

Abstract
Imprinted genes play an important role in fetal and placental development. Using quantitative bisulfite pyrosequencing assays, we determined the DNA methylation levels at two paternally methylated (H19 and MEG3) and four maternally methylated (LIT1, NESP55, PEG3, and SNRPN) imprinted regions in fetal muscle samples from abortions and stillbirths. Two of 55 (4%) spontaneous abortions and 10 of 57 (18%) stillbirths displayed hypermethylation in multiple genes. Interestingly, none of 34 induced abortions had extreme methylation values in multiple genes. All but two abortions/stillbirths with multiple methylation abnormalities were male, indicating that the male embryo may be more susceptible to excess methylation. Hypermethylation of multiple imprinted genes is consistent with stochastic failures of the mechanism, which normally protects the hypomethylated allele from de novo methylation after fertilization. Two of six informative abortions/stillbirths with H19 hypermethylation revealed significant biallelic expression of the autocrine growth factor IGF2. In two other cases hypermethylation of MEG3 was associated with transcriptional down-regulation. We propose that primary epimutations resulting in inappropriate methylation and expression patterns of imprinted genes may contribute to both normal human variation and disease, in particular spontaneous pregnancy loss.
Pregnancy loss is a common medical problem. Approximately 25% of reproductive-aged women attempting pregnancy have spontaneous abortions, and at least 10% of all clinical pregnancies are lost.1,2 The predominant cause of pregnancy loss in the first trimester are aneuploidies, mainly attributable to chromosome errors during maternal meiosis.3,4 In addition, multiple other etiologies have been described, including single gene defects (ie, trombophilic mutations), endocrine and immunological factors, uterine anatomical abnormalities, environmental exposures, and infections. Despite modern diagnostics of chromosomal and nonchromosomal factors, a large proportion of cases remains unexplained.
Epigenetic changes of the genetic information are not produced by changes in the DNA sequence itself, but by reversible modifications of DNA and chromatin structure. The most thoroughly studied epigenetic modification is methylation of CpG dinucleotides in cis-regulatory sequences, which is associated with specific histone modifications leading to a condensed chromatin structure and transcriptional repression.5,6 Genomic imprinting is a parent-specific epigenetic modification in which allele-specific expression depends on male versus female germline transmission. Most imprinted genes display differentially methylated regions (DMRs), which are thought to function as imprinting control centers.7,8
Parent-specific methylation patterns (genomic imprints) are established during gametogenesis. In the zygote and early embryo genome-wide demethylation waves erase most of these germline patterns, followed by de novo methylation and establishment of somatic methylation patterns around the time of implantation.9–11 Only 100 to 200 imprinted genes among our approximately 25,000 genes escape this methylation reprogramming after fertilization and maintain their parent-specific methylation and activity. Many imprinted genes are active in the placenta and/or fetus and play an important role in resource acquisition of the fetus.12,13
Animal studies have shown that genome-wide alterations of methylation reprogramming lead to developmental arrest and embryo loss in the preimplantation period.14 More subtle reprogramming defects in individual genes may be compatible with further development and cause abnormal phenotypes.15,16 It has been estimated that such primary epimutations occur at a much (one or two orders of magnitude) higher rate than somatic DNA mutations.17,18 Most epimutations may be stochastic errors during establishment and/or maintenance of the appropriate methylation patterns, however environmental and genetic factors are also known to affect the regulation of methylation reprogramming.14,19,20 It is well known that epimutations affecting methylation imprints can cause Beckwith-Wiedemann syndrome (BWS) and other imprinting disorders.19,21 However, the pathological significance of epimutations is not restricted to these rare syndromes.22–24 Here, we have analyzed the methylation patterns of two paternally and four maternally methylated genes in human abortions and stillbirths.
Materials and Methods
Tissue Samples
Muscle samples were obtained from aborted fetuses and stillbirths who underwent diagnostic examination at the Department of Pediatric Pathology of Mainz University Medical Center. Use of “excess” tissue materials for scientific analyses was approved by the local ethics committee (Aerztekammer Rheinland-Pfalz, decision no. 837.103.04 [4261]). Gestational age of each fetus was determined by foot length measurements (in mm) and last menstrual period. The sex was identified by examination of the external genitalia. Biopsies from the Musculus quadriceps were dissected within 24 hours to 48 hours after abortion and stored at −80°C until further analysis. The DNeasy Blood and Tissue Kit (Qiagen, Hilden Germany) was used for genomic DNA isolation. Total RNAs were isolated using Trizol reagent (Invitrogen, Karlsruhe, Germany).
Of the 146 studied cases, 55 were spontaneous abortions (from 12 to 20 weeks gestation), 57 stillbirths (from 21 to 42 weeks gestation), and 34 induced abortions (from 12 to 24 weeks gestation). Placenta and fetus of all cases were analyzed pathomorphologically by an experienced pediatric pathologist. Of the 112 spontaneous pregnancy losses (spontaneous abortions and stillbirths), 63 had a normal karyotype and 49 an unknown karyotype. Because we wished to study epigenetic causes of pregnancy loss, we focused on chromosomally normal cases. Twenty-four (21%) spontaneous pregnancy losses displayed an abnormal phenotype; no fetal abnormalities were detected in the majority of cases. The 34 induced abortions included eight cases with unbalanced karyotypes; the remaining 26 cases (without known chromosome abnormalities) also exhibited different malformations, including skeletal dysplasias, heart defects, brain malformations, and/or neural tube defects.
Gene-Specific Methylation Analyses
The DMRs of two paternally methylated (H19 and MEG3) and four maternally methylated (LIT1, NESP55, PEG3, and SNRPN) genes that can cause developmental defects when abnormally regulated in animal models and/or humans were analyzed by bisulfite pyrosequencing. Bisulfite treatment of genomic DNA was performed with the EpiTect Bisulfite Kit (Qiagen). Bisulfite pyrosequencing was performed on a PSQ96MA Pyrosequencing System (Biotage, Uppsala, Sweden) with the PyroGold SQA reagent kit (Biotage). Polymerase chain reaction (PCR) and sequencing primers for bisulfite pyrosequencing (Table 1) were designed using the Pyrosequencing Assay Design Software (Biotage). The Pyro Q-CpG software (Biotage) was used for data analysis. To demonstrate the reliability of our quantitative methylation assays, we performed duplicate tests for a subset of fetal muscle samples. The mean methylation difference between duplicate measurements was 2.6% for H19 (13 samples tested), 2.3% for MEG3 (16 samples), 2.4% for LIT1 (13 samples), 2.4% for NESP55 (12 samples), 1.9% for PEG3 (12 samples), and 2.0% for SNRPN (29 samples).
Table 1.
Genes and Primers for Bisulfite Pyrosequencing
| Gene | Primer | Sequence | Amplicon length (bp) | Localization (bp) | Number of CpGs |
|---|---|---|---|---|---|
| H19 | Outer forward | 5′-TTTTTGGTAGGTATAGAGTT-3′ | |||
| Outer reverse | 5′-AAACCATAACACTAAAACCC-3′ | ||||
| Nested forward | 5′-TGTATAGTATATGGGTATTTTTGGAGGTTT-3′ | Chromosome 11 | |||
| Nested reverse* | 5′-TCCTATAAATATCCTATTCCCAAATAACC-3′ | 231 | 1977,647–1977,878 | 4 | |
| Sequencing | 5′-TGGTTGTAGTTGTGGAAT-3′ | ||||
| MEG3 | Forward | 5′-GATTTTTTTTATATATTGTGTTTG-3′ | Chromosome 14 | ||
| Reverse* | 5′-CTCATTTCTCTAAAAATAATTAACC-3′ | 220 | 100,361,907–100,362,129 | 3 | |
| Sequencing | 5′-GTGTTTGAATTTATTTTGTTTGG-3′ | ||||
| LIT1 | Forward | 5′-AATTAGTAGGTGGGGGG-3′ | Chromosome 11 | ||
| Reverse* | 5′-CTAAAAAACTCCCTAAAAATC-3′ | 122 | 2677,751–2677,873 | 2 | |
| Sequencing | 5′-GGGGGTAGTYGGAG-3′ | ||||
| NESP55 | Outer forward | 5′-TTTTTTATTTTATAGGGTGTATTT-3′ | |||
| Outer reverse | 5′-AAAATAAAATACTTAAACACCAC-3′ | ||||
| Nested forward | 5′-TTTTTGTAGAGTTAGAGGGTAGGT-3′ | Chromosome 20 | |||
| Nested reverse* | 5′-AAAAAAAACAACTCAAAATCTACC-3′ | 343 | 56,848,506–56,848,849 | 3 | |
| Sequencing | 5′-GTGTTTAAGAGGATGGAT-3′ | ||||
| PEG3 | Forward | 5′-GGTGTAGAAGTTTGGGTAGTTG-3′ | Chromosome 19 | ||
| Reverse* | 5′-CTCACCTCACCTCAATACTAC-3′ | 153 | 62,043,756–62,043,909 | 4 | |
| Sequencing | 5′-TGTTTATTTTGGGTTGGT-3′ | ||||
| SNRPN | Forward* | 5′-AGGGAGTTGGGATTTTTGTATT-3′ | Chromosome 15 | ||
| Reverse | 5′-CCCAAACTATCTCTTAAAAAAAAC-3′ | 237 | 22,751,105–22,751,342 | 2 × 3 = 6 | |
| Sequencing 1 | 5′-ACACAACTAACCTTACCC-3′ | 3 | |||
| Sequencing 2 | 5′-CCAACCTACCTCTAC-3′ | 3 |
This table provides the primer sequences for bisulfite pyrosequencing of the six studied genes, the length and chromosomal localization (ensembl 54, May 2009) of the amplified segments, and the number of CpG sites in the amplicons. Biotinylated primers are indicated by an asterisk.
Expression Analysis
cDNAs were synthesized with random primers and SuperScript III reverse transcriptase (Invitrogen) from total RNAs of six samples that were heterozygous for a transcribed SNP, rs10770125, in IGF2. PCR products for quantification of allele-specific expression by pyrosequencing (QUASEP)25 were generated from cDNA using FastStart TaqDNA Polymerase (Roche, Mannheim, Germany) and normal hot start PCR (Table 2). Pyrosequencing was performed on a PSQ96MA Pyrosequencing System with the PyroGold SQA reagent kit. The PSQ96MA 2.1.1 software (Biotage) was used for data analysis. Quantitative real-time RT-PCR for MEG3 (Table 2) was performed on an Applied Biosystems (Darmstadt, Germany) 7500 Fast Real-Time PCR system using standard protocols. Relative quantification was performed with the Relative Expression Software Tool (REST, version 2.0.7),26 using BRD1 (QuantiTect Primer QT00196924, Qiagen) and ERCC3 (QT00080276) as endogenous controls.
Table 2.
Primers for QUASEP (IGF2) and Quantitative Real-Time RT-PCR (MEG3)
| Gene | Primer | Sequence | cDNA amplicon length (bp) | cDNA localization (bp) |
|---|---|---|---|---|
| IGF2 | Forward | 5′-ACTAGAGTACAGGGGCCGAAGAGT-3′ | ENST00000300632 | |
| Reverse* | 5′-GGGCTGGAATCCACCTCCT-3′ | 63 | 466–529 | |
| Sequencing | 5′-GAGTCACCACCGAGC-3′ | |||
| MEG3 | Forward | 5′-ATCCGTCCACCTCCTTG-3′ | ENST00000451743 | |
| Reverse | 5′-GAGCATAGCAAAGGTCAGG-3′ | 277 | 115–392 |
This table provides the primer sequences of an intron-spanning QUASEP assay for IGF2, which is under control of the analyzed H19 DMR, and quantitative real-time RT-PCR analysis of MEG3, which is controlled by the analyzed MEG3 DMR. The biotinylated QUASEP primer is indicated by an asterisk.
Statistical Methods
The methylation level, measured in percentage of maximal possible methylation of six representative imprinted genes, was obtained from 146 fetal muscle samples. Quantitative data were analyzed with SPSS version 17.0.1. Box plots were generated using the default parameters of SPSS. The bottom and the top of the box indicate the 25th and 75th percentile, respectively. The T bars extend from the boxes to at most 1.5 times the height of the box. Outliers are samples that do not lie within these T bars, extreme outliers have values more extreme than three times the box length away from the median. The χ2 test was used to compare the number of outliers in spontaneous abortions, stillbirths, and induced abortions. A P value of less than 5% was considered significant. Scatter plots were used to study a possible relationship between DNA methylation and gestational age. A linear regression (Pearson correlation, R2) was calculated for each studied gene to infer the correlation between the dependent (methylation percentage) and the independent (age) variable.
Results
We performed box plot analyses of six functionally important DMRs in fetal muscles of 146 abortions and stillbirths (Figure 1). The analyzed regions represent both primary imprints (H19, LIT1, PEG3, and SNRPN), which are established in the germline and stably maintained after fertilization, as well as secondary imprints (MEG3 and NESP55), which are established in the early embryo. In a given sample, a gene-specific methylation imprint was designated as normal, if the average methylation percentage of all analyzed CpGs in the DMR fell in the T bars of the box plot for all analyzed fetal muscles. In the 55 spontaneous abortions, 15 of 330 (4.6%) analyzed DMR methylation values (for the six studied genes) represented outliers including four (1.2%) extreme outliers (Table 3). In the 57 stillbirths, the number of potentially abnormal methylation values was significantly higher (χ2 test; P < 0.01): 35 of 342 (10.2%) methylation values were outliers and 8 (2.3%) extreme outliers. In contrast, the 34 induced abortions exhibited only 2 of 204 (1.0%) outliers and no extreme outlier, which is significantly (P = 0.03, respectively, P < 0.001) lower than in spontaneous abortions and stillbirths. We excluded that the higher rate of extreme methylation values in the stillbirth group, compared with abortions, was a consequence of a skewed age distribution. Linear regression analyses did not reveal significant effects of gestational age on DMR methylation (data not shown). It is interesting to note that 48 (92%) outliers represented hypermethylated and only four (8%) hypomethylated DMRs (Figure 1).
Figure 1.
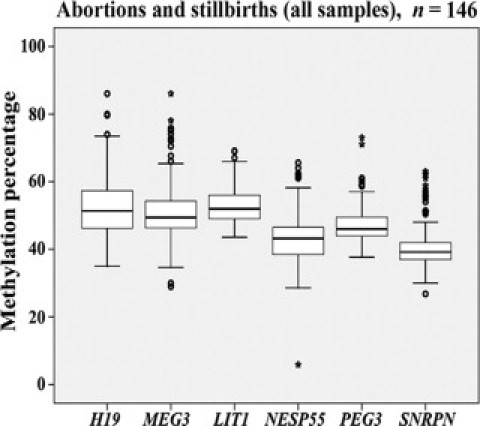
Methylation values of imprinted genes in 146 fetal muscle samples from abortions and stillbirths. The box plots show the distribution of DMR methylation values for H19, MEG3, LIT1, NESP55, PEG3, and SNRPN, respectively. The median is represented by horizontal lines. The bottom of the box indicates the 25th percentile, the top the 75th percentile. Outliers are shown as open circles, extreme outliers as stars.
Table 3.
Extreme Methylation Values in Fetal Muscle Samples
| All fetal muscles, n = 146 |
Spontaneous abortions, n = 55 |
Stillbirths, n = 57 |
Induced abortions, n = 34 |
||||
|---|---|---|---|---|---|---|---|
| Gene | Median methylation | Number of outliers | Extreme outliers | Number of outliers | Extreme outliers | Number of outliers | Extreme outliers |
| H19 | 51% | 0 | 0 | 4 | 0 | 0 | 0 |
| MEG3 | 49% | 4 | 1 | 7 | 3 | 1 | 0 |
| LIT1 | 52% | 1 | 0 | 2 | 0 | 0 | 0 |
| NESP55 | 43% | 2 | 1 | 5 | 0 | 1 | 0 |
| PEG3 | 46% | 3 | 1 | 5 | 1 | 0 | 0 |
| SNRPN | 39% | 5 | 1 | 12 | 4 | 0 | 0 |
| 15/330 | 4/330 | 35/342 | 8/342 | 2/204 | 0/204 | ||
| 4.6% | 1.2% | 10.2% | 2.3% | 1.0% | 0% | ||
This table presents the median methylation values and the number of extreme methylation values for six imprinted genes in fetal muscle samples from spontaneous abortions, stillbirths, and induced abortions.
In abnormally methylated samples usually more than one of the six analyzed genes was affected. Two of 55 (4%) spontaneous abortions and 10 of 57 (18%) stillbirths displayed hypermethylated DMRs in two to five genes (Table 4). In contrast, none of 34 induced abortions displayed multiple outliers. Eight of 12 (66%) abortions/stillbirths with multiple methylation abnormalities showed an impairment of villus maturation in the placenta; one fetus suffered from a lymphangioma, whereas nothing abnormal was detected in the remaining 11 fetuses. It is interesting to note that 10 of the 12 (83%) samples with multiple methylation abnormalities were male, whereas of all analyzed fetal samples 54% were male and 46% female. To exclude the possibility that the significant (χ2 test; P = 0.05) excess of male abortions/stillbirths with methylation abnormalities was attributable to sex-specific differences, we performed separate box plot analyses for the 78 male and the 66 female samples (data not shown). Although the median methylation values differed slightly between male and female fetuses (53% versus 51% for H19, 53% versus 50% for MEG3, 53% versus 52% for LIT1, 45% versus 41% for NESP55, 48% versus 47% for PEG3, and 42% versus 40% for SNRPN), the results obtained with the male and the female data sets were similar to those obtained with all samples. In both the male and the female group, extreme methylation values were more frequent in stillbirths than in spontaneous abortions. Eleven of the 12 abortions/stillbirths with multiple outliers in the box plot analysis of all fetal muscles also displayed abnormal methylation values in the male group and the female group analysis, respectively.
Table 4.
Fetal Muscle Samples with Hypermethylation of Multiple Imprinted Genes
| Methylation values |
Phenotype |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample number | Weeks gestation | H19 | MEG3 | LIT1 | NESP55 | PEG3 | SNRPN | Number of outliers | Placenta | Fetus | Karyotype/Genital sex |
| Spontaneous abortions | |||||||||||
| 2 | 17 | N | N | + | N | + | N | 2 | CHA | NAD | 46,XY |
| 118 | 20 | N | ++ | N | N | ++ | + | 3 | RC | NAD | 46,XY |
| Stillbirths | |||||||||||
| 1 | 35 | + | ++ | N | + | N | ++ | 4 | VMI | NAD | Male |
| 6 | 37 | + | + | N | + | + | + | 5 | VMI | NAD | 46,XY |
| 7 | 32 | N | N | N | + | N | + | 2 | VMI | NAD | 46,XY |
| 16 | 22 | N | + | + | N | + | + | 4 | VMI | NAD | Female |
| 30 | 36 | + | ++ | N | + | ++ | ++ | 5 | VMI | NAD | 46,XX |
| 53 | 28 | + | ++ | N | N | + | ++ | 4 | VMI | NAD | 46,XY |
| 93 | 22 | N | + | N | N | N | + | 2 | VMI | NAD | 46,XY |
| 102 | 37 | N | + | N | + | N | ++ | 3 | VMI | NAD | 46,XY |
| 104 | 26 | N | + | N | + | + | ++ | 4 | CHA | NAD | 46,XY |
| 134 | 24 | N | N | + | N | + | N | 2 | NAD | Lymphangioma | Male |
This table presents abortions and stillbirths with extreme methylation values in at least two studied genes. Box plot analysis was performed for all 146 fetal muscle samples. “N” indicates normal methylation. Hypermethylated outliers are indicated by “+” and extreme outliers by “++.” The observed placental phenotypes include chorionamnionitis (CHA), regressive changes (RC), and villus maturation impairment (VMI). NAD means nothing abnormal detected.
Hypermethylation of the H19 DMR may lead to ectopic expression of IGF2 from the normally hypomethylated maternal allele.27 We performed QUASEP of six informative abortions/stillbirths with H19 hypermethylation. Samples no. 53 (with 74% H19 methylation) and 30 (80%) displayed significant biallelic IGF2 expression (70% C allele versus 30% T allele and 61% C versus 39% T, respectively; Table 5). Abnormal methylation of the normally hypomethylated maternal MEG3 allele may lead to gene silencing.28 We compared the MEG3 mRNA expression levels of three normally methylated (48% to 50%) and two hypermethylated fetal muscle samples (no. 27 with 64% and no. 118 with 75% MEG3 methylation). REST analysis of triplicate measurements of the Ct values (Table 5) revealed a significant (P < 0.01) down-regulation of MEG3 expression in sample no. 27 (relative expression 4%) and a trend (P = 0.07) toward down-regulation in sample no. 118 (relative expression 30%), compared with the normally methylated samples.
Table 5.
Expression Analysis of Hypermethylated Fetal Muscle Samples
| QUASEP analysis of IGF2 |
|||
|---|---|---|---|
| Sample number | H19 DMR methylation | C allele | T allele |
| 1 | 86% | 100 ± 0% | 0 ± 0% |
| 6 | 80% | 100 ± 0% | 0 ± 0% |
| 7 | 71% | 2.2 ± 0.3% | 97.8 ± 0.3% |
| 19 | 73% | 1.7 ± 0.1% | 98.3 ± 0.1% |
| 30 | 80% | 61.1 ± 1.2% | 38.9 ± 1.2% |
| 53 | 74% | 69.8 ± 2.8% | 30.2 ± 2.8% |
| Sample number | MEG3 DMR methylation | Real-time RT-PCR of MEG3 Ct value |
|---|---|---|
| 70 (control) | 48% | 16.1 ± 0.1 |
| 77 (control) | 50% | 18.5 ± 0.2 |
| 29 (control) | 50% | 18.2 ± 0.1 |
| 27 | 64% | 27.1 ± 0.3 |
| 118 | 75% | 26.2 ± 0.2 |
The upper part of this table presents the QUASEP results (mean and standard deviation of duplicate tests) of fetal muscles with H19 hypermethylation. Two samples (nos. 30 and 53) express both IGF2 alleles, indicative of loss of imprinting. The bottom part presents quantitative measurements of MEG3 expression (mean and standard deviation of triplicate tests) in two samples (nos. 27 and 118) with MEG3 hypermethylation versus three controls.
Discussion
Many imprinted genes play essential roles in mammalian development, notably in the regulation of placental, fetal, and/or postnatal growth. Paternally expressed genes such as LIT1 and PEG3 tend to enhance growth, whereas maternally expressed genes such as H19 restrict growth.7,8,12,13 It is generally assumed that a parent-specific expression mechanism has been established during mammalian evolution to settle the conflicting interests between the paternal and maternal genomes on placental/fetal growth.29 Their monoallelic expression makes imprinted genes particularly susceptible to genetic or epigenetic dysregulation. The methylation abnormalities that were observed in 4% of spontaneous abortions and 18% of stillbirths are consistent with somatic mosaicism that is the presence of hypermethylated and normally methylated cells in the same fetus. If an epimutation occurred in the germline, all cells of the individual should be affected, leading to clear DMR hypomethylation (<10%) or hypermethylation (>90%). In contrast, if an epimutation occurred during early embryogenesis, it affects only a subset of cells. Our results suggest a postzygotic gain of methylation at multiple imprinted genes, regardless of whether the hypomethylated allele is derived from the paternal or the maternal germline and regardless of whether it is a primary or secondary imprint. This may be attributable to relaxation of the so-far unknown mechanism that normally protects the hypomethylated allele from de novo methylation in the morula to blastocyst stage.11 The preimplantation embryo is much less protected from environmental factors than the germline and, thus, may be particularly susceptible to epigenetic alterations.14,19
Two stillbirths with H19 hypermethylation showed biallelic IGF2 expression. Loss of IGF2 imprinting has been found in many different tumor types.30 Importantly, IGF2 was also dysregulated in normal mucosa and peripheral blood lymphocytes of colorectal cancer patients,31 indicating that relaxation of imprinting may predispose to tumorigenesis and, by extrapolation, to other multifactorial phenotypes. About 10% of blood samples from normal populations show biallelic IGF2 expression.32 Similar to the situation in tumor cells,28 in our study hypermethylation of the MEG3 DMR was associated with transcriptional repression in fetal tissue samples. Although the methylation and expression patterns of imprinted genes may show considerable variation, one can envision a link between epigenetic alterations at multiple imprinted loci during critical stages of placental/fetal development and pregnancy loss. We propose a multifactorial threshold model for pregnancy loss. If extreme methylation values attributable to primary epimutations in imprinted and/or other developmentally important genes exceed a critical threshold, the phenotype may become manifest. Similar to other multifactorial diseases, additional genetic and environmental factors also play a role.
In this study, we focused on methylation abnormalities of imprinted genes, which may serve as an indicator for more profound epigenetic defects. It is plausible to assume that the observed epimutations are not restriced to imprinted genes but also affect other developmentally important (non-imprinted) loci. A substantial fraction (>20%) of the spontaneous abortions and stillbirths had malformations that are difficult to explain by imprinting disorders. Interestingly, none of the 12 fetuses with multiple methylation abnormalities displayed detectable malformations; one fetus suffered from a mediastinal lymphangioma. Epimutations are known to play a role in tumorigenesis.22,30 On the other hand, most placentae of pregnancies with multiple methylation abnormalities displayed villus maturation impairment or other pathological features. Overall, epigenetic defects were predominantly associated with unexplained pregnancy loss, in particular in the second half of pregnancy.
The majority (>80%) of abortions/stillbirths with multiple methylation abnormalities were male. This is consistent with the observed tendency toward 1% to 4% higher methylation levels at selected loci in male fetuses, compared with females. There are more male than female embryos at conception and a male excess of fetal deaths, which is little understood.33 In Japan, the male-to-female ratio of fetal deaths increased since the 1970s, reaching more than two in the late 1990s.34 Maternal exposure to severe life events before and/or during the time of conception has been associated with a differential loss of male pregnancies.35 Epigenetic mechanisms are thought to mediate environmental influences on gene regulation and to modulate disease risk.20,22–24 In this context it is interesting to speculate that an increased vulnerability of the male embryo to methylation reprogramming defects contributes to the greater male to female risk of fetal mortality.
Our study is limited by the fact that we could analyze only one fetal tissue and that it is difficult to collect adequate controls. Fetal muscle biopsies from healthy pregnancies are ethically out of discussion. Chorionic villus samples (ie, excess materials from prenatal diagnosis) could be easily obtained, but are not suitable for studying imprinted gene regulation in the fetus. In contrast to somatic tissues, imprinting in the placenta appears to be primarily controlled by histone modifications independent of DNA methylation.36 Possibly because of their more direct contact with the environment, trophectoderm and placenta may also be much more susceptible to changes in DNA methylation patterns than embryonic or fetal tissues.37 At present, we cannot exclude the formal possibility that the observed methylation patterns represent normal variation during fetal development. Moreover, it is possible that the observed extreme methylation values in aborted fetuses are not the cause but a consequence of aberrant development. In any case, this is the first study presenting baseline data on imprinted gene methylation in fetal tissues and a first step in detailing what kind of variation to expect.
Footnotes
Supported by research grant HA 1374/8–1 from the German Research Foundation.
References
- 1.Brown S. Miscarriage and its associations. Semin Reprod Med. 2008;26:391–400. doi: 10.1055/s-0028-1087105. [DOI] [PubMed] [Google Scholar]
- 2.Warren JE, Silver RM. Genetics of pregnancy loss. Clin Obstet Gynecol. 2008;51:84–95. doi: 10.1097/GRF.0b013e318161719c. [DOI] [PubMed] [Google Scholar]
- 3.Boue A, Boue J, Gropp A. Cytogenetics of pregnancy wastage. Adv Hum Genet. 1985;14:1–57. doi: 10.1007/978-1-4615-9400-0_1. [DOI] [PubMed] [Google Scholar]
- 4.Eiben B, Bartels I, Bähr-Porsch S, Borgmann S, Gatz G, Gellert G, Goebel R, Hammans W, Hentemann M, Osmers R, Rauskolb R, Hansmann I. Cytogenetic analysis of 750 spontaneous abortions with the direct-preparation method of chorionic villi and its implications for studying genetic causes of pregnancy wastage. Am J Hum Genet. 1990;47:656–663. [PMC free article] [PubMed] [Google Scholar]
- 5.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 6.Vaissière T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res. 2008;659:40–48. doi: 10.1016/j.mrrev.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Bartolomei MS, Tilghman SM. Genomic imprinting in mammals. Annu Rev Genet. 1997;31:493–525. doi: 10.1146/annurev.genet.31.1.493. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson-Smith AC, Surani MA. Imprinting and the epigenetic asymmetry between parental genomes. Science. 2001;293:1086–1089. doi: 10.1126/science.1064020. [DOI] [PubMed] [Google Scholar]
- 9.Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- 10.Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature. 2000;403:501–502. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- 11.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 12.Miozzo M, Simoni G. The role of imprinted genes in fetal growth. Biol Neonate. 2002;81:217–228. doi: 10.1159/000056752. [DOI] [PubMed] [Google Scholar]
- 13.Constância M, Kelsey G, Reik W. Resourceful imprinting. Nature. 2004;432:53–57. doi: 10.1038/432053a. [DOI] [PubMed] [Google Scholar]
- 14.Shi W, Haaf T. Aberrant methylation patterns at the two-cell stage as an indicator of early developmental failure. Mol Reprod Dev. 2002;63:329–334. doi: 10.1002/mrd.90016. [DOI] [PubMed] [Google Scholar]
- 15.Khosla S, Dean W, Brown D, Reik W, Feil R. Culture of preimplantation mouse embryos affects fetal development and the expression of imprinted genes. Biol Reprod. 2001;64:918–926. doi: 10.1095/biolreprod64.3.918. [DOI] [PubMed] [Google Scholar]
- 16.Young LE, Fernandes K, McEvoy TG, Butterwith SC, Gutierrez CG, Carolan C, Broadbent PJ, Robinson JJ, Wilmut I, Sinclair KD. Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat Genet. 2001;27:153–154. doi: 10.1038/84769. [DOI] [PubMed] [Google Scholar]
- 17.Bennett-Baker PE, Wilkowski J, Burke DT. Age-associated activation of epigenetically repressed genes in the mouse. Genetics. 2003;165:2055–2062. doi: 10.1093/genetics/165.4.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goyal R, Reinhardt R, Jeltsch A. Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Res. 2006;34:1182–1188. doi: 10.1093/nar/gkl002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horsthemke B. Epimutation in human disease. Curr Top Microbiol Immunol. 2006;310:45–59. doi: 10.1007/3-540-31181-5_4. [DOI] [PubMed] [Google Scholar]
- 20.Sutherland JE, Costa M. Epigenetics and the environment. Ann NY Acad Sci. 2003;983:151–160. doi: 10.1111/j.1749-6632.2003.tb05970.x. [DOI] [PubMed] [Google Scholar]
- 21.Temple IK. Imprinting in human disease with special reference to transient neonatal diabetes and Beckwith-Wiedemann syndrome. Endocr Dev. 2007;12:113–123. doi: 10.1159/000109638. [DOI] [PubMed] [Google Scholar]
- 22.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 23.Hatchwell E, Greally JM. The potential role of epigenomic dysregulation in complex human disease. Trends Genet. 2007;23:588–595. doi: 10.1016/j.tig.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Foley DL, Craig JM, Morley R, Olsson CA, Dwyer T, Smith K, Saffery R. Prospects for epigenetic epidemiology. Am J Epidemiol. 2009;169:389–400. doi: 10.1093/aje/kwn380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruf N, Dunzinger U, Brinckmann A, Haaf T, Nürnberg P, Zechner U. Expression profiling of uniparental mouse embryos is inefficient in identifying novel imprinted genes. Genomics. 2006;87:509–519. doi: 10.1016/j.ygeno.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 26.Pfaffl MW, Horgan GW, Dempfle L. Relative Expression Software Tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:E36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sasaki H, Ishihara K, Kato R. Mechanisms of Igf2/H19 imprinting: DNA methylation, chromatin and long-distance gene regulation. J Biochem. 2000;127:711–715. doi: 10.1093/oxfordjournals.jbchem.a022661. [DOI] [PubMed] [Google Scholar]
- 28.Astuti D, Latif F, Wagner K, Gentle D, Cooper WN, Catchpoole D, Grundy R, Ferguson-Smith AC, Maher ER. Epigenetic alteration at the DLK1-GTL2 imprinted domain in human neoplasia: analysis of neuroblastoma, phaeochromocytoma and Wilms' tumour. Br J Cancer. 2005;92:1574–1580. doi: 10.1038/sj.bjc.6602478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore T, Haig D. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 1991;7:45–49. doi: 10.1016/0168-9525(91)90230-N. [DOI] [PubMed] [Google Scholar]
- 30.Jelinic P, Shaw P. Loss of imprinting and cancer. J Path. 2007;211:261–268. doi: 10.1002/path.2116. [DOI] [PubMed] [Google Scholar]
- 31.Cui H, Horon IL, Ohlsson R, Hamilton SR, Feinberg AP. Loss of imprinting in normal tissue of colorectal cancer patients with microsatellite instability. Nat Med. 1998;4:1276–1280. doi: 10.1038/3260. [DOI] [PubMed] [Google Scholar]
- 32.Sakatani T, Wei M, Katoh M, Okita C, Wada D, Mitsuya K, Meguro M, Ikeguchi M, Ito H, Tycko B, Oshimura M. Epigenetic heterogeneity at imprinted loci in normal populations. Biochem Biophys Res Commun. 2001;283:1124–1130. doi: 10.1006/bbrc.2001.4916. [DOI] [PubMed] [Google Scholar]
- 33.Kraemer S. The fragile male. BMJ. 2000;321:1609–1612. doi: 10.1136/bmj.321.7276.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mizuno R. The male/female ratio of fetal deaths and births in Japan. Lancet. 2000;356:738–739. doi: 10.1016/S0140-6736(00)02637-4. [DOI] [PubMed] [Google Scholar]
- 35.Hansen D, Moller H, Olsen J. Severe periconceptional life events and the sex ratio in offspring: follow up study based on five national registers. BMJ. 1999;319:548–549. doi: 10.1136/bmj.319.7209.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis A, Mitsuya K, Umlauf D, Smith P, Dean W, Walter J, Higgins M, Feil R, Reik W. Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat Genet. 2004;36:1291–1295. doi: 10.1038/ng1468. [DOI] [PubMed] [Google Scholar]
- 37.Fortier AL, Lopes FL, Darricarrère N, Martel J, Trasler JM. Superovulation alters the expression of imprinted genes in the midgestation mouse placenta. Hum Mol Genet. 2008;17:1653–1665. doi: 10.1093/hmg/ddn055. [DOI] [PubMed] [Google Scholar]