

Abstract
Riboflavin synthase catalyzes the transfer of a 4-carbon fragment between two molecules of the substrate, 6,7-dimethyl-8-ribityllumazine, resulting in the formation of riboflavin and 5-amino-6-ribitylamino-2,4(1H,3H)-pyrimidinedione. Earlier, a pentacyclic adduct formed from two substrate molecules was shown to be a catalytically competent intermediate, but the mechanism of its formation is still poorly understood. The present study shows that the recombinant N-terminal domain of riboflavin synthase from Escherichia coli interacts specifically with the exomethylene-type anion of 6,7-dimethyl-8-ribityllumazine but not with any of the tricyclic adduct-type anions that dominate the complex anion equilibrium in aqueous solution. Whereas these findings can be implemented into previously published mechanistic hypotheses, we also present a novel, hypothetical reaction sequence that starts with the transfer of a hydride ion from the 6,7-dimethyl-8-ribityllumazine exomethylene anion to an electroneutral 6,7-dimethyl-8-ribityllumazine molecule. The pair of dehydrolumazine and dihydrolumazine molecules resulting from this hydride transfer is proposed to undergo a 4+2 cycloaddition affording the experimentally documented pentacyclic intermediate. In contrast to earlier mechanistic concepts requiring the participation of a nucleophilic agent, which is not supported by structural and mutagenesis data, the novel concept has no such requirement. Moreover, it requires fewer reaction steps and is consistent with all experimental data.
Keywords: Riboflavin synthase, biosynthesis of riboflavin, reaction mechanism, hydride transfer, cycloaddition
Introduction
Riboflavin synthase catalyzes the transfer of a 4-carbon unit between two molecules of the substrate, 6,7-dimethyl-8-ribityllumazine (1, Scheme 1), which results in the formation of riboflavin (4) and 5-amino-6-ribitylamino-2,4(1H,3H)-pyrimidinedione (5).1-4 The unusual reaction can proceed under surprisingly mild conditions, even without catalysis (boiling an aqueous solution of 1 under an inert atmosphere). The enzyme-catalyzed and the uncatalyzed reactions are both regiospecific.5-7
Scheme 1.
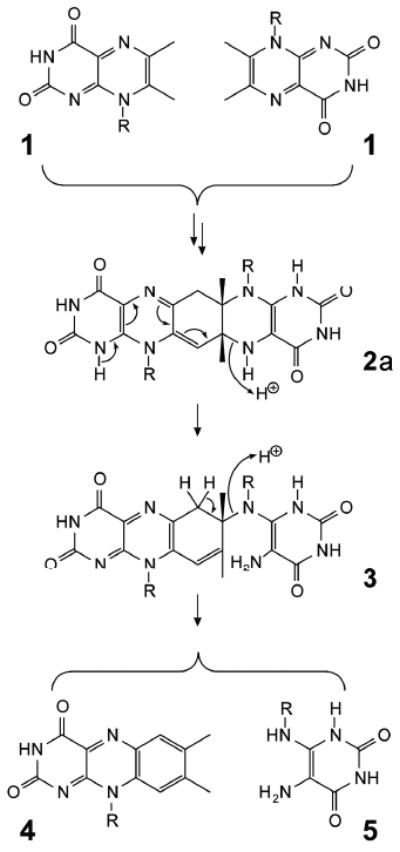
The pentacyclic adduct 2a has been identified as a catalytically competent intermediate of riboflavin synthase;8, 9 R, D-ribityl.
The riboflavin synthase substrate 1 and its structural analogs are characterized by unusual CH acidity of the position 7 methyl group.10 Thus, deprotonation of 6,7,8-trimethyllumazine (6; pKa, 8.9) affords the exomethylene anion 7 (Fig. 1). In case of the riboflavin synthase substrate 1 (apparent pKa, 7.9), the exomethylene species 8 is only present in trace amounts, whereas the dominant components are two tricyclic diastereomer pairs (9 – 12, Fig. 1) which arise by nucleophilic attack of the position 2′ and 3′ hydroxy groups, respectively, at C-7 of the pyrazine ring.
Figure 1.
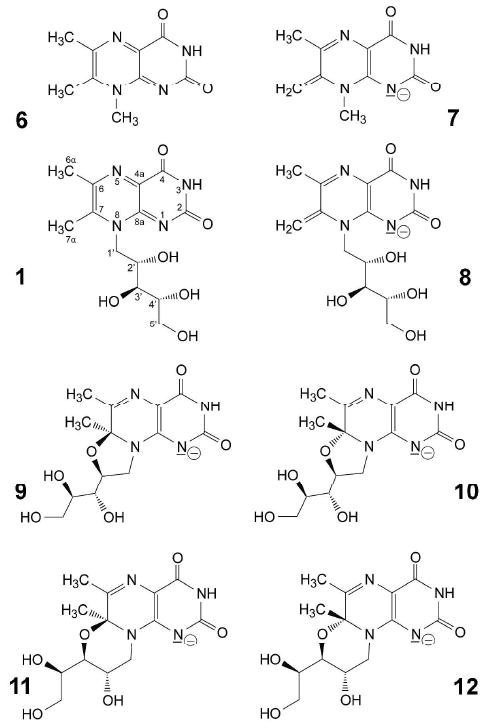
Structures of lumazine derivatives. 1, 6,7-dimethyl-8-ribityllumazine; 8, 7-exomethylene anion of 1; 9 – 12, tricyclic adduct anions of 1; 6, 6,7,8-trimethyllumazine; 7, 6,7,8-trimethyllumazine anion.
An intermediate of the riboflavin synthase-catalyzed reaction was first detected spectroscopically by single-turnover experiments and was subsequently isolated and identified as the pentacyclic compound 2a, which is a covalent adduct of two molecules of the enzyme substrate 1 (Scheme 1).8,11,12 The pentacyclic compound was shown to be a catalytically competent reaction intermediate which can be cleaved by riboflavin synthase in two different ways affording either two molecules of 1 (reverse reaction) or one molecule each of 4 and 5 (forward reaction).12 The forward reaction (cleavage of 2a under formation of riboflavin) is easily explained as a sequence of two elimination steps via the hypothetical intermediate 3 (Scheme 1). On the other hand, the reversible formation of 2 from 1 continues to be somewhat of an enigma. A hypothetical reaction sequence8 that combines the more recently discovered intermediate 2a (Scheme 2) with hypothetical reaction steps proposed around 1970 by Plaut, Wood and their coworkers7,13 appears rather cumbersome for a reaction that can proceed under mild conditions, even without catalysis (the mechanism originally proposed by Beach and Plaut,10,13 prior to the discovery of the pentacyclic intermediate 2a, is shown in the Supplementary Scheme S1).
Scheme 2.
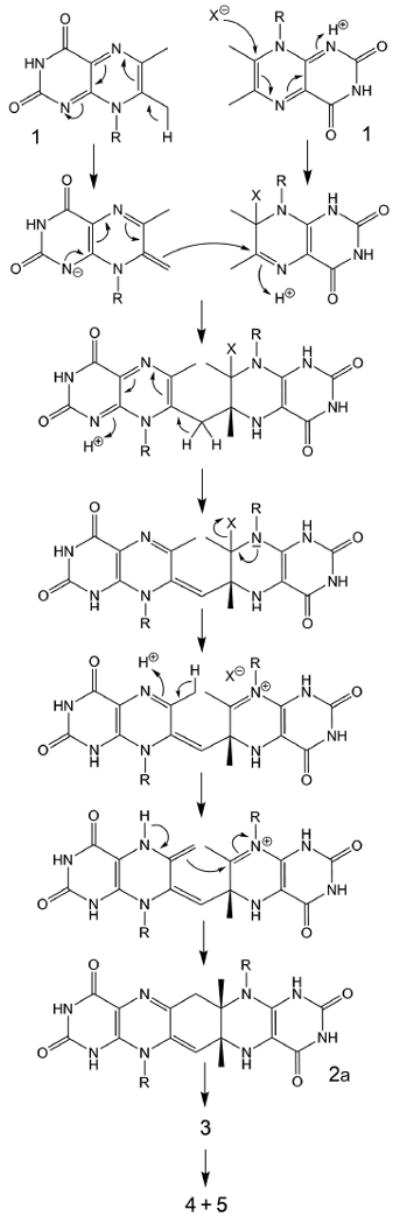
A hypothetical mechanism for the formation of riboflavin from 1. R, D-ribityl; X, unknown nucleophile.
Riboflavin synthase of Escherichia coli is a homotrimer of 25 kDa subunits. Each subunit folds into two similar domains, in line with internal sequence similarity.9,14,15 Whereas pairs of domains are related by pseudo-c2 symmetry, the homotrimer per se is devoid of trimeric symmetry, and spectroscopic studies of the enzyme/ligand interaction are hampered by the multiplicity of signals resulting from the topological non-equivalence of the six folding domains.16-18 For this reason, the NMR studies reported in this paper were performed with a recombinant N-terminal domain of riboflavin synthase of E. coli, which forms a c2 symmetric homodimer that can bind two molecules of 1 at topologically equivalent sites19-21.
In this paper, we show that the homodimeric N-terminal domain binds and stabilizes the exomethylene anion 8. Furthermore, we propose a simple mechanism for the formation of the pentacyclic intermediate 2a via hydride transfer between two substrate molecules and a consecutive 4+2 cycloaddition.
Results
At neutral pH, the optical spectra of 1 and 6 in aqueous solution and of 1 in complex with N-terminal riboflavin synthase domain are all similar (Fig. 2). Under alkaline conditions, the optical spectrum of protein-bound 1 resembles that of 7 and differs substantially from that of uncomplexed 1.
Fig. 2.
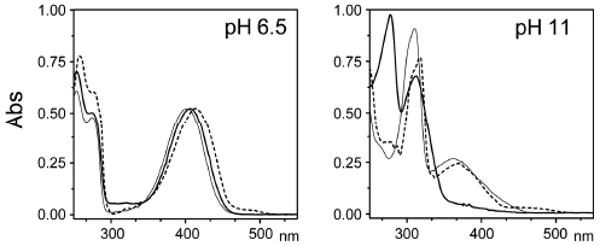
Absorption spectra of lumazine derivatives under neutral and alkaline conditions. Thick line: free 1, thin line: free 6, dashed line: 1 complex with N-terminal domain of riboflavin synthase.
The difference in the absorption spectra of 1 and 6 in alkaline aqueous solution is well understood. Whereas deprotonation of 6 affords the exomethylene anion 7, the deprotonation of 1 affords a complex mixture of anions that is dominated by the tricyclic anion species 9 – 12, and where the exomethylene anion 8 is only present in small amounts.7,22-25 The optical spectra shown in Fig. 2 suggested that the N-terminal domain of riboflavin synthase can bind the exomethylene anion 8 but none of the tricyclic anions 9 – 12. In search of unequivocal confirmation, we decided to study the protein/ligand complex by NMR under neutral and alkaline conditions.
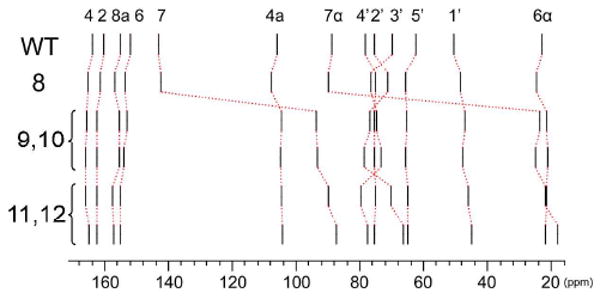
Fig. 3 shows 13C NMR spectra of the N-terminal riboflavin synthase domain (wild type) in complex with [U-13C13]-1 at different pH values. Different 13C signal sets (each comprising 13 signals and obviously representing all carbon atoms of the bound ligand) were observed at pH 6.9 and 9.0. The spectrum at pH 8.5 shows a mixture of these respective components. Hence, the neutral and the alkaline species observed are not in rapid exchange. The neutral and alkaline signal sets each comprise one respective singlet-type signal, whereas the other signals appear as multiplets. The singlet signals of the neutral and alkaline spectra, respectively, are easily assigned as C-2 of the bound substrate, and the multiplets can be assigned on basis of 13C13C coupling multiplicity. Despite that, we decided to obtain more rigorous assignments by comprehensive isotopologue editing. Specifically, we obtained 13C NMR spectra of protein/ligand complexes using [6α,6,7,7α-13C4]-1 and [7α-13C1]-1 (Fig. 4).
Fig. 3.
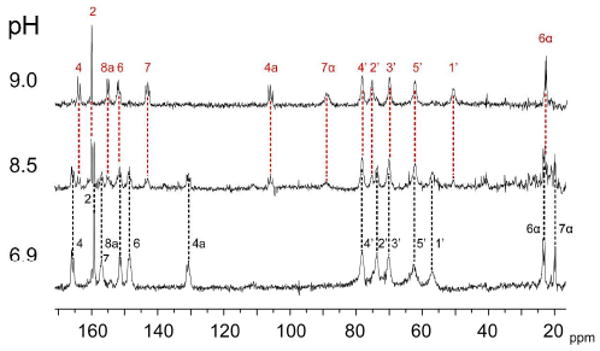
13C NMR spectra of [U-13C13]-1 in complex with the N-terminal domain of riboflavin synthase (wild type).
Fig. 4.
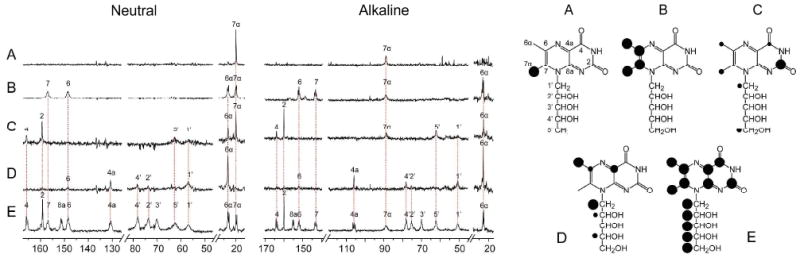
Left, 13C NMR spectra of isotopologues of 1 in complex with the N-terminal domain of riboflavin synthase (wild type) at pH 6.9 (neutral) and pH 11.0 (alkaline). Right, labeling patterns of 1 isotopologue samples used; A, [7α-13C1]-1; B, [6α,6,7,7α-13C4]-1; C, 1 biosynthesized from [1-13C1]glucose; D, 1 biosynthesized from [2-13C1]glucose; E, [U-13C13]-1; the size of the dots indicates 13C enrichment.
We also made use of isotopologue abundance editing, a technique that we have pioneered earlier for assignment of ligand signals in protein/ligand complexes.26-28 Briefly, we used samples of 1 that had been prepared by fermentation of a recombinant E. coli strain with [1-13C1]glucose or [2-13C1]glucose as the sole carbon source. The resulting samples have been reported earlier to represent mixtures of various isotopologues present at different abundances (Fig. 4 C, D). The relative intensities of 13C signals in spectra of the isotopologue mixtures in complex with the protein can provide abundant assignment information as shown in more detail in previous studies.26,27 It should be noted that the multipronged assignment approach based on specifically labelled samples of 1, randomly labelled isotopologue mixtures of 1 and 13C13C coupling patterns from multiply 13C-labelled 1 provides unequivocal assignments for all ligand carbon atoms. Most notably, C-7α is firmly assigned by the spectra obtained with [7α-13C1]-1, and C-7 is firmly assigned by the experiments with [6α,6,7,7α-13C4]-1.
In summary, these experiments provide a massively overdetermined set of 13C assignments for all ligand carbon atoms at neutral and alkaline pH that are summarized in Fig. 5 and Supplementary Table S1 (together with additional data obtained with mutant proteins and with data on the ligand in neutral aqueous solution). Most notably, at alkaline pH, the signal of C-7α of the protein-bound ligand is shifted to lower field by about 69 ppm, to a position at about 89 ppm. Major pH-associated chemical shift changes are also observed for C-4a (shifted upfield by about 25 ppm), C-7 (shifted upfield by about 14 ppm) and C-1′ (shifted upfield by about 6 ppm).
Fig. 5.
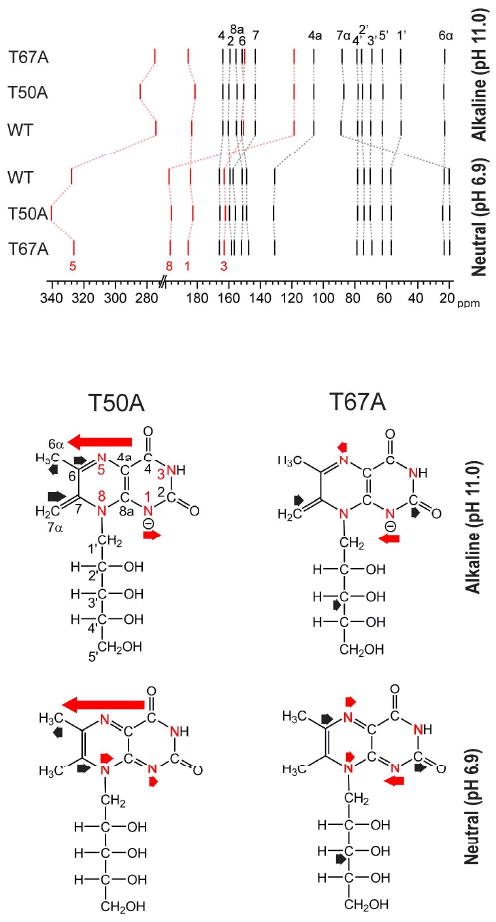
Top, 13C chemical shifts (black) and 15N chemical shifts (red) of 1 in complex with the N-terminal domain of riboflavin synthase [wild type (WT) or mutant as indicated]. In the bottom part of the figure, the impact of point mutations on the chemical shifts of neutral 1 and its exomethylene anion (8) is shown by arrows (13C shift changes, black; 15N shift effects, red).
A detailed study of the 13C chemical shifts of the various anionic species of 1 that are in a state of equilibrium in alkaline aqueous solution has been performed earlier by Bown et al.22 These data are summarized in Fig. 6, using the same display format as in Fig. 5. Also included in this Figure are the chemical shift values of 1 in complex with the riboflavin synthase N-domain at alkaline pH (repeated from Fig. 5). It is immediately obvious from this compilation that the 13C shifts of 1 in complex with the protein under alkaline conditions match those of the exomethylene-type anion but not of any other known anionic species of 1. The most diagnostic feature is the 13C resonance at about 89 ppm that we can unequivocally assign as an exomethylene group arising by deprotonation of the 7α methyl group of 1.
Fig. 6.
13C Chemical shifts of lumazine derivatives. WT, exomethylene anion 8 in complex with the N-terminal domain of riboflavin synthase; 8, exomethylene anion 8 in aqueous solution22; 9 - 12, tricyclic anions 9 – 12 in aqueous solution (data from Ref.22; cf. Fig. 2); pH, 11.0.
We have also obtained 15N NMR spectra of the recombinant riboflavin synthase domain in complex with various 15N-labeled samples of 6,7-dimethyl-8-ribityllumazine at neutral and alkaline conditions. These data are also summarized in Fig. 5. Major upfield shifts were noted for the signals of N-5 and N-8 upon transition from neutral to alkaline pH, and these can be safely assumed to reflect the transition from the neutral molecule to the exomethylene anion of the bound ligand. Notably, 15N assignments had not been previously reported for the exomethylene anion of 1 (in fact, they would not be easily obtained for the free ligand in solution because of sensitivity limitations, since the exomethylene form is a minor species under these conditions). In summary, the transition from the neutral 1 to anionic 8 causes major upfield shifts of C-4a, N-5 and N-8. These changes are tentatively interpreted as an increase in electron density at these respective positions and suggest that the negative charge is delocalized predominantly to the pyrazine ring of the bicyclic lumazine system.
Photometric titration of 1 in complex with the protein affords a pKa of 8.0 for the bound ligand, whereas 1 in aqueous solution has an apparent pKa of 7.9, and 6 in aqueous solution has a pKa of 9.8 (both characterizing protonation-deprotonation equilibrium of each respective compound) (Table 1, Supplementary Fig. S1). The increased apparent acidity of 1, by comparison with 6, is probably due to the higher stability of the tricyclic anions 9 – 12 as compared to the exomethylene anion, which is only present as a trace component in the anion mixture in aqueous solution. The pKa values suggest that the complexation with the N-terminal domain of riboflavin synthase stabilizes the exomethylene anion to a significant degree. This raises the question of specific interactions between the protein-bound lumazine derivative and its specific active site environment.
Table 1.
Properties of lumazine derivatives.
| Sample | pKa | Absorbance maxima, nm | Isosbestic points, nm | |
|---|---|---|---|---|
| pH 6.9 | pH 11.0 | |||
| 1 (aqueous solution) | 7.9 | 407 | 312 | 346 |
| 1 in complex with wild type riboflavin synthase | 8.0 | 413 | 318, 370 | 378 |
| 1 in complex with riboflavin synthase mutant T50A | 10.1 | 409 | 317, 376 | 369 |
| 1 in complex with riboflavin synthase mutant T67A | 8.9 | 418 | 315, 375 | 375 |
| 6 (aqueous solution) | 9.8 | 401 | 311, 360 | 369 |
As shown by X-ray crystallography, the N-terminal riboflavin synthase domain can bind a structural analog of the substrate 1 in a shallow surface depression (Fig. 7).9,20,21 One side of the heterocyclic molecule is solvent-exposed. The protein contacts the heterocyclic moiety predominantly via backbone elements that are believed to engage in hydrogen bonding. The γ hydroxy group of threonine 50 has been proposed to serve as a hydrogen bond donor for N-5, and the γ hydroxy group of threonine 67 is believed to serve as a hydrogen bond donor for N-1 of bound ligands. Moreover, that hydroxy function is also a candidate for an acceptor-type hydrogen bond interaction with the position 3′ hydroxy group of the ribityl side chain.
Fig. 7.
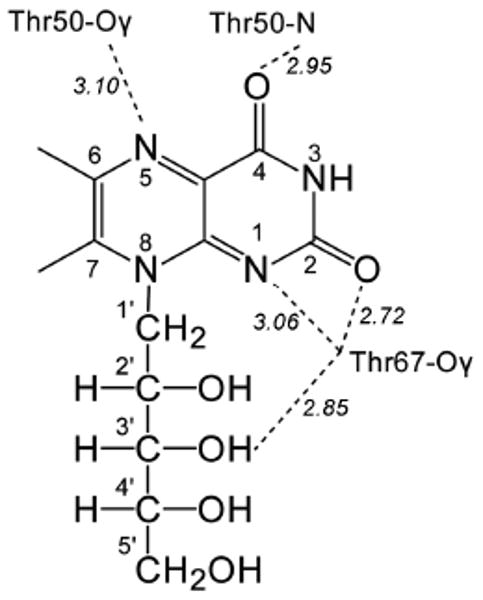
Hydrogen bond contacts of 1 in complex with the N-terminal domain of riboflavin synthase (based on the structure of riboflavin in complex with the N-terminal domain of riboflavin synthase, pdb code 1PKV21). Hydrogen bonding distances (Å) are shown in italic.
In order to analyze the proposed hydrogen bonding network in more detail, we replaced the threonine residues 50 and 67 with alanine residues in the N-terminal riboflavin synthase domain of E. coli. pKa values for 1 in complex with the various mutant proteins were obtained by photometric titration (Table 1). The replacement of threonine residues 50 or 67 with alanine decreased the acidity of protein-bound 1 by one to two orders of magnitude.
NMR experiments were performed using 13C- and 15N-labelled isotopologues of 1 in complex with the wild type and mutant proteins under neutral and acidic conditions. Chemical shifts modulation caused by the amino acid replacements are summarized in Fig. 5 and Table S1. The replacement of threonine 50 by alanine shifts the 15N NMR signal of N-5 to lower field (cf. Table S1 B). The neutral ligand 1 and the ligand anion 8 in complex with the protein are similarly affected. This is well in line with the presence of a hydrogen bond between the side chain hydroxy group of threonine 50 and N-5 of the ligand (Fig. 7). The chemical shift changes accompanying the replacement of threonine 67 by alanine are smaller, both at neutral and alkaline pH. Notably, that amino acid substitution causes a slight downfield shift of C-3′ carrying a hydroxy group that has been proposed to be involved in hydrogen bond interaction with the side chain hydroxy group of threonine 67.21 A similar pattern of 13C chemical shifts is also observed when riboflavin was used as ligand (Supplementary Fig. S3, cf. Supplementary Table S2).
Discussion
In alkaline aqueous solutions of 1, the exomethylene anion 8 is only present as a trace component (less than 1% as estimated by optical absorbance spectroscopy) in the complex equilibrium mixture comprising at least five anionic species (Fig. 1). On the other hand, the exomethylene form is the only anionic species detectably binding to the N-terminal domain dimer of riboflavin synthase.
The pKa of 6,7,8-trimethyllumazine (6) is 9.8, as compared to the apparent pKa of 7.9 for 1 (Table 1). However, since the exomethylene anion accounts for 100% of anions in case of 6,7,8-trimethyllumazine (6) and less than 1% in case of 1, the values signify that the CH acidity of the position 7 methyl group is about the same in both compounds. The higher apparent acidity of 1 is not caused by a change in CH acidity, but by the higher stability of the tricyclic anions.
The data obtained with mutant proteins support the notion that the exomethylene anion is specifically stabilized by hydrogen bonding with the side chain hydroxy groups of threonine residues 50 and 67. Mutations that abolish the hydrogen-bonding capacity of these amino acids increase the pKa of the ligand (equivalent to reduced CH acidity of the position 7 methyl group). The data suggest that the free enthalpy of the exomethylene anion is lowered to a larger degree by binding to the wild type protein as compared to that of the neutral molecule.
The N-terminal domain of riboflavin synthase has been shown to serve as the acceptor site for C-4 transfer between the two substrate molecules.9 The stabilization of the exomethylene anion 8 by the N-terminal domain is well in line with the requirements of the hypothetical reaction mechanism shown in Scheme 2. However, as pointed out earlier, the previously published mechanistic hypotheses, including the specific variation on that theme that is shown in Scheme 2, require the obligatory participation of a nucleophile (designated X in Scheme 2), but protein structure analysis in conjunction with mutagenesis studies has failed to identify an amino acid residue that would qualify for that function.9,20,21,29 Although a water molecule could be implicated, as a last-ditch defense, to serve as the elusive nucleophile X in Scheme 2, it appears timely to explore alternative mechanistic concepts. Such an alternative, centered around the concept of hydride transfer between the exomethylene anion and a second substrate molecule and a subsequent 4+2 cycloaddition is presented in the Scheme 3.
Scheme 3.
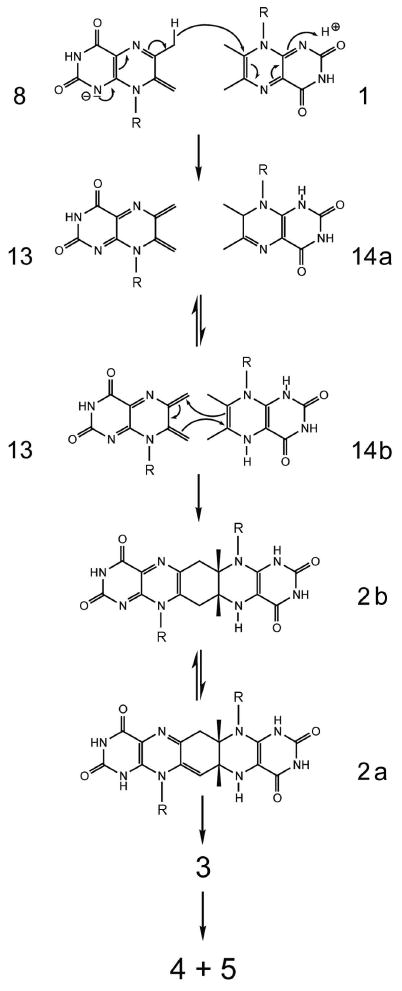
Proposed mechanism for riboflavin synthase via hydride transfer. Tautomerization reactions (tautomer pairs 2a/2b and 14a/14b, respectively) are indicated by double arrows.
Specifically, we propose that the exomethylene anion 8 bound at the N-terminal domain of riboflavin synthase donates a hydride ion to C-7 of a neutral 1 molecule that is bound at the C-terminal domain of an adjacent subunit, thus affording (1H,7H)-dihydro-6,7-dimethyl-8-ribityllumazine (14a) and the dehydrolumazine derivative 13. Tautomerization of 14a may yield (1H,5H)-dihydro-6,7-dimethyl-8-ribityllumazine (14b) which could then undergo a 4+2 cycloaddition affording the pentacyclic intermediate 2a. These proposed reaction steps are discussed in more detail below.
-
The proposed hydride transfer between anionic 8 and neutral 1 resembles the Cannizzaro reaction affording an alcohol and a carboxylate anion by dismutation of an aromatic aldehyde where an anionic species resulting from the addition of a hydroxy group of one aldehyde molecule donates a hydride ion to a neutral aldehyde molecule. Notably, the Cannizzaro reaction proceeds extremely fast in aqueous solution at room temperature. It should also be noted that hydride transfer, in general, is one of the most common elementary processes in enzyme catalysis.
In parallel with riboflavin, 1 can undergo 2-electron reduction affording a dihydro derivative.30 The pyrazine moiety of 8 is electron-rich as shown by its 13C and 15N chemical shifts (cf. Fig. 4). At the active site of E. coli riboflavin synthase, the two bound substrate molecules are ideally placed to enable the transfer of hydride from the position 6 methyl group of the exomethylene anion 8 (bound at the N-terminal domain) to C-7 carbon of 1 (bound at the C-terminal domain).9 The distance to be traversed is quite short, and vanishing and emerging π orbitals have favourable orientations.
The two-electron reduction product of 1 exists as an equilibrium mixture of the tautomers 14a and 14b.30
The electronics favor an inverse-electron-demand 4+2 cycloaddtiion involving overlap of the HOMO of the dienophile 14b with the LUMO of the diene 13. This scenario for the reaction is favored by the electron-donating groups on the dienophile and the electron withdrawing groups on the diene. The diene is electron-poor due to conjugative interactions involving the two electronegative carbonyl groups, as well as the two N-1 and N-5 imine nitrogens. On the other hand, the dienophile 14b is electron-rich through donation of electrons by the N-5 enamine nitrogen atom. Electron donation from the N-8 ribitylamino nitrogen toward the dienophile is less important because it is part of a vinylogous amide system.
The mechanism proposed in Schemes 2 and 3 are both very well in line with the large isotope effects that were observed already in 1970 with 6α deuterium and tritium isotopologs of 1 as substrates of riboflavin synthase.31 Specifically, the hypothetical mechanism in Scheme 2 requires the abstraction of a proton from the 6α-methyl group of the lumazine molecule destined to serve as 4-carbon acceptor, and the novel hypothesis proposed in Scheme 3 requires the transfer of a hydride anion. In both hypothetical pathways, the heterolytic cleavage of the carbon-hydrogen could qualify as a rate-determining step; hence, hydrogen isotope effects fail to decide between the two hypotheses under discussion.
With the hypothesis in Scheme 3, the reverse and forward reactions affording two molecules of 1 or one respective molecule of each 4 and 5 are both catalyzed with similar velocity by riboflavin synthase.8,11 It should be mentioned that the two fragmentation pathways affording either 1 or a mixture of 4 and 5, respectively, are believed to depart from different tautomers of the pentacyclic intermediate (cf. Scheme 4). Notably, the tautomers 2a and 2b are endowed with different sets of predetermined fracture points (Scheme 4).
In contrast to the mechanism in Scheme 2, Supplementary Scheme S1, and related concepts, the novel hypothetical mechanism (Scheme 3) has no requirement for an amino acid side chain to act as a nucleophile (or for any non-protein nucleophile). This is advantageous since X-ray crystallography and mutation screening have failed to identify an amino acid residue that could serve as the elusive nucleophile in Scheme 2.9,29
By comparison with the reaction sequence shown in Scheme 2, the novel hypothesis is parsimonious with regard to the number of reaction steps conducive to the pentacyclic intermediate.
Scheme 4.
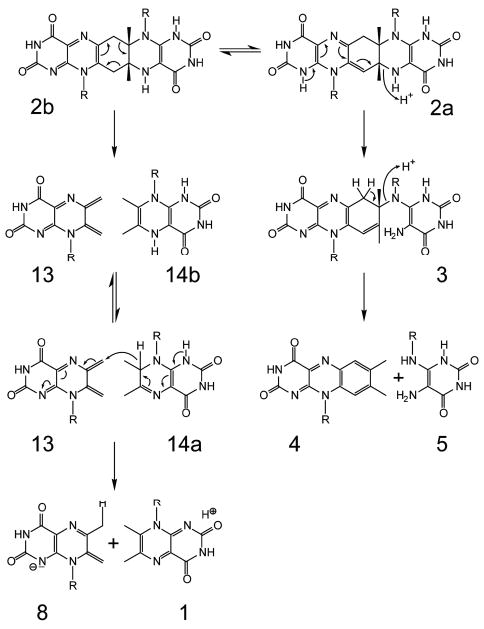
Proposed fragmentation of the experimentally documented pentacyclic intermediate (shown in the tautomeric forms 2a and 2b). The different fragmentation paths originating from the two tautomeric forms afford either 4 and 5 or 1 and 8 (i.e. the neutral and exomethylene anion form of the 6,7-dimethyl-8-ribityllumazine).
Experimental Section
Materials
Isotopologues of 1 and of riboflavin were prepared by published procedures.26,32,33
Microorganisms and plasmids
Bacterial strains and plasmids used in this study are summarized in the Supplementary Table S3.
Site directed mutagenesis
Site-directed mutagenesis was performed as described earlier.29 The plasmid pERN was used as template.19 Oligonucleotides used as primers are shown in the Supplementary Table S4.
Bacterial culture and protein purification
Recombinant N-terminal domain of riboflavin synthase from E. coli was prepared by published procedures.19 Briefly, frozen cell mass (5 g) was suspended in 50 mL of buffer A containing 50 mM sodium phosphate, pH 7, 300 mM sodium chloride, 10 mM imidazole and 0.02% (w/v) sodium azide. Lysozyme (5 mg) and DNAse (5 mg) were added, and phenylmethanesulfonylfluoride was added to a final concentration of 0.5 mM. The mixture was incubated at room temperature for 30 min with stirring and was then passed through a French Press. Cell debris was removed by centrifugation and the supernatant was placed on a nickel chelating column (2 × 15 cm) that had been equilibrated with buffer A. The column was washed with 5 column volumes of buffer A and was then developed with a linear gradient of 10 to 500 mM imidazole. Fractions were combined, dialyzed against 50 mM sodium phosphate, pH 7, containing 1 mM DTT, and were then concentrated by ultrafiltration.
Ligand Exchange
A solution containing 30 mg of N-terminal riboflavin synthase domain in 50 mL of buffer A was placed on a nickel chelating column (2 × 5 cm) that was then washed with buffer A until the effluent was colorless (mutant protein with replacement of C48 required the addition of 8 M urea). Buffer A (20 mL) containing 1.5 to 2 mg of isotope-labeled 1 or riboflavin was recycled through the column overnight. The column was then washed with buffer A and the reconstituted protein was eluted with 500 mM imidazole.
NMR Spectroscopy
Samples contained 50 mM sodium phosphate (pH 6.9, 8.5, 9.2, or 11.0), 10% D2O, 20 μM [1-13C1]glucose (as internal standard) and 1.5 to 2 mM protein loaded with isotopically labeled 1 as indicated. NMR spectra were recorded at 290 K using a DRX 500 spectrometer (Bruker Instruments, Karlsruhe, Germany). Composite pulse decoupling was used for 13C NMR measurements.
Supplementary Material
Acknowledgments
This work was supported by the Fonds der Chemischen Industrie and the Hans Fischer Gesellschaft e.V., by NIH grant GM51469 and a Korea Research Foundation Grant funded by the Korean Government.
References
- 1.Plaut GW. J Biol Chem. 1960;235:PC41–42. [PubMed] [Google Scholar]
- 2.Plaut GW. J Biol Chem. 1963;238:2225–2243. [PubMed] [Google Scholar]
- 3.Wacker H, Harvey RA, Winestock CH, Plaut GW. J Biol Chem. 1964;239:3493–3497. [PubMed] [Google Scholar]
- 4.Fischer M, Bacher A. Arch Biochem Biophys. 2008;474:252–265. doi: 10.1016/j.abb.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Rowan T, Wood HCS. Proc Chem Soc. 1963:21–22. [Google Scholar]
- 6.Beach R, Plaut GW. Tetrahedron Lett. 1969;40:3489–3492. doi: 10.1016/s0040-4039(01)88428-8. [DOI] [PubMed] [Google Scholar]
- 7.Paterson T, Wood HC. J Chem Soc [Perkin 1] 1972;8:1051–1056. doi: 10.1039/p19720001051. [DOI] [PubMed] [Google Scholar]
- 8.Illarionov B, Eisenreich W, Bacher A. Proc Natl Acad Sci USA. 2001;98:7224–7229. doi: 10.1073/pnas.131610698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerhardt S, Schott AK, Kairies N, Cushman M, Illarionov B, Eisenreich W, Bacher A, Huber R, Steinbacher S, Fischer M. Structure (Camb) 2002;10:1371–1381. doi: 10.1016/s0969-2126(02)00864-x. [DOI] [PubMed] [Google Scholar]
- 10.Beach R, Plaut GWE. Biochemistry. 1970;9:760–770. doi: 10.1021/bi00806a009. [DOI] [PubMed] [Google Scholar]
- 11.Illarionov B, Haase I, Bacher A, Fischer M, Schramek N. J Biol Chem. 2003;278:47700–47706. doi: 10.1074/jbc.M305050200. [DOI] [PubMed] [Google Scholar]
- 12.Illarionov B, Haase I, Fischer M, Bacher A, Schramek N. Biol Chem. 2005;386:127–136. doi: 10.1515/BC.2005.016. [DOI] [PubMed] [Google Scholar]
- 13.Plaut GW, Beach RL. Substrate specificity and stereospecific mode of action of riboflavin synthase. In: Singer TP, editor. Flavins and Flavoproteins. Elsevier Scientific Publishing Company; 1976. pp. 737–746. [Google Scholar]
- 14.Schott K, Kellermann J, Lottspeich F, Bacher A. J Biol Chem. 1990;265:4204–4209. [PubMed] [Google Scholar]
- 15.Liao DI, Wawrzak Z, Calabrese JC, Viitanen PV, Jordan DB. Structure (Camb) 2001;9:399–408. doi: 10.1016/s0969-2126(01)00600-1. [DOI] [PubMed] [Google Scholar]
- 16.Cushman M, Patrick DA, Bacher A, Scheuring J. J Org Chem. 1991;56:4603–4608. [Google Scholar]
- 17.Scheuring J, Lee J, Cushman M, Patel H, Patrick DA, Bacher A. Biochemistry. 1994;33:7634–7640. doi: 10.1021/bi00190a017. [DOI] [PubMed] [Google Scholar]
- 18.Scheuring J, Fischer M, Cushman M, Lee J, Bacher A, Oschkinat H. Biochemistry. 1996;35:9637–9646. doi: 10.1021/bi9600916. [DOI] [PubMed] [Google Scholar]
- 19.Eberhardt S, Zingler N, Kemter K, Richter G, Cushman M, Bacher A. Eur J Biochem. 2001;268:4315–4323. doi: 10.1046/j.1432-1327.2001.02351.x. [DOI] [PubMed] [Google Scholar]
- 20.Truffault V, Coles M, Diercks T, Abelmann K, Eberhardt S, Luttgen H, Bacher A, Kessler H. J Mol Biol. 2001;309:949–960. doi: 10.1006/jmbi.2001.4683. [DOI] [PubMed] [Google Scholar]
- 21.Meining W, Eberhardt S, Bacher A, Ladenstein R. J Mol Biol. 2003;331:1053–1063. doi: 10.1016/s0022-2836(03)00844-1. [DOI] [PubMed] [Google Scholar]
- 22.Bown DH, Keller PJ, Floss HG, Bacher A. J Org Chem. 1986;51:2461–2467. [Google Scholar]
- 23.Pfleiderer W, Mengel R, Hemmerich P. Chem Ber. 1971;104:2273–92. [Google Scholar]
- 24.Pfleiderer W, Bunting JW, Perrin DD, Nuebel G. Chem Ber. 1966;99:3503–3523. [Google Scholar]
- 25.Beach R, Plaut GWE. J Org Chem. 1971;36:3937–3943. [Google Scholar]
- 26.Illarionov B, Fischer M, Lee CY, Bacher A, Eisenreich W. J Org Chem. 2004;69:5588–5594. doi: 10.1021/jo0493222. [DOI] [PubMed] [Google Scholar]
- 27.Illarionov B, Lee CY, Bacher A, Fischer M, Eisenreich W. J Org Chem. 2005;70:9947–9954. doi: 10.1021/jo051662f. [DOI] [PubMed] [Google Scholar]
- 28.Eisenreich W, Joshi M, Illarionov B, Richter G, Romisch-Margl W, Muller F, Bacher A, Fischer M. FEBS J. 2007;274:5876–5890. doi: 10.1111/j.1742-4658.2007.06111.x. [DOI] [PubMed] [Google Scholar]
- 29.Illarionov B, Kemter K, Eberhardt S, Richter G, Cushman M, Bacher A. J Biol Chem. 2001;276:11524–11530. doi: 10.1074/jbc.M008931200. [DOI] [PubMed] [Google Scholar]
- 30.Macheroux P, Ghisla S, Hastings JW. Flavins and Flavoproteins. Walter de Gruyter&Co.; Berlin - New York: 1994. Bacterial luciferase: bioluminescence emission using lumazines as substrates; pp. 839–842. [Google Scholar]
- 31.Plaut GWE, Beach RL, Aogaichi T. Biochemistry. 1970;9:771–785. doi: 10.1021/bi00806a010. [DOI] [PubMed] [Google Scholar]
- 32.Sedlmaier H, Muller F, Keller PJ, Bacher A. Z Naturforsch C. 1987;42:425–429. doi: 10.1515/znc-1987-0416. [DOI] [PubMed] [Google Scholar]
- 33.Romisch W, Eisenreich W, Richter G, Bacher A. J Org Chem. 2002;67:8890–8894. doi: 10.1021/jo026105x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.