

Abstract
Sporadic corticobasal syndrome (CBS) has been associated with diverse pathological substrates, but frontotemporal lobar degeneration with TDP-43 immunoreactive inclusions (FTLD-TDP) has only been linked to CBS among progranulin mutation carriers. We report the clinical, neuropsychological, imaging, genetic, and neuropathological features of GS, a patient with sporadic corticobasal syndrome. Genetic testing revealed no mutations in the microtubule associated protein tau (MAPT) or progranulin (PGRN) genes, but GS proved homozygous for the T allele of the rs5848 PGRN variant. Autopsy showed ubiquitin and TDP-43 pathology most similar to a pattern previously associated with PGRN mutation carriers. These findings confirm that FTLD-TDP should be included in the pathological differential diagnosis for sporadic CBS.
Keywords: corticobasal degeneration, TDP-43, frontotemporal lobar degeneration, progranulin
Introduction
Corticobasal syndrome (CBS) refers to a clinical presentation featuring progressive asymmetric bradykinesia, rigidity, and dystonia accompanied by cortical signs such as apraxia, alien limb phenomena, cortical sensory loss, myoclonus, and mirror movements (3). This constellation of symptoms and deficits was initially linked to corticobasal degeneration (CBD), a distinct pathological entity with characteristic tau-immunoreactive neuronal and glial inclusions and robust subcortical white matter tauopathy (9). Autopsy series, however, have demonstrated that diverse pathologies can underlie sporadic CBS, including Alzheimer’s disease (AD), Pick’s disease, progressive supranuclear palsy (PSP), neurofilament inclusion body disease(43), dementia with Lewy bodies, Creutzfeldt-Jakob disease (4, 24, 25, 43), and FTLD with ubiquitin-immunoreactive inclusions (FTLD-U) before TDP-43 was linked to this subtype (18, 19, 24) or in patients with TDP-43 negative FTLD-U (23). Conversely, patients with CBD pathology often present with syndromes other than CBS, such as progressive nonfluent aphasia, a PSP-like syndrome, and behavioral variant frontotemporal dementia (bvFTD) (17, 32, 37). These observations underscore the need to distinguish CBS, the syndrome, from CBD, the pathologic entity. Current consensus neuropathological nomenclature for FTLD (27) recognizes CBD as a histopathological subtype of FTLD with tau immunoreactive inclusions (FTLD-tau).
Historically, most patients with inherited CBS have harbored microtubule associated protein tau (MAPT) mutations, resulting in FTLD-tau pathology strongly resembling CBD (5). More recently, however, inherited CBS has been linked to progranulin (PGRN) mutations, which cause FTLD with ubiquitin- and TAR DNA-binding protein 43 (TDP-43) immunoreactive inclusions (FTLD-TDP). Pathological TDP-43 is hyperphosphorylated, ubiquitinated, and cleaved to produce C-terminal fragments, and neurons with cytoplasmic TDP-43 inclusions show characteristic loss of normal (nuclear) TDP-43 localization (30). Although FTLD-TDP has emerged as an important cause of inherited CBS, it remains uncertain whether TDP-43 pathology can cause sporadic CBS in patients who lack a PGRN mutation.
Two closely related classification schemes have emerged to describe the pathologic heterogeneity of FTLD-TDP (26, 36). Mackenzie et al. (26) divided FTLD-TDP into three subtypes based on appearance and distribution of TDP-43 pathology in cortex and hippocampus. Type 1 featured many short dystrophic neurites (DNs), neuronal cytoplasmic inclusions (NCIs), and lentiform neuronal intranuclear inclusions (NIIs) within cortical layer II, and variable numbers of NCIs in dentate gyrus granule cells. Type 2 displayed abundant long DNs but few NCI or NII except in the dentate gyrus, where scattered NCIs were identified. Type 3 consisted of numerous NCIs but few DNs and rare or no NIIs. Sampathu et al (36) devised a similar scheme, with abundant long DNs in Type 1 (Mackenzie Type 2), numerous NCIs in superficial and deep layers in Type 2 (Mackenzie Type 3), and ring or comma-shaped NCIs, variable NIIs, and short DNs in superficial layers in Type 3 (Mackenzie Type 1). Nearly all patients with PGRN mutations show Mackenzie Type 1 (Sampathu Type 3) pathology (26, 36). Recently, Rademakers et al reported that patients with FTLD-TDP homozygous for a common PGRN polymorphism, the T-allele of variant rs5848, often feature a Mackenzie Type 1 TDP-43 pathological pattern, similar to PGRN mutation carriers (31). The authors hypothesized that the T-allele of rs5848 increases microRNA binding efficiency to the 3’ untranslated region (UTR) of PGRN mRNA, leading to enhanced suppression of PGRN translation and reduced levels of functional progranulin (31).
In this report, we describe GS, a patient with sporadic CBS due to FTLD-TDP. This novel clinicopathological coupling, though reminiscent of patients with PGRN mutations, occurred in a patient who lacked a PGRN mutation.
Materials and Methods
Subjects
GS presented at age 63 years and was studied over three years as part of a longitudinal dementia research program. Twenty-eight age-matched healthy women (mean age = 63.0 years, s.d. = 6.2) underwent a neurological and neuropsychological assessment, were diagnosed as clinically normal at a consensus case conference, and served as controls for the imaging and neuropsychological analyses. Controls were selected to match GS for age from a pool of 54 female subjects with neuropsychological data and an available MRI scan within 90 days of clinical assessment. All subjects (or their surrogates) provided written informed consent prior to participation. The study was approved by the University of California San Francisco Committee on Human Research.
Neuropsychological Testing
A neuropsychological evaluation was performed, as previously described (38), to assess language, memory, visuospatial, and executive function. Mood was assessed with the Geriatric Depression Scale. One control subject lacked a complete neuropsychological data set and was excluded from these analyses.
Neuroimaging
MRI scans were obtained on a 1.5-T Magnetom VISION system (Siemens Inc., Iselin, NJ) equipped with a standard quadrature head coil. Structural MRI sequences were obtained as previously described to produce high resolution T1-weighted images with whole-brain coverage (35). Cortical reconstruction and volumetric segmentation were performed with the Freesurfer image analysis suite (http://surfer.nmr.mgh.harvard.edu/). Imaging procedural details are described in prior publications (7, 8, 12-15). The Freesurfer processing stream includes motion correction and averaging of multiple volumetric T1-weighted images, removal of non-brain tissue using a hybrid watershed/surface deformation procedure (40), automated Talairach transformation (12, 15), intensity normalization (42), tessellation of the gray matter-white matter boundary, automated topology correction (41), and surface deformation (7). The resulting three-dimensional models of the cortical surface were then registered to an atlas using individual cortical folding patterns to match cortical geometry across subjects (14) and parcellated into regional subunits based on gyral and sulcal structure (8, 15). Using both intensity and continuity information from the MR volume and the cortical surface model, representations of cortical thickness were produced, calculated as the closest distance from the gray/white boundary to the gray/CSF border at each vertex on the tessellated surface (11). A single-subject statistical comparison of cortical thickness between GS and controls was conducted by registering each subject’s map of thickness values to a common surface, and using a general linear model (GLM) fit at each node using a built-in Freesurfer group analysis tool. The thickness maps for each subject were smoothed with a 10 mm Gaussian kernel, and the significance threshold was set to p < 0.01, uncorrected for multiple comparisons.
Genetic Analyses
For sequencing analyses, all 12 coding exons and the non-coding exon 0 of PGRN and exon 1 and exons 9–13 of MAPT were amplified by PCR using previously published primers and protocols (1, 20). PCR products were purified using AMPure (Agencourt Biosciences, Beverly, MA) then sequenced in both directions using the Big Dye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA). Sequencing reactions were purified using CleanSEQ (Agencourt Biosciences) and run on an ABI3730 Genetic Analyser (Applied Biosystems).
Total RNA was quantified according to previously published methods (6). Absolute expression values were log2 transformed to and normalized using quantile normalization. The normal range was established in 199 healthy controls (mean age 64.2, s.d. 10.5 yrs).
PGRN ELISA assay
GS’s plasma PGRN expression level was analyzed using the human Progranulin ELISA kit (Adipogen Inc., Seoul, Korea) using a 1:100 dilution of the plasma samples in 1x diluent following manufacturer’s instructions. Recombinant human PGRN provided with the ELISA kit was used as a standard. A total of 70 healthy controls (mean age 66.3, s.d. 11.0 yrs) as well as previously analyzed FTLD patients with and without PGRN mutations were also included in the analyses (10).
Neuropathology
The fresh brain was removed and cut into 8-10 mm thick coronal slabs. These slabs were alternately fixed, in freshly made 4% paraformaldehyde for 72 hours, or rapidly frozen. Tissue blocks covering dementia-related regions of interest were dissected from the fixed slabs, and basic and immunohistochemical stains were applied following standard diagnostic procedures developed for patients with dementia (21). Immunohistochemistry was performed using antibodies to: ubiquitin (anti-rabbit, 1:1000, DAKO North America, Carpinteria, CA), TDP43 (anti-rabbit, 1:2000, Proteintech Group, Chicago, IL), hyperphosphorylated tau (CP-13 antibody, courtesy of P. Davies), beta-amyloid (anti-mouse, 1:250, Millipore, Billerica, MA), glial fibrillary acidic protein (GFAP; anti-rabbit, 1:250, DAKO North America, Carpinteria, CA), alpha synuclein (anti-mouse, 1:1000, Millipore, Billerica, MA), alpha-internexin (anti-mouse, 1:200, Invitrogen, Carlsbad, CA), and fused in sarcoma (FUS, anti-rabbit, 1:200, Sigma Aldrich, St. Louis, MO). All immunohistochemical runs included positive control sections to exclude technical factors as a cause of absent immunoreactivity.
Results
Clinical History
GS was a 63-year-old right-handed woman referred to UCSF for progressive, asymmetric rigidity and dystonia of her left side. Shortly before the assessment, she had stopped working as an accountant because of difficulty with her left hand, which prevented her from typing or driving. Her first symptoms had emerged six years prior, when she developed numbness in her left fifth digit. She was diagnosed with carpal tunnel syndrome and treated with repeated steroid injections without improvement. Over the following five years, her entire left hand became numb. During the year prior to presentation, she noted left hand twisting and stiffness, which progressed to constant dystonic hand posturing, rendering the hand useless. She reported that her left arm “appeared to have a mind of its own”. It interfered when she tried to apply facial cream with her right hand; at times she slammed doors on her left arm, as it did not always follow her body through doorways. While driving, her left hand turned the steering wheel in an unintended direction. She could no longer use tools or utensils with the left hand. She acknowledged the left arm as her own and was frustrated by its lack of coordination. Gait instability emerged due to difficulty with the left leg, especially when it did not follow the right leg. She reported a near fall on an escalator because her right foot started up the escalator but her left foot failed to follow. She had also noticed difficulties in the right hand, with smaller, illegible handwriting. Her speech became slurred. She reported difficulty navigating while driving. Language and memory were preserved and she could manage most executive tasks. She delegated financial matters to her husband due to her inability to use a computer or write clearly.
Past medical history was unremarkable. Family history was negative for neurological disease except for a paternal grandfather with late-life tremor.
On initial neurological examination, GS had a normal affect. Spontaneous speech and language production and comprehension were normal, but she displayed oral-buccal apraxia. Square wave jerks were present in primary gaze, and there was mild lateral gaze restriction with saccadic breakdown of smooth pursuits. Voluntary side-to-side tongue movements were slow, uncoordinated, and mimicked by her left hand. There was mild tongue tremor, dysarthria and hypophonia. Limb power was full throughout, but increased tone with rigidity and cogwheeling was present in the left upper extremity. There was mild rigidity in the right upper extremity. She had a resting tremor and intermittent myoclonus in her left arm and mild bilateral postural tremor. Her left hand was dystonically closed, and she could not open it. Tone in the lower extremities was increased on the left but normal on the right. No limb apraxia or alien limb phenomena were noted. Sensation to pinprick, vibration, and joint position sense were normal. She had significant bilateral cortical sensory deficits (agraphesthesia and astereognosis), worse on the left. Reflexes were normal and symmetric and both plantar responses were flexor. There was no dysmetria on finger-to-nose testing. Gait was mildly wide-based, and toe and heel walking exaggerated dystonic posturing in both upper extremities, worse on the left. The left leg occasionally dragged behind her. She had mild retropulsive instability.
The subsequent year brought worsening rigidity and gait disturbance. Her right upper extremity became clumsy, with involuntary grasping. Dysphagia and dysarthria worsened, attenuating verbal output. She became depressed with intermittent confusion. She became mildly impulsive, leading to occasional falls, and some social disinhibition emerged. The following year, she became mute and immobile with ophthalmoparesis. Her right side worsened, rendering both hands useless. She developed pyramidal weakness and decreased primary sensation in the left arm. She developed aspiration pneumonia and died in March of 2008.
Neuropsychological Testing
GS’s first neuropsychological evaluation occurred in July, 2005 (Table 1). Overall, the testing revealed mild speech abnormalities and deficits in visual-spatial and executive function, with sparing of verbal memory and naming. At follow-up, evolving motor and speech difficulties precluded a complete battery, but worsening visual-spatial and working memory deficits were noted, with continued sparing of verbal memory, naming, and reading skills.
Table 1.
Neuropsychological Assessment at presentation and after 1 year
| Controls (N=27) | GS July 2005 | GS Sept. 2006 | |
|---|---|---|---|
| MMSE (30) | 30 (0.6) | 29 | 23/25 |
| CVLT-II Long Delay Free Recall (16) | 13.4 (2.8) | 10 | n/a |
| CVLT-Short Form 10-minute Free Recall (9) | 8 (1.1) | n/a | 8 |
| Modified Rey Complex Figure Recall (17) | 11.9 (2.5) | 6 *** | could not assess |
| 15-Item Boston Naming Test (15) | 14.6 (0.9) | 15 | 15 |
| Category Fluency (Animals) | 23.9 (5.9) | 18 | could not assess |
| Modified Rey Complex Figure Copy (17) | 15.9 (1.2) | 10 **** | could not assess |
| VOSP Spatial Location Task (10) | 8.7 (2.5) | 4 **** | 6 **** |
| Digits Backward (Max. Span) | 5.0 (1.2) | 5 | 4* |
| Stroop Interference Task | 50.0 (0.5) | 35 ** | could not assess |
| Modified Trail Making Test (120”) | 31.2 (20) | 120”/2 errors **** | could not assess |
| Letter Fluency (D words) | 17.0 (4.3) | 11* | could not assess |
Values listed are mean (s.d.). In first column, numbers listed in parentheses = maximum attainable score possible. n/a = not available.
=-1.0<z<-1.5;
-1.5<z<-2.0;
-2.0<z<-2.5;
-2.5<z<-3.0.
Neuroimaging
A brain MRI obtained six years after symptom onset revealed bilateral posterior frontal and anterior parietal atrophy, significantly worse on the right (Figure 1). Affected regions included primary and supplementary sensorimotor cortices (Fig. 1a). The precuneus, posterior cingulate and orbitofrontal cortices were spared. Freesurfer analysis revealed significant right frontal cortical thickness reductions, within the banks of the precentral and superior frontal sulci, superior frontal gyrus including medial supplementary motor area, and precentral gyrus (Fig 1b). Significant parietal atrophy was also evident, most severely affecting the superior parietal lobule and postcentral gyri. Though less severe, cortical thickness reductions were also identified in homologous left hemisphere regions (Fig. 1b), and the overall pattern was consistent with gross observations at autopsy (Fig. 1c).
Figure 1. Neuroimaging features.

(a) Coronal (top) T1-weighted MRI at presentation. (b) Freesurfer comparison of GS to 28 healthy age-matched women shown at two levels of statistical significance revealed atrophy in right precentral sulcus, superior frontal gyrus, precentral gyrus, medial supplementary motor area, superior parietal lobule and postcentral gyrus. Foci of reduced cortical thickness were also apparent in homologous left hemisphere regions. (c) Dorsal view of fresh brain exhibiting marked, right greater than left, perirolandic atrophy. Arrow indicates Central Sulcus. Scale bar represents 5 cm.
Genetic and expression studies
No mutations were identified in PGRN and MAPT by extensive sequencing analyses. Genetic analyses further showed homozygosity for the extended MAPT H1 haplotype and homozygosity for the T-allele of the rs5848 genetic variant in PGRN. Apolipoprotein E genotype was E2/E3.
The whole blood progranulin mRNA level in GS was 9.33, well within spectrum seen in controls (mean 9.37, s.d. 0.32). A PGRN plasma ELISA assay detected a PGRN level of 169 ng/ml, also within the healthy control range (mean 228 ng/ml, s.d. 50 ng/ml, range 138-376 ng/ml).
Neuropathology
The fresh brain weighed 920 grams. Gross examination revealed severe atrophy of the frontal and parietal lobes, bilaterally, worse on the right than the left (Figure 1c and 2a-b). Marked cerebral cortical thinning was evident over the posterior frontal and anterior parietal lobes, worse on the right than the left. The temporal lobes and hippocampi showed no atrophy. Microscopically, the sampled region most devastated was the right postcentral gyrus (Fig. 2c-e, Table 2). The pattern of regional degeneration, highly consistent with the clinical deficits and previous descriptions of CBS and CBD, is summarized in Table 2. Hematoxylin & eosin (H & E) staining revealed rare ballooned neurons in frontal and parietal cortex; none were identified with tau or silver stains.
Figure 2. Asymmetric, focal perirolandic degeneration.
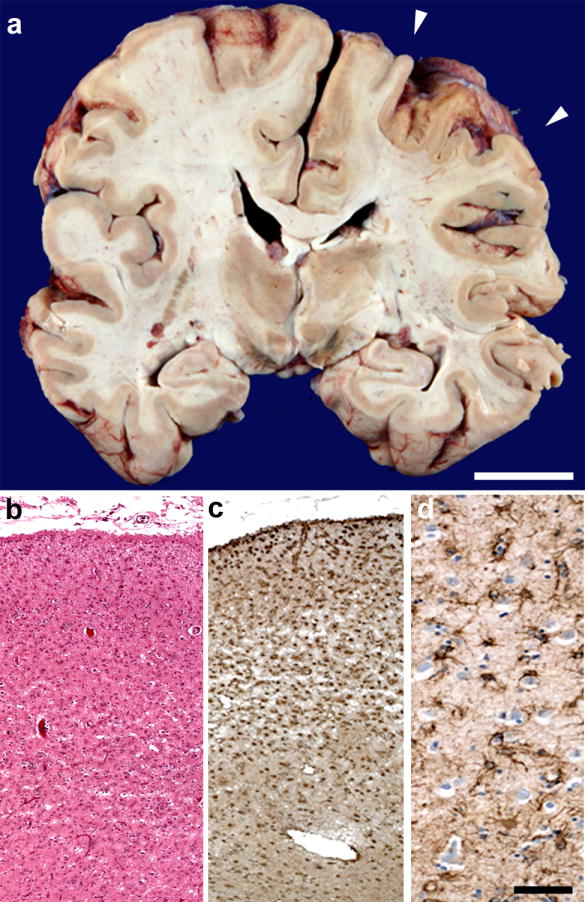
(a) Coronal view through postcentral gyrus (arrowheads) further highlights asymmetric atrophy. (b) Microscopically, right postcentral gyrus showed profound microvacuolation and gliosis, which included marked reactive astrogliosis (c-d). Scale bar in (a) represents 2 cm. Scale bar in (d) applies to (b-d) and represents 200 uM (b, c) and 50 uM (d). Hematoxylin and eosin, (b). Antibody to glial fibrillary acidic protein (c-d).
Table 2.
Semi-quantitative ratings of microvacuolation, gliosis, and ubiquitin and TDP-43 pathology.
| H & E | Ubiquitin | TDP-43 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L/R Microvacuolation | L/R Gliosis | L/R NIIs | L/R NCIs | L/R DNs | L/R GCIs | L/R Dots/threads | L/R NIIs | L/R NCIs | L/R DNs | L/R GCIs | L/R dots/threads | |
| aMCC | ++/++ | ++/++ | 0/0 | ++/++ | +/+ | 0/0 | ++/+++ | 0/0 | +/+++ | +/+++ | 0/+ | ++/0 |
| Middle insula | +/++ | ++/++ | 0/0 | +/+++ | ++/+++ | +/+ | +/0 | 0/0 | ++/+++ | ++/++ | +/+ | 0/0 |
| Pre-SMA | ++/+++ | ++/+++ | +/0 | ++/+++ | ++/++ | +/0 | +++/+++ | +/0 | +++/++ | ++/+++ | +/+ | +++/+ |
| Precentral gyrus | ++/+++ | ++/+++ | 0/0 | ++/++ | ++/+ | +/0 | ++/++ | 0/0 | ++/++ | +++/+ | +/+ | +/0 |
| Angular gyrus | 0/+ | +/++ | +/0 | +/++ | +/+ | 0/0 | ++/++ | +/0 | +/++ | +/++ | 0/+ | ++/0 |
| Claustrum | NA | +/+ | 0/0 | 0/+ | +/+ | 0/0 | +/0 | 0/0 | +/++ | ++/+ | 0/0 | 0/0 |
| Putamen | NA | +++/+++ | 0/0 | +/+ | ++/+ | 0/0 | +/+ | 0/0 | +/+ | ++/+ | 0/0 | ++/+ |
| Globus pallidus | NA | +/0 | 0/0 | 0/0 | 0/0 | 0/0 | +/0 | 0/0 | 0/+ | +/+ | 0/+ | +/0 |
| Entorhinal cortex | 0/0 | 0/0 | 0/0 | 0/0 | +/+ | 0/0 | +/+ | 0/0 | 0/0 | 0/+ | 0/0 | 0/0 |
| Dentate gyrus | N/A | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| CA3-4 | N/A | 0/-- | 0/-- | 0/-- | 0/-- | 0/-- | +/-- | 0/-- | 0/-- | 0/-- | 0/-- | 0/-- |
| CA2 | N/A | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| CA1/subiculum | N/A | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | +/0 | 0/0 | 0/0 | 0/+ | 0/0 | 0/0 |
H & E = Hematoxylin and eosin, Ub = Ubiquitin, TDP-43 = TAR DNA-binding protein 43, L/R = Left/Right, NCIs = Neuronal cytoplasmic inclusions, DNs = Dystrophic neurites, GCIs = Glial cytoplasmic inclusions, NIIs = Neuronal intranuclear inclusions, aMCC = anterior midcingulate cortex, PCg = Postcentral gyrus, EntoRC/PeriRC = Entorhinal/perirhinal cortex, Ub = Ubiquitin, SMA = Supplementary motor area, NA = Not assessed. 0 = NSPA, + = mild, ++ = moderate, +++ = severe, -- = region damaged/not available.
In affected cortical regions, TDP-43 immunohistochemistry revealed frequent short, angular neuropil threads, most likely DNs, and round or comma-shaped NCIs. Both DNs and NCIs were most prominent in layers 2 and 3 (Figure 3, Table 2). Abundance of TDP-43 pathology correlated with the severity of microvacuolation and gliosis, except in the right postcentral gyrus, where profound neuronal loss rendered NCIs and DNs less frequent. Figure 3 provides representative examples of the cortical pathology in selected frontal and parietal regions. Temporal neocortical regions showed the mildest Ub and TDP-43 pathology. A few large Layer 5 neurons with skein-like NCIs were detected in left primary motor cortex. Sparse TDP-43 pathology was seen in hippocampus and entorhinal cortex. Notably, the dentate gyrus showed no TDP-43-immunoreactive NCIs (Fig. 3i). More frequent NCIs and threads were seen in substantia nigra, midbrain tegmentum and periaqueductal gray. The hypoglossal nucleus showed a normal population of motor neurons, without NCIs. The basal ganglia showed only sparse Ub and TDP-43 pathology (Fig. 4a-d), but robust TDP-43 pathology was detected in subcortical white matter (Fig 4e-f), with numerous threads and coiled oligodendroglial inclusions (29)]. NIIs (Figure 3g) were distinctly rare. In general, ubiquitin immunoreactive neuropil threads and inclusions followed a similar pattern as those seen with TDP-43 staining, though ubiquitin stained most areas less avidly than did TDP-43. All regions showing TDP-43/ubiquitin pathology stained negatively for tau. Overall, the pathological findings were most consistent with FTLD-TDP Type 1 (Mackenzie et al (26))/Type 3 (Sampathu et al (36)).
Figure 3. FTLD-TDP microscopic pathology.

Ubiquitin (Ub) and TDP-43 immunoreactive inclusions including neuronal cytoplasmic inclusions (NCIs), dystrophic neurites (DNs), glial cytoplasmic inclusions (GCIs) and neuronal intranuclear inclusions (NIIs) were most similar to FTLD-TDP Type 1 (Mackenzie et al(26)). (a-d) In frontal and parietal cortex, short DNs and numerous small, round or comma-shaped NCIs in superficial layers predominated. (e-h) At higher magnification, note NCIs in pre-supplementary motor cortex (preSMA) and a lentiform NII (g) in right angular gyrus (ANG). (i) Inclusions immunopositive for TDP-43 or ubiquitin (not shown) were not observed in the dentate gyrus. Scale bar in (h) represents 20 uM and applies to (e), (f) and (h). Scale bar in (i) represents 10 um in (g), 20 um in (a-d) and 50 um in (i).
Figure 4. Subcortical and white matter pathology.

Mild Ub (a, c) and TDP-43 (b, d) pathology consisting primarily of dystrophic neurites in caudate (a, b) and putamen (c, d). Significant subcortical WM pathology in right supplementary motor area (e, f). Ub (e) and TDP-43 (f) immunoreactive coiled inclusions in oligodendroglia accompanied by white matter thread pathology. Scale bars represent 100 uM in (a) and (b); 50 uM in (d) and (e) and 20uM in (c) and (f).
There were no neuritic plaques (NP), neurofibrillary tangles (NFTs), globose tangles, or Pick bodies noted with silver stains. Sparse tau immunopositive neuropil threads and NFTs were seen in hippocampal CA1, and moderate to frequent neuropil threads and NFTs were observed in entorhinal cortex, corresponding to Braak Stage 2. No tau immunoreactivity was seen elsewhere. Lewy bodies were not detected. No FUS or alpha-internexin immunoreactivity was seen in medial temporal lobe or premotor cortex.
Discussion
GS suffered from a progressive neurodegenerative illness typical of CBS. Her complex sensorimotor and cognitive deficits correlated with focal perirolandic pathology involving primary and secondary sensorimotor cortices and extending into lateral frontal and parietal association areas. Although right-predominant, the disease advanced into homologous bilateral cortical and subcortical regions that together constitute a large-scale functional network shown to degenerate in CBS (39). Neuropsychology and neuroimaging revealed conspicuous temporal lobe sparing, confirmed microscopically at autopsy.
Although previous reports have linked CBS to PGRN mutation-associated FTLD-TDP (2, 28) and to FTLD-U (18, 19, 24), to our knowledge GS represents the first reported patient with CBS/FTLD-TDP in the known absence of a PGRN mutation. Therefore, our findings add FTLD-TDP to the pathological differential diagnosis for sporadic CBS. As CBS treatment trials begin to emerge, discriminating biomarkers will be needed to sort patients according to underlying pathology. Potential biomarkers include genetic polymorphisms that modify disease gene expression, like homozygosity for the rs5848 T-allele identified in GS, but larger studies are required to determine whether sporadic CBS due to FTLD-TDP occurs more commonly in patients with this or other disease modifying genes. A recent study found no increased risk for frontotemporal dementia in T allele homozygotes (34), but it remains to be determined whether this genotype alters the clinical phenotype in patients with sporadic FTLD-TDP.
CBS pathological heterogeneity highlights important issues regarding the relationship between clinical syndrome and molecular pathogenesis. Strikingly, GS’s TDP-43 pattern and distribution shared several features with typical CBD pathology. As in CBD, the burden of NCIs and neuropil threads was severe in primary and secondary sensorimotor cortices but spared the temporal and occipital lobes. Like patients with CBD, GS showed extensive subcortical white matter pathology, with frequent threads and coiled body-like oligodendroglial inclusions similar to those described previously in FTLD-TDP (29). The pattern of TDP-43 pathology correlated well with clinical and radiological deficits, which were typical of CBS. As seen in most patients with CBS due to CBD, genetic testing revealed homozygosity for the extended MAPT H1 haplotype, but the significance of this finding in GS remains uncertain. Overall, GS’s pathological features reinforce the view that the location of pathology, rather than its molecular identity, is what determines the clinical syndrome.
FTLD-TDP represents an emerging pathological spectrum that is still being actively defined (16). GS’s cortical pathology was characterized by short, angulated neuropil threads and numerous NCIs in superficial more than deep layers. Her frontoparietal distribution (22, 33) and histological features most strongly resemble Mackenzie Type 1 (Sampathu Type 3) FTLD-TDP pathology, the subtype associated with mutations in PGRN\. Although the role of NIIs in defining this FTLD-TDP subtype remains unsettled (16), GS lacked the NII frequency described in some previous reports (26). Finally, in contrast to all Mackenzie subtypes (which included characterization of the dentate gyrus), GS showed no dentate gyrus granule cell inclusions, perhaps merely reflecting her lack of temporal lobe involvement.
Because CBS had only been associated with TDP-43 among PGRN mutation carriers, we suspected a de novo PGRN mutation in GS. Sequencing of the PGRN gene, however, revealed no mutation, though GS did prove homozygous for a newly identified possible risk factor polymorphism, the T-allele of the rs5848 PGRN variant. This polymorphism was previously suggested to improve microRNA binding efficiency to the PGRN 3’ UTR, resulting in reduced PGRN expression (31), but in GS whole blood PGRN mRNA and plasma protein levels were normal. Therefore, patient-specific factors that occasionally drive FTLD-TDP into the dorsal frontoparietal CBS network remain mysterious. Disease-modifying genetic interactions could influence not only which molecular pathology accrues in a brain, but also where it accrues. Future correlative clinical, radiological, pathological, and genetic approaches will be required to approach this key issue in neurodegenerative disease research.
Acknowledgments
We thank Giovanni Coppola for assistance with the progranulin mRNA analysis. This study was supported by National Institute on Aging (NIA, grants P01 AG019724-08, P50 AG023501-06). We thank the patient and her family for their participation in this study.
Footnotes
DISCLOSURES MC Tartaglia reports no disclosures.
M Sidhu reports no disclosures.
V Laluz reports no disclosures.
C Racine reports no disclosures.
GD Rabinovici reports no disclosures.
K Creighton reports no disclosures.
A Karydas reports no disclosures.
R Rademakers reports no disclosures.
EJ Huang reports no disclosures.
BL Miller reports no disclosures.
SJ DeArmond reports no disclosures.
WW Seeley reports no disclosures.
References
- 1.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 2.Benussi L, Binetti G, Sina E, Gigola L, Bettecken T, Meitinger T, Ghidoni R. A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol Aging. 2008;29:427–435. doi: 10.1016/j.neurobiolaging.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 3.Boeve BF. Corticobasal Degeneration: The Syndrome and the Disease. In: Litvan I, editor. Atypical Parkinsonian Disorders. Totawa: Humana Press; 2005. pp. 309–334. [Google Scholar]
- 4.Boeve BF, Maraganore DM, Parisi JE, Ahlskog JE, Graff-Radford N, Caselli RJ, Dickson DW, Kokmen E, Petersen RC. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology. 1999;53:795–800. doi: 10.1212/wnl.53.4.795. [DOI] [PubMed] [Google Scholar]
- 5.Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, Morbin M, Primavera A, Carella F, Solaro C, Grisoli M, Savoiardo M, Spillantini MG, Tagliavini F, Goedert M, Ghetti B. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol. 1999;58:667–677. doi: 10.1097/00005072-199906000-00011. [DOI] [PubMed] [Google Scholar]
- 6.Coppola G, Karydas A, Rademakers R, Wang Q, Baker M, Hutton M, Miller BL, Geschwind DH. Gene expression study on peripheral blood identifies progranulin mutations. Ann Neurol. 2008;64:92–96. doi: 10.1002/ana.21397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis. I. Segmentation and surface reconstruction. Neuroimage. 1999;9:179–194. doi: 10.1006/nimg.1998.0395. [DOI] [PubMed] [Google Scholar]
- 8.Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, Albert MS, Killiany RJ. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 9.Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SS, Tabaton M, Vonsattel JP, Wakabayashi K, Litvan I. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61:935–946. doi: 10.1093/jnen/61.11.935. [DOI] [PubMed] [Google Scholar]
- 10.Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R, Bisceglio G, Rovelet-Lecrux A, Boeve B, Petersen RC, Dickson DW, Younkin SG, Deramecourt V, Crook J, Graff-Radford NR, Rademakers R. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 2009;132:583–591. doi: 10.1093/brain/awn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci U S A. 2000;97:11050–11055. doi: 10.1073/pnas.200033797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 13.Fischl B, Sereno MI, Dale AM. Cortical surface-based analysis. II: Inflation, flattening, and a surface-based coordinate system. Neuroimage. 1999;9:195–207. doi: 10.1006/nimg.1998.0396. [DOI] [PubMed] [Google Scholar]
- 14.Fischl B, Sereno MI, Tootell RB, Dale AM. High-resolution intersubject averaging and a coordinate system for the cortical surface. Hum Brain Mapp. 1999;8:272–284. doi: 10.1002/(SICI)1097-0193(1999)8:4<272::AID-HBM10>3.0.CO;2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischl B, van der Kouwe A, Destrieux C, Halgren E, Segonne F, Salat DH, Busa E, Seidman LJ, Goldstein J, Kennedy D, Caviness V, Makris N, Rosen B, Dale AM. Automatically parcellating the human cerebral cortex. Cereb Cortex. 2004;14:11–22. doi: 10.1093/cercor/bhg087. [DOI] [PubMed] [Google Scholar]
- 16.Forman MS, Trojanowski JQ, Lee VM. TDP-43: a novel neurodegenerative proteinopathy. Curr Opin Neurol. 2008;17:548–555. doi: 10.1016/j.conb.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorno-Tempini ML, Murray RC, Rankin KP, Weiner MW, Miller BL. Clinical, cognitive and anatomical evolution from nonfluent progressive aphasia to corticobasal syndrome: a case report. Neurocase. 2004;10:426–436. doi: 10.1080/13554790490894011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grimes DA, Bergeron CB, Lang AE. Motor neuron disease-inclusion dementia presenting as cortical-basal ganglionic degeneration. Mov Disord. 1999;14:674–680. doi: 10.1002/1531-8257(199907)14:4<674::aid-mds1019>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 19.Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M, Bak TH, Kril JJ, Halliday GM. Clinicopathological correlates in frontotemporal dementia. Ann Neurol. 2004;56:399–406. doi: 10.1002/ana.20203. [DOI] [PubMed] [Google Scholar]
- 20.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 21.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1095–1097. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve BF, Knopman DS, Petersen RC, Davies P, Duara R, Graff-Radford NR, Uitti RJ, Rademakers R, Adamson J, Baker M, Hutton ML, Dickson DW. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol. 2007;66:142–151. doi: 10.1097/nen.0b013e31803020cf. [DOI] [PubMed] [Google Scholar]
- 23.Josephs KA, Lin WL, Ahmed Z, Stroh DA, Graff-Radford NR, Dickson DW. Frontotemporal lobar degeneration with ubiquitin-positive, but TDP-43-negative inclusions. Acta Neuropathol. 2008;116:159–167. doi: 10.1007/s00401-008-0397-8. [DOI] [PubMed] [Google Scholar]
- 24.Kertesz A, Martinez-Lage P, Davidson W, Munoz DG. The corticobasal degeneration syndrome overlaps progressive aphasia and frontotemporal dementia. Neurology. 2000;55:1368–1375. doi: 10.1212/wnl.55.9.1368. [DOI] [PubMed] [Google Scholar]
- 25.Kleiner-Fisman G, Bergeron C, Lang AE. Presentation of Creutzfeldt-Jakob disease as acute corticobasal degeneration syndrome. Mov Disord. 2004;19:948–949. doi: 10.1002/mds.20140. [DOI] [PubMed] [Google Scholar]
- 26.Mackenzie IR, Baborie A, Pickering-Brown S, Du Plessis D, Jaros E, Perry RH, Neary D, Snowden JS, Mann DM. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 2006;112:539–549. doi: 10.1007/s00401-006-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117:15–18. doi: 10.1007/s00401-008-0460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masellis M, Momeni P, Meschino W, Heffner R, Jr, Elder J, Sato C, Liang Y, St George-Hyslop P, Hardy J, Bilbao J, Black S, Rogaeva E. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain. 2006;129:3115–3123. doi: 10.1093/brain/awl276. [DOI] [PubMed] [Google Scholar]
- 29.Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA, Van Deerlin VM, Clark CM, Grossman M, Miller BL, Trojanowski JQ, Lee VM. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol. 2007;66:177–183. doi: 10.1097/01.jnen.0000248554.45456.58. [DOI] [PubMed] [Google Scholar]
- 30.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 31.Rademakers R, Eriksen JL, Baker M, Robinson T, Ahmed Z, Lincoln SJ, Finch N, Rutherford NJ, Crook RJ, Josephs KA, Boeve BF, Knopman DS, Petersen RC, Parisi JE, Caselli RJ, Wszolek ZK, Uitti RJ, Feldman H, Hutton ML, Mackenzie IR, Graff-Radford NR, Dickson DW. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet. 2008;17:3631–3642. doi: 10.1093/hmg/ddn257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raggi A, Marcone A, Iannaccone S, Ginex V, Perani D, Cappa SF. The clinical overlap between the corticobasal degeneration syndrome and other diseases of the frontotemporal spectrum: three case reports. Behav Neurol. 2007;18:159–164. doi: 10.1155/2007/218189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rohrer JD, Warren JD, Omar R, Mead S, Beck J, Revesz T, Holton J, Stevens JM, Al-Sarraj S, Pickering-Brown SM, Hardy J, Fox NC, Collinge J, Warrington EK, Rossor MN. Parietal lobe deficits in frontotemporal lobar degeneration caused by a mutation in the progranulin gene. Arch Neurol. 2008;65:506–513. doi: 10.1001/archneur.65.4.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rollinson S, Snowden JS, Neary D, Morrison KE, Mann DM, Pickering-Brown SM. TDP-43 gene analysis in frontotemporal lobar degeneration. Neurosci Lett. 2007;419:1–4. doi: 10.1016/j.neulet.2007.03.044. [DOI] [PubMed] [Google Scholar]
- 35.Rosen HJ, Gorno-Tempini ML, Goldman WP, Perry RJ, Schuff N, Weiner M, Feiwell R, Kramer JH, Miller BL. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology. 2002;58:198–208. doi: 10.1212/wnl.58.2.198. [DOI] [PubMed] [Google Scholar]
- 36.Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, Bruce J, Grossman M, Trojanowski JQ, Lee VM. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169:1343–1352. doi: 10.2353/ajpath.2006.060438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanchez-Valle R, Forman MS, Miller BL, Gorno-Tempini ML. From progressive nonfluent aphasia to corticobasal syndrome: a case report of corticobasal degeneration. Neurocase. 2006;12:355–359. doi: 10.1080/13554790600977218. [DOI] [PubMed] [Google Scholar]
- 38.Seeley WW, Crawford R, Rascovsky K, Kramer JH, Weiner M, Miller BL, Gorno-Tempini ML. Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Arch Neurol. 2008;65:249–255. doi: 10.1001/archneurol.2007.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62:42–52. doi: 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Segonne F, Dale AM, Busa E, Glessner M, Salat D, Hahn HK, Fischl B. A hybrid approach to the skull stripping problem in MRI. Neuroimage. 2004;22:1060–1075. doi: 10.1016/j.neuroimage.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 41.Segonne F, Pacheco J, Fischl B. Geometrically accurate topology-correction of cortical surfaces using nonseparating loops. IEEE Trans Med Imaging. 2007;26:518–529. doi: 10.1109/TMI.2006.887364. [DOI] [PubMed] [Google Scholar]
- 42.Sled JG, Zijdenbos AP, Evans AC. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging. 1998;17:87–97. doi: 10.1109/42.668698. [DOI] [PubMed] [Google Scholar]
- 43.Yokota O, Tsuchiya K, Terada S, Ishizu H, Uchikado H, Ikeda M, Oyanagi K, Nakano I, Murayama S, Kuroda S, Akiyama H. Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol. 2008;115:561–575. doi: 10.1007/s00401-007-0329-z. [DOI] [PubMed] [Google Scholar]