

Abstract
A total synthesis of the Aspidosperma alkaloids (+)-fendleridine (1) and (+)-1-acetylaspidoalbidine (2) is detailed, providing access to both enantiomers of the natural products and establishing their absolute configuration. Central to the synthetic approach is a powerful intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of a 1,3,4-oxadiazole in which the pentacyclic skeleton and all the stereochemistry of the natural products are assembled in a reaction that forms three rings, four C–C bonds, five stereogenic centers including three contiguous quaternary centers, and introduces the correct oxidation state at C19 in a single synthetic operation. The final tetrahydrofuran bridge is subsequently installed in one-step, enlisting an intramolecular alcohol addition to an iminium ion generated by nitrogen assisted opening of the cycloadduct oxido bridge, with a modification that permits release of useful functionality (a ketone) at the cleavage termini.
Introduction
Fendleridine (1) and 1-acetylaspidoalbidine (2) are the parent members of the aspidoalbine family of alkaloids.1 Fendleridine (aspidoalbidine, 1) was first isolated in 1964 from the Venezuelan tree Aspidosperma fendleri WOODSON by Burnell (Figure 1),2 and later in 1979 from the bark of the Venezuelan tree Aspidosperma rhombeosignatum MARKGRAF by Medina and co-workers.3 In addition, fendleridine (1) represents the parent compound to 1-acetylaspidoalbidine (2) that was first disclosed in 1963 by Djerassi, having been isolated from Vallesia dichotoma RUIZ et PAV in Peru,4 and that has been referred to as dehydroxyhaplocidine5 in a more recent isolation. Unique to the aspidoalbines is the oxidized C19 N,O-ketal embedded in the characteristic Aspidosperma alkaloid pentacyclic ring system typically linked to an oxidized C5 substituent. More highly oxidized Aspidosperma alkaloids have been disclosed bearing the hexacyclic core structure of 1 and 2 that incorporate further hydroxylation of the aromatic ring, incorporate a five-membered lactone versus tetrahydrofuran, or incorporate further unsaturation in the six-membered rings.6
Figure 1.

Natural product structures.
The only total synthesis of fendleridine (1) disclosed to date was reported in 1976 by Ban and co-workers, and relied on a C19 oxidation and subsequent cyclization to install the C19 N,O-ketal (Hg(OAc)2 in 5% aq. AcOH) forming the tetrahydrofuran ring bridging an alcohol on the two carbon side chain at C5 to C19,7 confirming the originally proposed structure. A year earlier, Ban disclosed the first total synthesis of 1-acetylaspidoalbidine (2) from a common intermediate,8 and inferred that it could not be readily de-acetylated to provide fendleridine (1). Ban subsequently reported an improved formal synthesis of 2 in 1987,9 and an impressive formal synthesis of 1-acetylaspidoalbidine (2) was disclosed in 1991 by Overman, where entry into an advanced pentacyclic intermediate was accomplished using a signature aza-Cope-Mannich rearrangement.10 In these efforts, fendleridine (1) and 1-acetylaspidoalbidine (2) were prepared in racemic form, and the assignment of their absolute configuration remain to be unambiguously established.11
In the course of the development of an approach to the total synthesis of members of the Aspidosperma alkaloids including vindoline12 and its extension to the total synthesis of vinblastine13,14 and vincristine,14 we introduced a powerful tandem [4 + 2]/[3 + 2] cycloaddition cascade reaction of 1,3,4-oxadiazoles that is especially suited for the preparation of their pentacyclic ring system.15 Herein, we report the extension of these studies in the total synthesis of 1 and 2 that is even more ideally suited for implementation of the cascade cycloaddition reaction, taking advantage of the cycloadduct intrinsic oxidation state at C19 for closure of the tetrahydrofuran ring and with a modification that permits introduction of useful functionality following cleavage of the cycloadduct oxido bridge (Figure 2).
Figure 2.

Key cycloaddition cascade and retrosynthetic analysis.
The key reaction cascade is initiated by an intramolecular [4 + 2] cycloaddition reaction of a 1,3,4-oxadiazole with a tethered dienophile,16,17 where the intrinsic regioselectivity is dictated by the linking tether. Following the initiating [4 + 2] cycloaddition, loss of N2 from the initial cycloadduct provides an intermediate 1,3-dipole, which is stabilized by the substitution at the dipole termini. The intrinsic dipole/dipolarophile regioselectivity of the ensuing 1,3-dipolar cycloaddition is reinforced by the linking tether, and the relative stereochemistry is dictated by an exclusive endo indole [3 + 2] cycloaddition, where the dipolarophile is sterically directed to the face opposite the newly formed fused lactam.15,18 In total, four C–C bonds, three rings, five relative stereogenic centers including a C19 N,O-ketal, and the complete natural product skeleton are assembled in a single step. Subsequent adjustment of the C3 substituent and acid-catalyzed oxido bridge cleavage, with intermediate generation and reaction of an iminium ion flanked by two quaternary centers, leads to introduction of the sixth tetrahydrofuran ring, with release of a stable cyanohydrin precursor to a C3 ketone.
Results and Discussion
The required 1,3,4-oxadiazole 5 bearing the tethered indole dipolarophile was prepared from 1-benzyltryptamine (7,19 Scheme 1) in a 3-step sequence. Treatment of 7 with 1,1-carbonyldiimidazole (CDI) afforded urea 8 (97%), which was reacted with methyl oxalyl hydrazide 920 to provide 10. Dehydration of 10 by treatment of TsCl and Et3N afforded the oxadiazole 5 (81% for 2-steps).
Scheme 1.

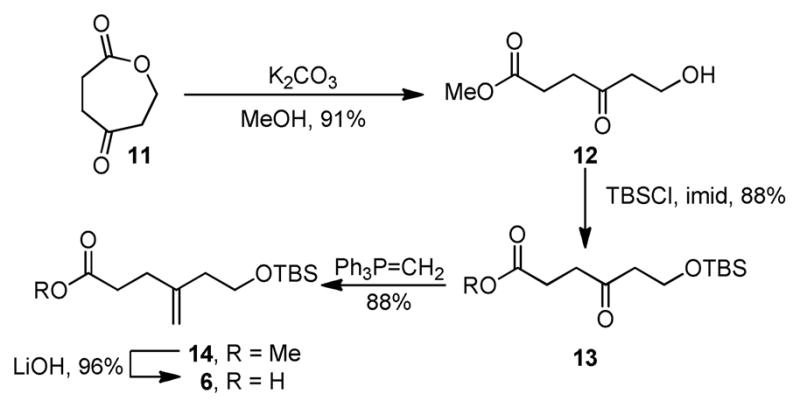
The acyl chain carboxylic acid 6 containing the tethered initiating dienophile was prepared in 4-steps from oxepane-2,5-dione (11,21 Scheme 2), which was subjected to methanolysis to afford methyl ester 12 (91%), and subsequent silyl ether protection of the resulting primary alcohol yielding 13 (88%). Wittig olefination of 13 to provide 14 (88%), and hydrolysis of the methyl ester afforded 6 (96%).
Scheme 2.
Coupling of 1,3,4-oxadiazole 5 with 6 was effected by treatment with EDCI (1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride) and DMAP to provide 4 (74%), the precursor for the [4 + 2]/[3 + 2] cycloaddition cascade (Scheme 3). The tandem cycloaddition reaction occurred cleanly at 180°C in o-dichlorobenzene to afford 3 in yields as high as 71% (55–71%) as a single diastereomer.22 The relative stereochemistry of 3 was assigned based on NMR and was in accord with expectations, which was later confirmed through X-ray analysis of 20.23 Treatment of 3 with NH3 and MeOH effected clean conversion to the primary amide that underwent subsequent dehydration to afford the nitrile 16 (90%, 2-steps).
Scheme 3.

Subsequent introduction of the tetrahydrofuran ring with formation of the C19 N,O-ketal proved straightforward (Scheme 4). Cleavage of the t-butyldimethylsilyl ether with Bu4NF in acetic acid afforded the primary alcohol 17 that could be converted to the cyanohydrin 18 by further treatment with formic acid in methanol. More conveniently, treatment of 16 with HF/pyridine provided a direct single-step conversion to the stable cyanohydrin 18 (quantitative). The reversibly generated iminium ion derived from oxido bridge protonation and opening is flanked by two quaternary centers facilitating direct trap by the pendant alcohol. Diastereoselective ketone reduction conducted directly on the cyanohydrin 18 was effected with Na-selectride to yield alcohol 19 (85%). Although of no consequence since the alcohol is subsequently removed, the reduction proceeds by hydride delivery from the more hindered concave face of the ketone, which adopts a boat conformation,23a producing the secondary alcohol occupying an equatorial position in a six-membered ring also adopting a boat conformation.23b Presumably, this preferential axial approach of the large hydride reducing agent from the most hindered face avoids an apparently more significant destabilizing electrostatic interaction between the reagent and the adjacent axial indoline nitrogen.
Scheme 4.

Consistent with challenges inherent in the reduction of the ketone derived from 18, the use of NaBH4 in methanol required longer reaction times, resulted in poorer conversion and lower diastereoselectivity, and ketone often persisted in the reaction. Additionally, the protecting group on the indoline nitrogen proved important at this stage of the synthesis. Initially, a carboxybenzyl (CBz) protecting group was utilized in our efforts, but afforded the transesterified oxazolidinone derived from intramolecular displacement of benzyl alcohol when the alcohol was exposed to basic conditions. These limitations were addressed through the use of the N-benzyl group to protect the indoline nitrogen.
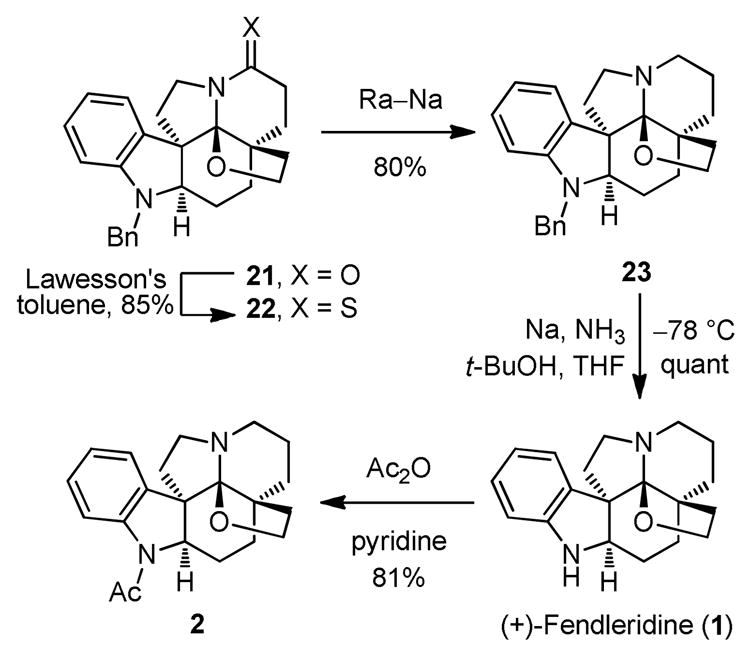
With the alcohol in hand, subsequent conversion of 19 to the methyldithiocarbonate 20 (4.7 equiv of NaH, 4.0 equiv of CS2, THF, 0°C, 1 h followed by 3.0 equiv of MeI, 25°C, 2 h, 92%) was readily effected (Scheme 4). Separation of the enantiomers of 20 (α= 1.39) was carried out on a semipreparative Daicel ChiralCel OD column (2 × 25 cm, 30% i-PrOH-hexane, 7 mL/min flow rate) to provide natural-(−)-20 (tR = 19.2 min) and ent-(+)-20 (tR = 26.5 min). The natural enantiomer (shown), whose absolute configuration was established with a single crystal X-ray structure determination of (−)-20 bearing a heavy atom (S),23b was converted to and matched the absolute configuration of (+)-1-acetylaspidoalbidine (2) whose optical rotation was previously reported.4,5 Deoxygenation of 20 was accomplished under Barton–McCombie conditions (Bu3SnH, cat. AIBN, toluene, 100°C, 1 h, 77%) to provide 21.
Treatment of amide 21 with Lawesson’s reagent24 cleanly afforded the thiolactam 22 (85%, Scheme 5). Although initial efforts focused on a single step desulfurization and cleavage of the N-benzyl protecting group through extended treatment with Ra–Ni, its removal from the final product proved difficult. Consequently, the desulfurization and debenzylation were accomplished in a two-step sequence. The thioamide was removed first by reductive desulfurization of 22 upon treatment with Ra–Ni to yield 23 (80%). The N-benzyl group on 23 was then removed by the reaction with Na and t-BuOH in THF–NH3,25 cleanly affording (+)- and ent-(−)-fendleridine (1, 100%), and requiring no further purification. Treatment of each enantiomer of fendleridine (2) with Ac2O in pyridine cleanly afforded (+)- and ent-(−)-1-acetylaspidoalbidine (2, 81%), respectively. Both (+)-1 ([α]D25 +43 (c 1.1, CHCl3)) and (+)-2 ([α]D25 +38 (c 0.2, CHCl3) and +42 (c 0.2, MeOH)) were identical in all respects with the properties reported for the naturally derived materials,4,5 and the 1H NMR of synthetic 2 was in full agreement with a copy of the spectrum of authentic 2. The optical rotation for fendleridine (1) has not been reported, and that of 1-acetylaspidoalbidine (2) has been reported as +46 (CHCl3)4 and +1 (c 0.2, CHCl3).5 The origin of these differences is not known, but our synthetic 2 matches closely that reported in the original Djerassi work,4 and the consistent dextrorotatory sign of the rotations recorded with naturally-derived material was used for the absolute configuration assignments.
Scheme 5.
Conclusion
Full details of the development of the total synthesis of (+)- and ent-(−)-fendleridine (1), and (+)- and ent-(−)-1-acetylaspidoalbidine (2) and the assignment of their absolute configuration are disclosed by enlisting an intramolecular tandem [4 + 2]/[3 + 2] cycloaddition cascade of a 1,3,4-oxadiazole that allowed the assembly of the entire natural product skeleton and all necessary stereochemistry in a single step, while also directly providing the desired oxidation state at C19, and facilitating ring closure of the tetrahydrofuran.
Supplementary Material
Acknowledgments
We thank Professor Zèches–Hanrot for providing the 1H NMR spectrum of authentic natural 1-acetylaspidoalbidine and Dr. R. Chadha for the X-ray structure determination of 20. We gratefully acknowledge the financial support of the National Institutes of Health (CA042056) and the Skaggs Institute for Chemical Biology. AZ is a NSF and Skaggs Fellow.
Footnotes
Supporting Information Available: Full experimental details are provided. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Hesse M. Indolalkaloide in Tabellen-Ergänzungswerk. Springer-Verlag; Berlin: 1968. p. 77. [Google Scholar]
- 2.Burnell RH, Medina JD, Ayer WA. Can J Chem. 1966;44:28. [Google Scholar]
- 3.Medina JD, Genova LD. Planta Med. 1979;37:165. [Google Scholar]
- 4.(a) Brown KS, Budzikiewicz H, Djerassi C. Tetrahedron Lett. 1963;4:1731. [Google Scholar]; (b) Walser A, Djerassi C. Helv Chim Acta. 1965;48:391. [Google Scholar]
- 5.Mitaine AC, Mesbah K, Richard B, Petermann C, Arrazola S, Moretti C, Zeches-Hanrot M, Le Men Olivier L. Planta Med. 1996;62:458. doi: 10.1055/s-2006-957939. [DOI] [PubMed] [Google Scholar]
- 6.Saxton JE. In: The Alkaloids. Cordell AG, editor. Vol. 51. 1998. p. 1. [Google Scholar]
- 7.Honma Y, Ohnuma T, Ban Y. Heterocycles. 1976;5:47. [Google Scholar]
- 8.Ban Y, Ohnuma T, Seki K, Oishi T. Tetrahedron Lett. 1975;16:727. [Google Scholar]
- 9.Yoshido K, Sakuma Y, Ban Y. Heterocycles. 1987;25:47. [Google Scholar]
- 10.Overman LE, Robertson GM, Robichaud AJ. J Am Chem Soc. 1991;113:2598. [Google Scholar]
- 11.For the total synthesis of the related alkaloid aspidophytine, see: He F, Boy Y, Altom JD, Corey EJ. J Am Chem Soc. 1999;121:6771.Sumi S, Matsumoto K, Tokuyama H, Fukuyama T. Tetrahedron. 2003;59:8571. doi: 10.1021/ol034445e.Mejia-Oneto JM, Padwa A. Org Lett. 2006;8:3275. doi: 10.1021/ol061137i.Marino JP, Cao G. Tetrahedron Lett. 2006;47:7711.Nicolaou KC, Dalby SM, Majumder U. J Am Chem Soc. 2008;130:14942. doi: 10.1021/ja806176w.
- 12.(a) Ishikawa H, Elliott GI, Velcicky J, Choi Y, Boger DL. J Am Chem Soc. 2006;128:10596. doi: 10.1021/ja061256t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Choi Y, Ishikawa H, Velcicky J, Elliott GI, Miller MM, Boger DL. Org Lett. 2005;7:4539. doi: 10.1021/ol051975x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ishikawa H, Boger DL. Heterocycles. 2007;72:95. [Google Scholar]; (d) Elliott GI, Velcicky J, Ishikawa H, Li Y, Boger DL. Angew Chem, Int Ed. 2006;45:620. doi: 10.1002/anie.200503024. [DOI] [PubMed] [Google Scholar]; (e) Yuan ZQ, Ishikawa H, Boger DL. Org Lett. 2005;7:741. doi: 10.1021/ol050017s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wolkenberg SE, Boger DL. J Org Chem. 2002;67:7361. doi: 10.1021/jo020437k. [DOI] [PubMed] [Google Scholar]
- 13.Ishikawa H, Colby DA, Boger DL. J Am Chem Soc. 2008;130:420. doi: 10.1021/ja078192m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishikawa H, Colby DA, Seto S, Va P, Tam A, Kakei H, Rayl TJ, Hwang I, Boger DL. J Am Chem Soc. 2009;131:4904. doi: 10.1021/ja809842b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Wilkie GD, Elliott GI, Blagg BSJ, Wolkenberg SE, Soenen DR, Miller MM, Pollack S, Boger DL. J Am Chem Soc. 2002;124:11292. doi: 10.1021/ja027533n. [DOI] [PubMed] [Google Scholar]; (b) Elliott GI, Fuchs JR, Blagg BSJ, Ishikawa H, Tao H, Yuan ZQ, Boger DL. J Am Chem Soc. 2006;128:10589. doi: 10.1021/ja0612549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Boger DL. Tetrahedron. 1983;39:2869. [Google Scholar]; (b) Boger DL. Chem Rev. 1986;86:781. [Google Scholar]
- 17.(a) Vasiliev NV, Lyashenko YE, Kolomiets AF, Sokolskii GA. Khim Geterostsikl Soedin. 1987:562. [Google Scholar]; (b) Thalhammer F, Wallfahrer U, Sauer J. Tetrahedron Lett. 1988;29:3231. [Google Scholar]; (c) Seitz G, Wassmuth H. Chem– Ztg. 1988;112:80. [Google Scholar]; (d) Warrener RN, Groundwater P, Pitt IG, Butler DN, Russell RA. Tetrahedron Lett. 1991;32:1985. [Google Scholar]
- 18.(a) Padwa A, Price AT. J Org Chem. 1998;63:556. doi: 10.1021/jo971424n. [DOI] [PubMed] [Google Scholar]; (b) Padwa A, Price AT. J Org Chem. 1995;60:6258. [Google Scholar]; (c) Padwa A, Lynch SM, Mejia-Oneta JM, Zhang H. J Org Chem. 2005;70:2206. doi: 10.1021/jo047834a. [DOI] [PubMed] [Google Scholar]; (d) Mejia-Oneta JM, Padwa A. Org Lett. 2004;6:3241. doi: 10.1021/ol048915w. [DOI] [PubMed] [Google Scholar]; (e) Mejia-Oneta JM, Padwa A. Tetrahedron Lett. 2004;45:9115. [Google Scholar]
- 19.Luo S, Fu X, Fang F, Zhuang Z, Xiong W, Jia X, Zhai H. Org Lett. 2006;8:115. doi: 10.1021/ol0526367. [DOI] [PubMed] [Google Scholar]
- 20.Christl M, Lanzendoerfer U, Groetsch MM, Ditterich E, Hegmann J. Chem Ber. 1990;123:2031. [Google Scholar]
- 21.Van der Ende AE, Kravitz EJ, Harth E. J Am Chem Soc. 2008;130:8706. doi: 10.1021/ja711417h. [DOI] [PubMed] [Google Scholar]
- 22.The cycloaddition of the corresponding free alcohol was also investigated, but was unsuccessful resulting in intramolecular trans-esterification.
- 23.(a) The boat conformation was established by X-ray and atomic coordinates for the ketone derived from cyanohydrin 18 (CCDC751262) have been deposited with the Cambridge Crystallographic Data Center. (b) The boat conformation as well as the structure, stereochemistry, and absolute configuration were established by X-ray and atomic coordinates for 20 (CCDC751263) have been deposited with the Cambridge Crystallographic Data Center.
- 24.Yde B, Yousif NM, Pederson U, Thomsen I, Lawesson SO. Tetrahedron. 1984;40:2047. [Google Scholar]
- 25.Overman LE, Shin Y. Org Lett. 2007;9:339. doi: 10.1021/ol062801y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.