

Abstract
Linear peptides derived from the membrane proximal region of the gp41 ectodomain are effective inhibitors of HIV type 1 (HIV-1)-mediated fusion events. These inhibitory peptides lack structure in solution, rendering mechanistic interpretation of their activity difficult. Using structurally constrained analogs of these molecules, we demonstrate that the peptides inhibit infectivity by adopting a helical conformation. Moreover, we show that a specific face of the helix must be exposed to block viral infectivity. Recent crystal structures show that the region of gp41 corresponding to the inhibitory peptides is helical and uses the analogous face to pack against a groove formed by an N-terminal coiled-coil trimer. Our results provide a direct link between the inhibition of HIV-1 infectivity by these peptides and the x-ray structures, and suggest that the conformation of gp41 observed by crystallography represents the fusogenic state. Other agents that block HIV-1 infectivity by binding to this groove may hold promise for the treatment of AIDS.
Envelope glycoproteins gp120 and gp41 of the type 1 HIV (HIV-1) mediate viral entry into target cells and dictate cell-specific tropism (1). GP120 interacts with target cells via CD4 and one of several coreceptors (2), after which gp41 promotes the fusion of viral and cellular membranes. The mechanism by which gp41 mediates membrane fusion has recently been the subject of intensive study. Evidence suggests that the process may involve the formation of a coiled-coil trimer; a similar process is thought to drive the transition from resting to fusogenic states in influenza hemagglutinin (3–5).
Two classes of linear peptides representing different regions of gp41 have been found to inhibit viral fusion. The first of these (e.g., DP107) was derived from a portion of gp41 near the N-terminal fusion peptide. DP107 is helical in solution and oligomerizes in a manner consistent with coiled-coil formation (6, 7). A more potent class of peptides, represented by DP178, was derived from the C-terminal region of the gp41 ectodomain (8, 9). Although this region was predicted to be α-helical (10), DP178 and related molecules lack discernible structure in solution (8).
The C-terminal peptides, though unstructured in isolation, form a highly stable helical bundle (apparent Tm of 90°C) when mixed in a 1:1 ratio with N-terminal (DP107) peptides (11–13). Recent crystallographic studies (14, 15) have have shown that a C-terminal segment of gp41, which overlaps DP178, forms an extended helix that packs in an antiparallel fashion against a groove created by a coiled-coil trimer containing the DP107 region. A model for inhibition has been proposed wherein DP178 binds to a similar groove on full-length gp41 on the virion surface (14, 15). Implicit in this model is the assumption that the bioactive conformation of DP178 is indeed helical. To date, however, there has been no direct information that links the conformation observed by x-ray to the mechanism of peptide inhibition during viral fusion events.
We describe here a set of constrained α-helical peptides that we have prepared to address the following questions. (i) Do DP178 and related peptides inhibit viral fusion by adopting a helical conformation? (ii) If so, must the face of the helix that binds to the N-terminal coiled-coil be exposed for inhibition to occur? (iii) Can a small subset of DP178, if properly structured, effectively inhibit viral infectivity?
MATERIALS AND METHODS
Peptide Design.
At the inception of this work, detailed information regarding the association of DP178 with DP107 was not available. Because different inhibitory peptides overlapping the DP178 sequence had been described, we wished to identify the portion of DP178 most likely to interact through helix association, and to determine whether a specific face of the helix would present a hydrophobic heptad repeat. Coiled-coil propensities (16) were calculated for 29 distinct gp160 sequences. The N-terminal 27 residues of DP178 maintained a high overall score with a consistent heptad register and were selected for the reference peptide.
Peptide Synthesis.
Linear peptides were synthesized according to standard solid phase techniques by using 9-fluorenylmethoxycarbonyl chemistry (17). Helix dipole effects were minimized by blocking the C-termini as amides and the N-termini as succinate groups. After formation of the lactam locks (ref. 18; A. C. Braisted and J.K.J., unpublished results), the peptides were cleaved from the resin and purified to homogeneity by using preparative reversed phase HPLC with water/acetonitrile/0.1% TFA gradients in the mobile phase. The identity of each peptide was confirmed by electrospray mass spectrometry: HIV24, calculated mass 3396.8, observed, 3396.0; HIV30, calculated mass 3413.7, observed, 3413.8; HIV31, calculated mass 3520.0, observed, 3520.7; HIV35, calculated mass 3330.8, observed, 3330.5.
Circular Dichroism (CD) Studies.
CD spectra were recorded on an AVIV 62DS CD spectrometer by using 0.05-cm pathlength cuvettes at 7°C. Spectra were gathered by averaging data from three runs spanning 250 nm to 190 nm in 1.0-nm increments, with 2-s averaging time at each wavelength. Peptide concentrations were approximately 200 μM in a solution of 10 mM Tris⋅HCl, pH 7.5, with 6% acetonitrile (vol/vol). For conversion of raw data to molar ellipticity values, precise concentrations were determined by measuring A276 and A280 (19); these values were confirmed by quantitative amino acid analysis.
HIV-1 Strains.
Cell culture adapted strain MN was a gift from M. Norcross (U.S. Food and Drug Administration, Washington, DC); primary isolate JRCSF was from I. Chen (University of California, Los Angeles); BZ167, a primary isolate from Brazil, was from S. Zolla-Pazner (Veteran’s Affairs Medical Center, New York); 301660 was from J. Bradac (National Institute of Allergy and Infectious Diseases, Bethesda); Th009, a primary isolate from Thailand, was also from J. Bradac, (National Institute of Allergy and Infectious Diseases).
HIV-1 Infectivity Assays.
Peripheral blood mononuclear cells from HIV-1-negative donors were stimulated with phytohemagglutinin in RPMI 1640 medium containing interleukin 2 (Boehringer Mannheim), glutamine, 20% heat-inactivated fetal calf serum, and 50 μg/ml gentamicin for 24 h. The medium was removed and the cells were incubated overnight in RPMI 1640 medium with glutamine, 20% heat-inactivated fetal calf serum, and gentamicin. Pretitered virus stocks were equilibrated with peptides for 1 h before being added to the peripheral blood mononuclear cells (2.5 × 105 cells per well in 96-well plates). Cells were incubated at 37°C for 3 days, rinsed to remove extracellular virus and peptides, and then supplemented with fresh medium and incubated at 37°C for an additional day. Cells were lysed, and p24 antigen was determined by ELISA (Coulter). Peptides were tested in triplicate at each concentration. Each assay also included the following controls (10 wells each on 96-well plates): uninfected cells without peptide as a negative control, infected cells without peptide as a positive control, and virus inoculum without cells to establish a baseline p24 level. Cytotoxicity was evaluated by incubating uninfected cells either in the presence (approximately 100 μM) or the absence of peptide. After 4 days, cells were counted and cell numbers were compared. No inhibition of cell growth was observed under these conditions.
RESULTS
Based on the coiled-coil analysis, a truncated form of DP178 (HIV35; Fig. 1) was selected as a reference. Because short peptides generally do not form stable α-helices in solution (20), we incorporated a covalent crosslink between amino acid side chains at positions i and i + 7 of the polypeptide chain to lock the intervening residues into an α-helical conformation (18). The helical presentation of side chains in positions a and d of the heptad was enforced by introducing a crosslink between pairs of residues occupying adjacent f positions on the opposite face of the helix. Analogs of HIV35 (Fig. 1) were prepared containing either one (HIV24) or two (HIV31) tethers to impart increasing helicity. A control peptide (HIV30) was prepared in which a tether was introduced between successive d residues to stabilize helicity while blocking potential binding interactions across the a–d face.‖
Figure 1.
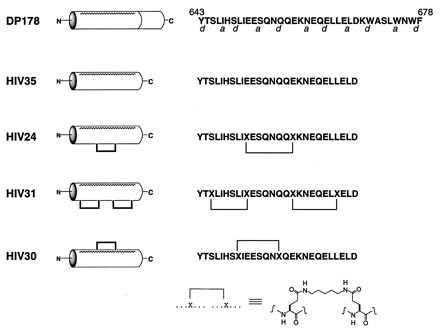
Sequences and schematic representations of the inhibitory peptides. The cylinders represent α-helices, with the stippled faces corresponding to the 4,3 hydrophobic repeat. Covalent constraints linking side chains at i and i + 7 are represented as dark lines.
CD analysis (Fig. 2a) confirms that the introduction of covalent locks markedly increases the helicity of the HIV35 analogs. The unconstrained peptide HIV35 has an almost featureless spectrum, similar to that reported for DP178 (11). The CD spectra of peptides containing a single constraint (HIV24 and HIV30) display maxima at 195 nm and minima at 209 and 222 nm, characteristic of α-helices. The intensity ratios of these regions of the spectra are skewed from ideality, suggesting that segments of the peptide backbone outside the constrained segment are disordered.** By contrast, the doubly constrained analog HIV31 appears to be largely helical by CD, giving the shape and intensity profile of a typical α-helix. HIV31 remains helical when heated from 7°C to 67°C and cooled back to 7°C (Fig. 2b), indicating that the peptide is structurally robust.
Figure 2.
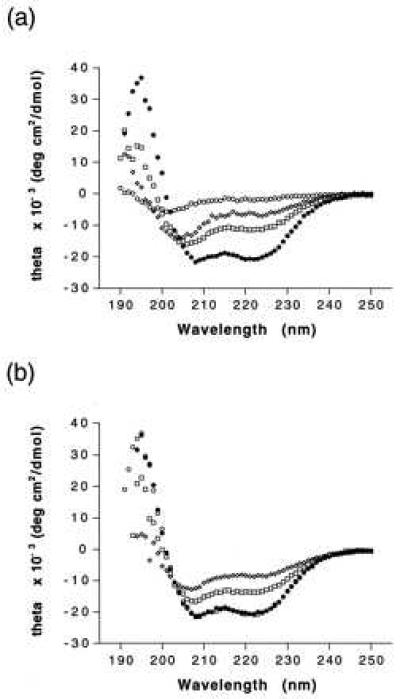
(a) CD spectra of peptides HIV24 (□), HIV30 (⋄), HIV31 (•), and HIV35 (○) at 7°C. (b) CD spectra of peptide HIV31 at 7°C (○), 37°C (□), 67°C (⋄), followed by cooling back to 7°C (•). All spectra were acquired in 10 mM Tris⋅HCl, pH 7.5.
When tested in viral infectivity assays, the peptides display a striking pattern of relative potency that extends across both syncitium-inducing and non-syncitium-inducing strains of HIV-1 (21). As shown in Fig. 3, truncating the hydrophobic C terminus of DP178 (HIV35) causes a dramatic drop in activity, which is partially restored when a single constraint (and partial α-helical character) is introduced (HIV24). Adding a second constraint (HIV31) imparts strong helical character and enhances the potency of the peptide to levels comparable to DP178. Thus, the additional stabilization afforded by preorganizing HIV31 into an active helical conformation offsets the loss of binding energy caused by deleting the C terminus. By contrast, inducing helicity in a manner that blocks the a–d face (HIV30) completely ablates activity.
Figure 3.
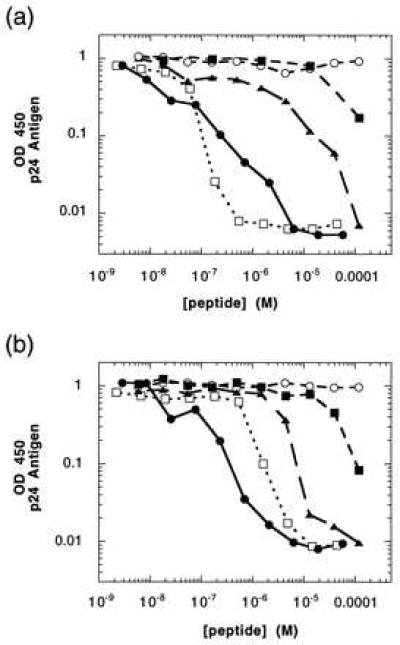
Effect of inhibitory peptides in primary infectivity assays by using peripheral blood mononuclear cells with virus JRCSF, a non-syncitium-inducing strain (a), and BZ167, a syncitium-inducing strain (b). ▴, HIV24; ○, HIV30; •, HIV31; ▪, HIV35; □, DP178.
A series of shorter constrained peptides was prepared to test whether a subset of DP178 could block infectivity. These peptides spanned positions 643–656, 649–662, 656–669, and 663–678 of HIV-1LAI, and were tethered between adjacent residues at the f positions of the heptad as indicated in Fig. 1. All of the shorter peptides, whether constrained or unconstrained, failed to show significant activity (data not shown).‡‡
DISCUSSION
The relative activities of HIV35, HIV24, and HIV31 demonstrate a clear correlation between helicity and inhibitory potency. The widely disparate activities of HIV30 and HIV24 indicate that peptide inhibition also requires exposure of the face of the helix containing the 4,3 hydrophobic repeat seen by crystallography to pack against the N-terminal trimer core of gp41. The lack of cytotoxicity observed for these peptides, coupled with previous observations that DP178 prevents syncitium formation (8), further suggests that these molecules function by interacting directly with gp41.
Our data, combined with prior model studies on isolated peptides and the recently published crystal structures, strongly support the hypothesis that the peptides inhibit membrane fusion by binding to a resting state of gp41 and preventing transformation to the fusogenic state (12). Peptide HIV31 is constrained to be helical, and is likely to interact as such with a cognate surface that is accessible in the resting state of gp41. X-ray analysis shows that the region of gp41 corresponding to HIV31 binds to the groove formed by the N-terminal coiled coil, burying the helical face shown here to be responsible for inhibitory function. We believe that this groove represents the cognate surface for the peptides. The inactivity of the truncated peptides suggests that a substantial portion of the surface of the groove must be occupied for inhibition to occur, or that simple mimicry of a subset of the native epitope may not provide sufficient binding energy for this site.
Fig. 4 outlines schematically a plausible mechanism by which the constrained helices prevent the assembly of the fusogenic state of gp41. The resting state of gp41 (Fig. 4 Upper) is presumed to be constitutively trimerized, featuring a coiled-coil bundle near the N terminus. The region corresponding to the C terminus of the ectodomain (which contains DP178) is not initially bound to the trimer bundle and has an unknown conformation. The binding of gp120 to either CD4, a coreceptor, or both, may induce a conformational shift that allows association of the C-terminal portion of gp41 with the N-terminal bundle (Fig. 4 Lower Right). The resulting antiparallel helical array observed in the x-ray structures is presumably the fusogenic state of gp41. Rearrangement to this state can be prevented if the trimer grooves are occupied by inhibitory peptides (Fig. 4 Lower Left).
Figure 4.
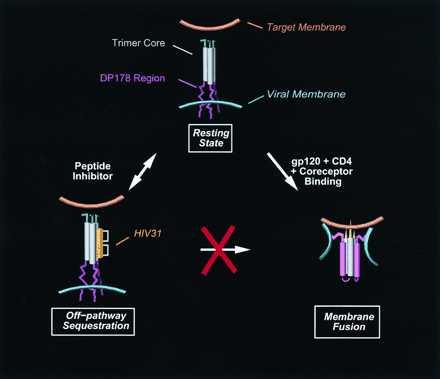
Proposed mechanisms for assembly of the fusogenic state of gp41 (Lower Right) and inhibition by constrained peptides (Lower Left).
In accord with reports from other laboratories (7–9), we have observed that DP178 displays similar inhibitory potency when tested against both cell culture-adapted strains (MN/H9) and primary isolates (301660 and Th009) (data not shown). Strain Th009 env is subtype E and is genetically distant from the North American subtype B env (e.g., JRCSF) (21). The potency of HIV24 is likewise retained when tested against Th009 (data not shown). Thus, genetically distant and phenotypically distinct subtypes of HIV-1 appear to employ a common fusion machinery that can be effectively blocked by peptides presenting a structurally defined epitope. Given that the groove to which this epitope is proposed to bind is formed by one of the most highly conserved regions in the HIV-1 genome (22), other pharmaceutical agents that target this groove may hold promise for the treatment of AIDS.
Acknowledgments
We thank Andrew Braisted for advice and reagents; Andrea Cochran for critically reading the manuscript; Nick Skelton for performing NMR analyses of various locked peptides; and Phil Berman and Gerry Nakamura for helpful discussions at the outset of the project.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviation: HIV-1, HIV type 1.
In the crystal structures, the C-terminal peptides pack against the N-terminal trimer by using the heptad face identified by the scoring algorithm, although their interaction more closely resembles an antiparallel helix bundle than a true coiled-coil.
NMR studies (N.J. Skelton, unpublished results; see also ref. 18) of similarly-constrained peptides have shown that molecules with only one constraint are fully helical within the locked region and frayed outside to varying degrees.
Subsequent to the inception of this work, crystal structures showed that a segment of gp41 N-terminal to DP178 contains a Trp-Trp-Ile cluster occupying a cavity on the surface of the N-terminal coiled coil (14, 15). An additional constrained peptide encompassing residues 631–644 of HIV-1LAI was prepared to test whether a structured presentation of this cluster was sufficient to inhibit infectivity. This molecule also failed to show activity.
References
- 1.Freed E O, Martin M A. J Biol Chem. 1995;270:23883–23886. doi: 10.1074/jbc.270.41.23883. [DOI] [PubMed] [Google Scholar]
- 2.D’Souza M P, Harden V A. Nat Med. 1996;2:1293–1300. doi: 10.1038/nm1296-1293. [DOI] [PubMed] [Google Scholar]
- 3.Carr C M, Kim P S. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- 4.Bullough P A, Hughson F M, Skehel J J, Wiley D C. Nature (London) 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 5.Wilson I A, Skehel J J, Wiley D C. Nature (London) 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 6.Wild C, Dubay J W, Greenwell T, Teaster Baird J, Oas T G, McDanal C, Hunter E, Matthews T. Proc Natl Acad Sci USA. 1994;91:12676–12680. doi: 10.1073/pnas.91.26.12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. Proc Natl Acad Sci USA. 1992;89:10537–10541. doi: 10.1073/pnas.89.21.10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wild C T, Shugars D C, Greenwell T K, McDanal C B, Matthews T J. Proc Natl Acad Sci USA. 1994;91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang S, Lin K, Strick N, Neurath A R. Nature (London) 1993;365:113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- 10.Gallaher W R, Ball J M, Garry R F, Griffin M C, Montelaro R C. AIDS Res Hum Retroviruses. 1989;5:431–440. doi: 10.1089/aid.1989.5.431. [DOI] [PubMed] [Google Scholar]
- 11.Lawless M K, Barney S, Guthrie K I, Bucy T B, Stephen R, Petteway J, Merutka G. Biochemistry. 1996;35:13697–13708. doi: 10.1021/bi9606962. [DOI] [PubMed] [Google Scholar]
- 12.Lu M, Blacklow S C, Kim P S. Nat Struct Biol. 1995;2:1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 13.Rabenstein M D, Shin Y-K. Biochemistry. 1996;35:13922–13928. doi: 10.1021/bi961743t. [DOI] [PubMed] [Google Scholar]
- 14.Weissenhorn W, Dessen A, Harrison S C, Skehel J J, Wiley D C. Nature (London) 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 15.Chan D C, Fass D, Berger J M, Kim P S. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 16.Lupas A, Dyke M V, Stock J. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 17.Fields G B, Noble R L. Int J Peptide Protein Res. 1990;35:161–214. doi: 10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- 18.Phalen J C, Skelton N J, Braisted A C, McDowell R S. J Am Chem Soc. 1997;119:455–460. [Google Scholar]
- 19.Edelhoch H. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 20.Marqusee S, Robbins V M, Baldwin R L. Proc Natl Acad Sci USA. 1989;86:5286–5290. doi: 10.1073/pnas.86.14.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Huang Y, He T, Cao Y, Ho D D. Nature (London) 1996;383:768. doi: 10.1038/383768a0. [DOI] [PubMed] [Google Scholar]
- 22.Gao F, Morrison S G, Robertson D L, Thornton C L, Craig S, Karlsson G, Sodroski J, Morgado M, Galvao-Castro B, Briesen H v, Beddows S, Weber J, Sharp P M, Shaw G M, Hahn B H. J Virol. 1996;70:1651– 1667. doi: 10.1128/jvi.70.3.1651-1667.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]