magnetic field, also provides an attractive alterative for smaller proteins. Two versions of the experiment are described for samples that are 13C labeled either uniformly or at alternate positions for removing one-bond 13C-13C couplings. In order to achieve both, high resolution and sensitivity, extensive non-uniform sampling was employed. Adding longitudinal relaxation enhancement agents can allow for shorter recycling delays, decreased measuring time, or enhanced sensitivity.
Keywords: NMR, Cα direct detection, resonance assignment, large molecules
Introduction
NMR is an established technique for studying structure and dynamics of macromolecules in solution. However, its use has severe limits when studying large and/or fast relaxing systems, such as multi-protein complexes, membrane proteins, or paramagnetic proteins. In traditional proton detected triple resonance experiments, the limitations originate primarily from relaxation-induced losses of signal intensity. These affect coherences not only during evolution or detection periods but also during magnetization transfers. The large 1H gyromagnetic ratio, which had been key to the performance of a large collection of 1H-detected triple-resonance experiments, is actually a “double-edged sword”. While, it provides a large Zeeman polarization at the beginning of an experiment, the stronger dipolar interaction resulting from the large magnetic moment also causes fast decay during the pulse sequences and detection. Since the peak heights are proportional to the integral over the detected free induction decay (FID) and not only to the initial amplitude, a slowly decaying signal of lower initial FID amplitude may have a superior signal-to-noise ratio (S/N) over a rapidly-relaxing signal of higher FID amplitude. Moreover, conventional 1H-detected experiments suffer from broad line widths of the detected signals leading to poor resolution. Thus, it is reasonable to consider 13C direct detection in these systems.
In the past, 13C direct detection has been demonstrated with carbonyls in uniformly 13C-labeled proteins1, 2 and was quite successful for small paramagnetic proteins. However, the carbonyl CSA becomes an efficient relaxation mechanism in large molecular weight systems, especially at high magnetic fields. In such cases, a Cα-detection experiment is expected to show better performance since Cα, which has a smaller CSA, benefits from slower transverse relaxation if the proteins are deuterated3. However, direct Cα detection is complicated due to the two strong one bond 13C-13C scalar couplings with C′ and Cβ causing crowded spectra and reducing sensitivity by splitting a Cα resonance into multiplets. Spectral complexity can be avoided by computational deconvolution4 or by selecting a single component within the split peaks using IPAP or S3E schemes3, when conventional uniform 13C labeling is used. The required spin manipulation, however, cause a loss in the signal intensity. The undesirable couplings can be eliminated by alternate 13C carbon labeling5, without complicated pulse programs and/or processing, as we described before6. Here, we present a 13Cα detection 3D CANCA experiment for both alternate and uniform 13C labeling that is amenable for studies of large systems at high field. The resulting spectrum provides sequential correlations across all dimensions and can thus be used single-handedly to assign all Cα and N backbone resonances.
Experimental Design
The Cα-direct detected 3D CANCA experiment provides a robust way to establish complete mainchain resonance assignment with simultaneous use of both Cα and N sequential connectivities. The 3D CANCA experiment correlates a given alpha carbon (Cαi) both with its attached nitrogen (Ni) and the nitrogen of the following residue (Ni+1). In another dimension, this alpha carbon is correlated with the Cα of both previous (Cαi-1) and following (Cαi+1) residues. This enables elongation of the chain of assigned residues simply by navigating along both dimensions using the so-called “stairway” assignment procedure7, 8. A Cα-Cα plane at the frequency of ω (Ni) (Figure 1B (cyan shadow) and Figure 1C (cyan planes)) will have four correlations of coordinates (ω(Cαi-1); ω(Cαi-1)), (ω(Cαi-1); ω(Cαi)), (ω(Cαi); ω(Cαi-1)), and (co(Cαi); ω(Cαi)). Thus, Cαi is directly correlated to its predecessor, Cαi-1. The chain is easily extended by inspecting the nitrogen dimension which displays another (ω(Cαi); ω(Cαi)) correlation at ω(Ni+1). At this nitrogen frequency, Cαi is correlated to its successor, Cαi+1. Unlike the previously published 2D NCA experiment, which exclusively relies on nitrogen chemical shifts for sequential connectivities6, the CANCA can establish sequential assignment via both Cα and N nuclei, eliminating ambiguities in case of nitrogen chemical shift degeneracies. Thus, the CANCA experiment alone provides the information for a complete sequence specific assignment, including proline residues.
Figure 1.
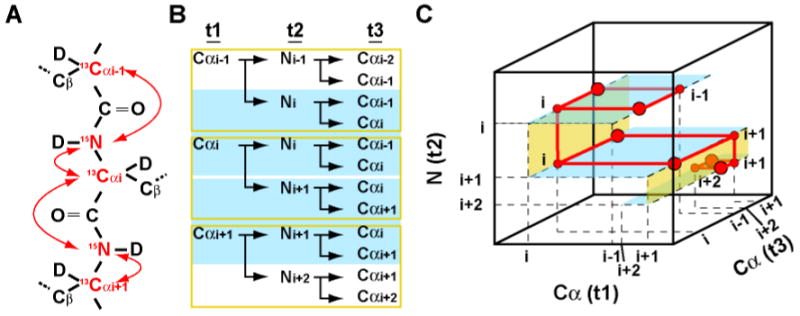
Coherences correlated by the 3D CANCA experiment. (A) Illustration of the coherences correlated with Cαi. The nuclei involved are colored in red and their scalar couplings are indicated by red arrows. (B) Signals and correlations observed in each plane of the N (cyan shadow) or the Cα (yellow box) dimensions. (C) Schematic representation of the 3D CANCA spectrum. Each Cα-Cα plane (colored in blue) has intra-residue and sequential carbon correlations. Sequential connections are also found in nitrogen dimension (yellow plane). Thus the assignment can be easily established by navigating between Cα-Cα planes up and down the “stairway” along the nitrogen dimension
Materials and Methods
All chemicals were purchased from Sigma (St. Louis, MO) unless otherwise noted. All stable-isotope-labeled materials were acquired from Cambridge Isotope laboratories (Cambridge, MA).
Expression and purification of the B domain of protein G (GB1)
The gene for 6His-tagged GB1, consisting of 64 amino acid residues, was cloned into the pET9d vector (Novagen, San Diego, CA) as previously described8. GB1 was expressed in commercially available BL21 (DE3) E.coli cells (Novagen) at 37°C and protein expression was induced for 6 hours at the same temperature. For [2-13C-glyceol] labeled samples, the cells were cultured in 2H15N M9 media containing 8.5 g/L Na2HPO4, 3 g/L KH2PO4, 0.5 g/L NaCl, 2mM MgCl2, 0.1 mM CaCl2, and 1 g/L of 15NH4Cl in D2O, which is supplemented with 2g/L [2-13C] glycerol and 2g/L NaH13CO3. For the uniformly 2H15N13C-labeled samples, the cells were cultured in 2H15N13C M9 media containing 1 g/L of 15NH4Cl, 2 g/L of 2H13C glucose in D2O, The protein was purified with Ni-NTA affinity chromatography as previously described8.
NMR Experiments
NMR spectra were recorded on a Bruker (Billerica, MA) Avance 500 spectrometer equipped with a triple-resonance carbon-cryogenic probe (TXO) designed for carbon detection experiments. All spectra were recorded at 15°C in buffer containing 10 mM sodium phosphate (pH 6.8), 100 mM NaCl and 20% w/v deuterated glycerol in D2O to induce a tumbling corresponding to a 90kDa protein at 25°C6. 3D CANCA experiments were recorded with a spectral width of 3511 Hz for carbon (centered at 55 ppm) and 1519 Hz for nitrogen (centered at 117 ppm). 1024 complexed data points were recorded for direct detection. 500 points out of 128 (Cα) ×128 (N) complex point were sparsely recorded in the indirect dimensions. The data was processed with a high-fidelity forward maximum entropy reconstruction algorithm developed in our laboratory, which conserves all measured time-domain data points and predict the missing data points by an iterative procedure9. The reconstructed time domain data were then multiplied by a cosine window function, zero filled and Fourier transformed. The resolution in the indirect dimensions after apodization and Fourier transformation was 27 Hz for Cα and 11 Hz for N. The recycling delay was optimized to be 1.25 × T1 (longitudinal relaxation time) = 4 sec or 1.2sec, with or without adding paramagnetic relaxation enhancement agents, respectively. All spectra were analyzed with the program Sparky10.
Pulse sequence
Figure 2 shows two alternative versions of the CANCA 3D experiment. Figure 2A displays the pulse sequence for samples with alternate 13C labeling (expressed by incorporating 2-13C glycerol or 1,3-13C glycerol) for removing one-bond 13C-13C scalar coupling. Figure 2B depicts the modifications that need to be implemented for uniformly 13C-enriched samples. The basic principle is common to both experiments and the pulse sequence of Figure 2A will first be described, and the modifications of Figure 2B will be explained later.
Figure 2.
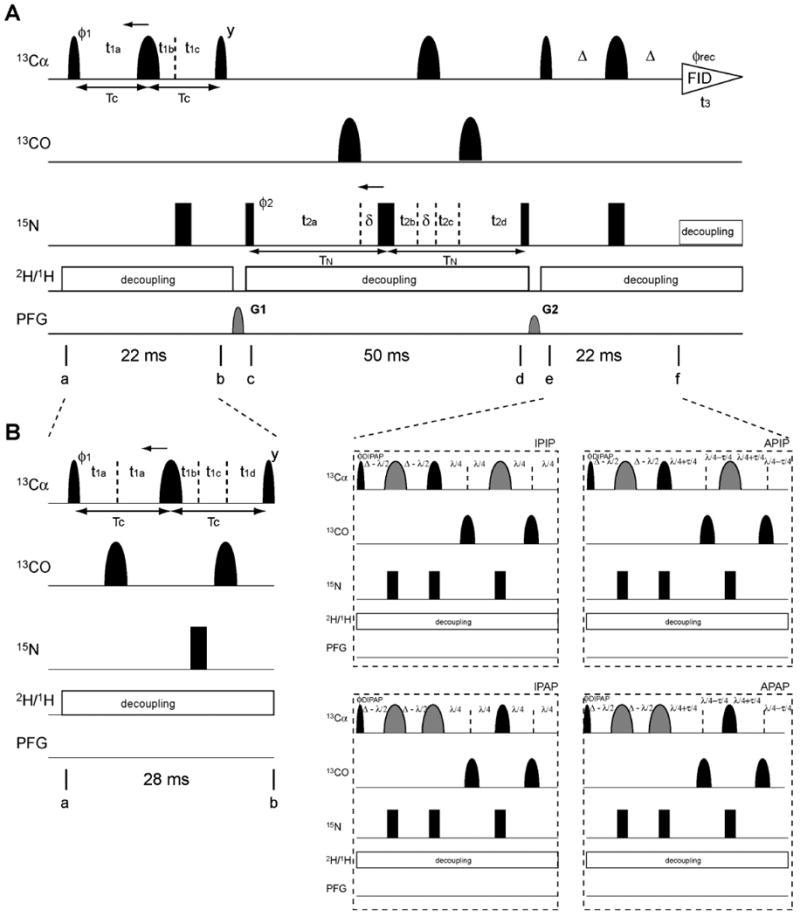
Pulse program of the 3D CANCA experiment optimized for (A) alternate and (B) uniform 13C-labeling. Narrow and wide black bars indicate non-selective π/2 and π pulses, respectively. Narrow and wide semi elliptical shapes on the carbon channel represent π/2 and π Gaussian cascades pulses (Q5 / 256 μs and Q3 / 205 μs, respectively)11 selective for the frequencies of aliphatic carbon nuclei. Wider Gaussian shapes in gray indicate Cα selective Gaussian cascades pulses (Q3 / 1557 μs). All pulses are applied along the x-axis unless otherwise indicated. (A) The delays were TC = Δ = 11 ms and TN = 25 ms, which is optimal for inter-residue connectivities. The short delay δ = 205 μs compensates for a 13C 180° pulse. The delays and increments for the 13C t1 semi-constant time (SCT) period (a-b) are: t1a = t1c = TC; t1b = 5 μs; Δt1a = -TC/n1max; Δt1c = 1/(2SWC)- TC/n1max; Δt1b = 1/(2SWC). If n1max×Δt1a < TC, a constant time (CT) is used. In this case, t1a = t1c = TC and t1b = 5 μs. Δt1a = - Δt1c = 1/(2SWC) and Δt1b = 0. The delays and increments for the 15N t2 SCT period (c-d) are: t2a = t2d = TN = 25 ms; t2b = t2c = 5 μs; Δt2a = -TN/n2max; Δt2b = Δt2d = 1/(2SWN)- TN/n2max; Δt2c = TN/n2max. If n2max×Δt2a < TN, a constant time is used. t2a = t2d = TN = 25 ms; t2b = t2c = 5 μs. Δt2a = Δt2b = Δt2d = Δt2c = 1/4(SWN). n1max and n2max are the values of the maximum Nyquist grid points in each dimension. In our experiment, both Cα and N are incremented in the SCT fashion (128 (Cα) × 128 (N) complex points, SWC = 3511 Hz, SWN = 1518 Hz). The phase cycle employed was ϕ1 = (x, -x), ϕ2 = (x, x, -x, -x), ϕrec = (y, -y, -y, y). Phase sensitive spectra in the 13C(t1) and 15N(t2) dimensions are obtained by incrementing the phases ϕ1 and ϕ2, respectively, in a States-TPPI manner12. The recycling delay is optimized based on longitudinal relaxation rates. The sine-shaped pulsed field gradients were all applied for 1.0 ms with maximum intensities of G1 = 19 G/cm (gz) and G2 = 16 G/cm (gz). Deuterium and proton decoupling are achieved by using WALTZ16 sequence (3.1 kHz and 1kHz, respectively). (B) The delays were TC = 14 ms, Δ = 11 ms and TN = 25 m, which is optimal for inter-residue connectivities. For IPAP, λ = 14.4 ms and τ = 9 ms were used. The short delay δ = 205 μs compensates for a 13C 180° pulse. The delays and increments for 13C t1 semi-CT period are: t1a = t1d = Tc/2; t1b = 3.3 ms; t1c = 3.8 ms; Δt1a = -Tc/n1max; Δt1b = 1/(2SWC)- 2TC/n1max; Δt1c = TC/n1max; δt1d = 1/(2SWC) - 2TC/n1max. If n1max×δt1a < TC, a constant time is used. t1a = t1d = Tc/2; t1b = 3.3 ms; t1c = 3.8 ms; δt1a = δt1c = δt1d = 1/4(SWC); δt1b = 0. The delays and increments for 15N t2 semi-CT or CT periods are the same as in (A). The phase cycle employed was ϕ1 = (x, -x), ϕ2 = (x, x, -x, -x), ϕDIPAP = (x, x, x, x, -x, -x, -x, -x) for IPIP and APAP, (-y, -y, -y, -y, y, y, y, y) for APIP, and (y, y, y, y, -y, -y, -y, -y) for IPAP, ϕrec = (x, -x, -x, x, -x, x, x, -x). Phase sensitive spectra in the 13C(t1) and 15N(t2) dimensions are obtained by incrementing the phases ϕ1 and ϕ2, respectively, in a States-TPPI manner. The recycling delay, pulsed-field gradients, deuterium and proton decoupling were the same as in (A).
A first INEPT module between points a and b converts a carbon Cαi single quantum coherence (SQC) into longitudinal two-spin order between this carbon and its two scalar-coupled nitrogens (Ni and Ni+1). Concomitantly, the coherence is encoded with the Cα chemical shift in either a semi-constant time (SCT) 13, 14 or a constant-time fashion, depending on the value of n1max. The density operator, σ, at point b is thus:
| (1) |
where only the real component of the indirect FID is depicted. SC(1J) = sin(π1JNCa2TC), SC(2J) = sin(π2JNCa2TC), while C stands for a cos of the corresponding arguments. To maximize the amplitude of antiphase coherences, 2TC is set to 1/4JCαN (∼ 22 ms)15-17. Between points c and d, nitrogen SQCs of residues i and i+1 are allowed to evolve under their respective chemical shifts, again either in a semi-constant time or in a constant-time fashion, depending on n2max. Simultaneously, they each evolve under both one-bond and two-bonds scalar couplings with Cα carbons. The terms of the density operator that are detected are:
| (2) |
In which SN(1J) = sin(π1JNCa2TN), SN(2J) = sin(π2JNCa2TN) and C stand for a cosine of the corresponding arguments. The duration of the delay TN is optimized by analogy to what is described for the hNcaNH experiment:18
| (3) |
Where TN* is the optimal value of the delay TN and R2 is the nitrogen single-quantum relaxation rate for a deuterated amide group. Inversion pulses are applied to carbonyl carbon nuclei to refocus the evolution under 1JNiC'i-1. A reversed INEPT (e-f) then converts nitrogen SQCs into the detected Cα single quantum coherences:
| (4) |
where Thus, the Cα of a given residue is correlated to those of its following and preceding residues, as well as to its own nitrogen and the nitrogen of its successor. This results in the sets of correlations that have been described above and that allow for a straightforward stairway assignment procedure.
For uniformly 13C enriched proteins, the modifications of Figure 2B need to be implemented. The initial value of the semi-constant time period 2TC is set to 1/1J(CαCβ) to minimize losses due to scalar couplings between alpha carbons and beta carbons. The evolution in t1 under this coupling is scaled from 1/SW(C) (for chemical shift evolution) to 1/SW(C) -2TC/nmax. In our implementation this results in a residual coupling of 8.1 Hz. Alternatively, a Cα selective pulse can be used for a relaxation optimized transfer, which leads to the shortening of the period “a-b” from 28 ms (1/1J(CαCβ) to 22 ms (1/41J(CαN). The reduction of the transfer duration results in about 20% of sensitivity gain for most residues. However, the signals from Gly, Ser, and some high-field Cα resonances become weaker as a draw back (data not shown). In addition, this Cα selective pulse does not eliminate evolution under 1J(CαCβ) during t1, which broadens the lines and may even split each signal into a doublet at high resolution. The Cα selective pulse can however be designed to only affect the Cβ of subset of residues and obtain additional information to identify the type of amino acid residues19. It is worth emphasizing that, if alternate 13C labeling is used, the first (a-b) and last (e-f) INEPT transfer periods can be set to the optimal (22 ms) without having any broadening caused by the unwanted scalar coupling evolution during t1. Thus, the highest sensitivity would be expected for those residues that are ∼100% 13Cα labeled in alternate sampling (Ala, Cys, Gly, His, Lys, Phe, Ser, Trp, Tyr, and Val) 6. Inversion pulses are applied to carbonyl carbon nuclei to refocus the evolution under 1J(CαC′). Multiplet patterns that would occur during detection due to 1J(CαCβ) and 1J(CαC′) are converted to single signals by using the double IPAP technique20.
Because the line widths of all signals are very small, the experiment is best exploited when using non-uniform sampling21. In our application, 500 out of 16384 indirect data points were sampled from a Nyquist grid of 128 (Cα) × 128 (N) complex points. Thus, only 3% of the indirect time domain points were recorded. The data were reconstructed with the forward maximum entropy (FM) algorithm developed in our laboratory9, 22. The final resolution in the indirect dimensions after Fourier transforms is 27 Hz for Cα and 11 Hz for N.
Results and discussion
The method was tested on a 4 mM solution of the B1 domain of protein G (GB1) uniformly enriched in 13C. To simulate the tumbling of a 90 kDa protein, 20% glycerol was added and the experiment was recorded at 285K (for a calibration of the correlation time, see Takeuchi et al6). Figure 3 shows an example of a “stairway” assignment for a small stretch of sequential residues. Each Cα-Cα plane shows the correlations between sequentially neighboring carbons at a given nitrogen frequency.
Figure 3.

Stairway sequential assignment of GB1 with the CANCA experiment recorded at conditions simulating a 90 kDa protein (20% glycerol, 285K). Red crosses indicate the correlations expected from square pattern in Cα-Cα plane. Blue arrows indicate the assignment walk in the “stairway” procedure from the Cα of residue 19 to the N of residue 23 (from top to bottom). Starting from Cαi in a Cα-Cα plane at ω (Ni), one can find the nitrogen frequency of the preceding residue ω (Ni+1) at the Cαi cross section in a Cα-N plane (strips on the side). The Cα-Cα plane at ω (Ni+1) gives the assignment of Cαi+1. Slices at the position of horizontal broken lines are shown in Cα-Cα planes.
As can be seen on the 1-dimensional traces of each panel, the S/N ratio is high and the lines are narrow due to the slow transverse relaxation of Cα. A strip along the indirect nitrogen dimension (shown at the right) at a given Cα chemical shift (diagonal peaks) allows finding the Cα-Cα plane containing the signals of the succeeding (or preceding) residues. Since the delays in the pulse program were set to optimize magnetization transfers for sequential Cαi-Cαi+1 (or Cαi-Cαi-1) correlations (50 ms in our case; see description of pulse program and Figure 2), “diagonal” Cαi-Cαi correlations tend to be missing or weak. However, the positions of these correlations can easily be obtained from the square cross-peak patterns in the Cα-Cα planes.
For the 4 mM uniformly 2H15N13C-labeled GB1 sample at conditions simulating a “90 kDa” protein all sequential connectivities (Cα and N) are observed in the CANCA experiment recorded within 2 days (Figure 4A). A uniformly 13C-labeled sample would primary be used to perform assignments, since Cα labeling efficiency in alternate 13C labeling is low for certain residues resulting in the loss (or reduction in intensity) of sequential Cα–Cα correlations for some residue types5. This is clearly exemplified by correlations involving Leu, in which Cα positions are not labeled when using 2-13C glycerol (Figure 4B). However, the alternate 13C-labeling can be used as complement in large molecular weight system, especially for residues with faster relaxation, since the labeling allows using the scheme of Figure 1A, which has a shorter Cα-transverse period, and is not involving Cα-selective pulses or double IPAP coherence selection scheme. In addition, no resonances suffer from imperfect inversion by the Cα-selective pulses (Gly, Ser, and some high-field Cα resonances) in contrast to what may happen with the scheme of Figure 1B. For the “90 kDa” GB1 sample, the median of S/N values for the 2-13C glycerol labeling experiment is ∼30% higher than that for the pulse sequence designed for and applied to the uniformly labeled sample (700 for 2-13C glycerol labeling experiment and 540 in uniformly labeling). Thus, the sensitivity of the 2-13C glycerol labeling experiment is superior to uniform labeling for most of the resonances. In addition, Ile, Leu and Val can readily be identified since these residues have a 13C in the Cβ position when using alternate 13C-labeling (Ile and Val with 2-13C glycerol labeling, and Leu with 1,3-13C glycerol labeling). Thus, cross peaks have opposite sign due to the Cα–Cβ coupling. For the “90 kDa” GB1 sample prepared with 2-13C glycerol labeling, all sequential connectivities are observed except for those involving Leu as discussed above (Figure 4B). The remaining correlations should readily be observed with this experiment when using 1, 3-13C glycerol for labeling.
Figure 4.
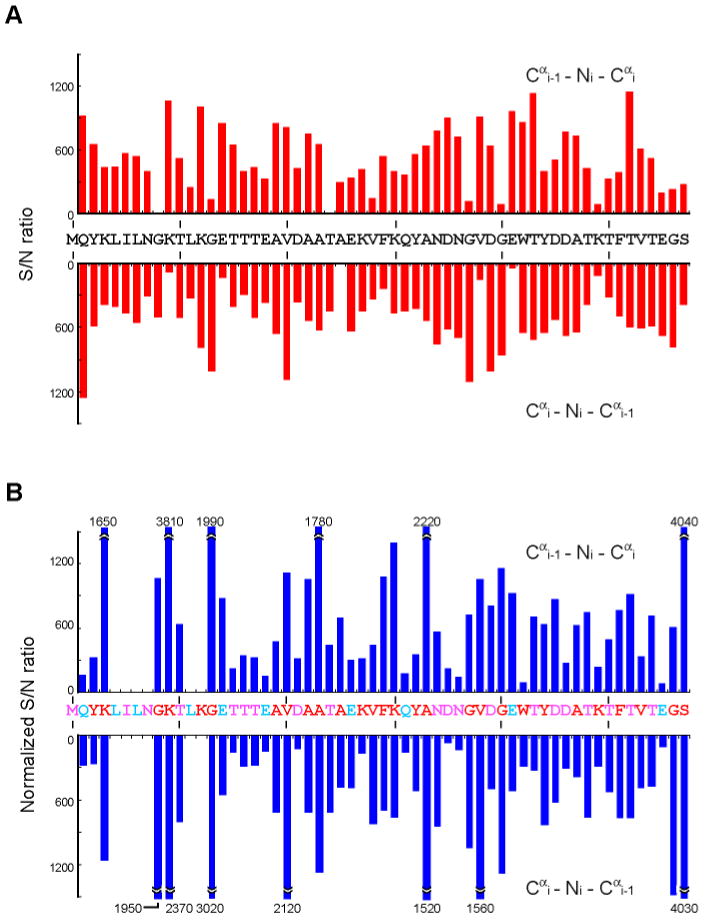
S/N ratio in CANCA experiments with (A) 4 mM uniformly 2H15N13C-labeled GB1 samples and (B) 5 mM 2-13C glycerol labeling at conditions simulating “90 kDa” proteins. Experiments were recorded for 2 days for uniformly 2H15N13C-labeled or 4 days for 2-13C glycerol labeling samples with the delays TN optimized for sequential connectivities. The S/N ratio for (B) was normalized against (A) to account for differences in concentrations and measuring times. Upper and lower bars at positions i indicate Cαi-1-Ni-Cαi and Cαi-Ni-Cαi-1 correlations, respectively. For (B), residues that show S/N ratio of more than 1500 were clipped and the actual values are indicated. Residues colored in red are more than 80 % 13Cα labeled with [2-13C] glycerol, while cyan residues have less than 20% incorporation. Pink residues are 40-70% 13Cα labeled. The noise level was estimated from the median of the absolute intensity values of 10000 randomly selected points in the spectrum.
We also recorded the Cα-Cα plane of a 3D CANCA experiment for the 52 kDa dimeric GST protein. For a 1mM sample of the [ul-2H,15N,13C] GST dimer (2mM monomer concentration) dissolved in D2O buffer. Without optimizing solution conditions we could observe ∼35% of the expected resonances in 5 days experiment. The observed resonances are reasonably narrow and well dispersed, indicating the applicability of the CANCA experiment to higher molecular weight protein systems (Supplement Figure 1). It is also clear that further improvements need to be introduced at the hardware level as well as in data acquisition and processing to enable a routine use of this experiment for very large proteins.
To compare the sensitivity of the CANCA experiment with other carbon detection experiments, we measured 1D traces of the CANCA and CANCO experiments. The latter was proposed previously for establishing sequential connectivities. The experiments were recorded at various temperatures in a viscous buffer using uniformly labeled GB1, and intensities of well-separated resonances from structured regions were compared (Supplement Figure 2). At conditions simulating the low molecular limit (∼6 kDa), CANCO is more sensitive than CANCA (CANCO:CANCA ∼ 1:0.6) as expected. However, the CANCA experiment shows comparable sensitivity at conditions simulating larger systems (>73 kDa) even with a 500 MHz magnet. However, it yields more useful sequential correlations than the CANCO. The CANCA experiment gives better 13C dispersion as well as additional sequential information in the nitrogen dimension compared to the CANCO experiment. Thus, CANCA has clear advantages for the higher molecular weight range. The advantages of CANCA would be more pronounced at higher magnetic fields. However, we would like to emphasize that the CANCA experiment is also useful for small proteins since it provides additional sequential information, and allows resolving ambiguities during the assignment process. For a system with less severe relaxation losses, the proton detected hNCAnH experiment provides sequential information in both indirect dimensions (N and CA), and is the method of choice given the increased polarization of proton nuclei.7 However, it is worth noting that the CANCA experiment is able to assign proline residues, which are missing in proton-detected experiments.
Since the longitudinal relaxation time (T1) of deuterated carbon is especially long in large molecular weight systems, the repetition delay has to be carefully set to have optimal sensitivity. The sensitivity of the experiment can be further enhanced by shortening T1 using paramagnetic reagents, such as Gd(DTPA-BMA) or Ni(DO2A) without significantly accelerating transverse relaxation rates23, 24. Indeed, addition of 3 mM Gd(DTPA-BMA) to the “90 kDa” sample decreased T1 values from 3.6 sec to 0.9 sec. This allowed faster recycling rates and acquisition of more scans per increment. It increased the sensitivity of the experiment by ∼50% (data not shown). It seemed possible that the paramagnetic relaxation enhancement would be less effective for the inaccessible core of very large proteins. We tested this with studies of the 52 kDa dimeric protein GST. We found, however, that the 13Cα T1 values of GST were also significantly shortened from approximately 3.6 sec to 1.3 sec, by addition of 4mM Gd (DTPA-BMA) (data not shown). This included residues from the core of the dimeric protein.
Conclusion
In summary, the 3D CANCA Cα-direct-detection experiment provides a robust way for establishing complete main-chain resonance assignment with simultaneous detection of Cα and N sequential connectivities. Using slowly relaxing Cα nuclei is especially beneficial for assigning high molecular weight proteins. Both conventional uniform 13C labeling or the alternate 13C labeling strategy5, 6 are suitable for the CANCA experiment, with complementary advantages as described above. The experiments described here were recorded on a 500 MHz spectrometer. The performance will be enhanced at higher magnetic fields. This is in contrast to C′ detecting experiments, which deteriorate at higher fields due the large C′ chemical shift anisotropy. Obtaining sequential information in both Cα and N dimensions is especially beneficial for resolving degeneracies, which makes this experiment attractive even for smaller molecular weight systems. An extensive sparse sampling coupled with FM reconstitution further enhances these features by achieving high resolution in indirect dimensions.
Supplementary Material
Acknowledgments
This work was supported by the NIH (grants AI37581, GM47467 and EB 002026).
Footnotes
Supporting Information Available. Cα-Cα plane of CANCA 3D experiments for 52 kDa GST protein and comparison of signal intensity in CANCA and CANC' experiment are shown in supplemental figures. The material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bermel W, Bertini I, Felli IC, Lee YM, Luchinat C, Pierattelli R. J Am Chem Soc. 2006;128:3918–9. doi: 10.1021/ja0582206. [DOI] [PubMed] [Google Scholar]
- 2.Bermel W, Bertini I, Felli IC, Kummerle R, Pierattelli R. Journal of the American Chemical Society. 2003;125:16423–16429. doi: 10.1021/ja037676p. [DOI] [PubMed] [Google Scholar]
- 3.Bermel W, Bertini I, Felli IC, Matzapetakis M, Pierattelli R, Theil EC, Turano P. J Magn Reson. 2007;188:301–10. doi: 10.1016/j.jmr.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Shimba N, Stern AS, Craik CS, Hoch JC, Dotsch V. J Am Chem Soc. 2003;125:2382–3. doi: 10.1021/ja027973e. [DOI] [PubMed] [Google Scholar]
- 5.LeMaster DM, Kushlan DM. J Am Chem Soc. 1996;118:9255–9264. [Google Scholar]
- 6.Takeuchi K, Sun ZY, Wagner G. J Am Chem Soc. 2008;130:17210–1. doi: 10.1021/ja806956p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frueh DP, Arthanari H, Koglin A, Walsh CT, Wagner G. Journal of the American Chemical Society. 2009 doi: 10.1021/ja9046685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frueh DP, Arthanari H, Wagner G. J Biomol NMR. 2005;33:187–96. doi: 10.1007/s10858-005-3204-z. [DOI] [PubMed] [Google Scholar]
- 9.Hyberts SG, Heffron GJ, Tarragona NG, Solanky K, Edmonds KA, Luithardt H, Fejzo J, Chorev M, Aktas H, Colson K, Falchuk KH, Halperin JA, Wagner G. J Am Chem Soc. 2007;129:5108–16. doi: 10.1021/ja068541x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kneller, T. D. G. a. D. G., University of California, San Francisco.
- 11.Emsley L, Bodenhausen G. Journal of Magnetic Resonance (1969) 1992;97:135–148. [Google Scholar]
- 12.Marion D, Ikura M, Tschudin R, Bax A. Journal of Magnetic Resonance (1969) 1989;85:393–399. [Google Scholar]
- 13.Grzesiek S, Bax A. Journal of Biomolecular NMR. 1993;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- 14.Logan TM, Olejniczak ET, Xu RX, Fesik SW. Journal of Biomolecular NMR. 1993;3:225–231. doi: 10.1007/BF00178264. [DOI] [PubMed] [Google Scholar]
- 15.Kay LE, Ikura M, Tschudin R, Bax A. Journal of Magnetic Resonance (1969) 1990;89:496–514. doi: 10.1016/j.jmr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Cavanagh, J.; Fairbrother, W. J.; III, A. G. P.; Skelton, N. J.; Rance, M., AcademicPress: San Diego 1996.
- 17.Sattler M, Schleucher Jg, Griesinger C. Progress in Nuclear Magnetic Resonance Spectroscopy. 1999;34:93–158. [Google Scholar]
- 18.Frueh DP, Sun ZY, Vosburg DA, Walsh CT, Hoch JC, Wagner G. J Am Chem Soc. 2006;128:5757–63. doi: 10.1021/ja0584222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun ZY, Frueh DP, Selenko P, Hoch JC, Wagner G. J Biomol NMR. 2005;33:43–50. doi: 10.1007/s10858-005-1284-4. [DOI] [PubMed] [Google Scholar]
- 20.Bermel W, Bertini I, Felli IC, Piccioli M, Pierattelli R. Progress in Nuclear Magnetic Resonance Spectroscopy. 2006;48:25–45. [Google Scholar]
- 21.Barna JCJ, Laue ED, Mayger MR, Skilling J, Worrall SJP. Journal of Magnetic Resonance (1969) 1987;73:69–77. [Google Scholar]
- 22.Hyberts S, Frueh D, Arthanari H, Wagner G. Journal of Biomolecular NMR. doi: 10.1007/s10858-009-9368-1. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eletsky A, Moreira O, Kovacs H, Pervushin K. J Biomol NMR. 2003;26:167–79. doi: 10.1023/a:1023572320699. [DOI] [PubMed] [Google Scholar]
- 24.Cai S, Seu C, Kovacs Z, Sherry AD, Chen Y. J Am Chem Soc. 2006;128:13474–8. doi: 10.1021/ja0634526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.