

Abstract
Severe pulmonary hypertension is irreversible and often fatal. Abnormal proliferation and resistance to apoptosis of endothelial cells (ECs) and hypertrophy of smooth muscle cells in this disease are linked to decreased mitochondria and preferential energy generation by glycolysis. We hypothesized this metabolic shift of pulmonary hypertensive ECs is due to greater hypoxia inducible-factor1α (HIF-1α) expression caused by low levels of nitric oxide combined with low superoxide dismutase activity. We show that cultured ECs from patients with idiopathic pulmonary arterial hypertension (IPAH-ECs) have greater HIF-1α expression and transcriptional activity than controls under normoxia or hypoxia, and pulmonary arteries from affected patients have increased expression of HIF-1α and its target carbonic anhydrase IX. Decreased expression of manganese superoxide dismutase (MnSOD) in IPAH-ECs paralleled increased HIF-1α levels and small interfering (SI) RNA knockdown of MnSOD, but not of the copper-zinc SOD, increased HIF-1 protein expression and hypoxia response element (HRE)-driven luciferase activity in normoxic ECs. MnSOD siRNA also reduced nitric oxide production in supernatants of IPAH-ECs. Conversely, low levels of a nitric oxide donor reduced HIF-1α expression in normoxic IPAH-ECs. Finally, mitochondria numbers increased in IPAH-ECs with knockdown of HIF-1α. These findings indicate that alterations of nitric oxide and MnSOD contribute to pathological HIF-1α expression and account for lower numbers of mitochondria in IPAH-ECs.
Severe pulmonary arterial hypertension is characterized by significant increases in pulmonary artery pressures to levels present in the systemic circulation. Pulmonary hypertension (PH) is a major determinant of morbidity and mortality in several pulmonary and heart diseases. The pathogenesis of severe pulmonary arterial hypertension has revolved around excessive vasoconstriction and/or abnormal pulmonary vascular remodeling. Recent experimental evidence has linked the pulmonary vascular disease in severe pulmonary arterial hypertension to an abnormal proliferative vascular cell phenotype, which is also characterized by resistance to endothelial and/or vascular smooth muscle cell apoptosis.1 The identification that there is a clonal expansion of endothelial cells (ECs) in idiopathic pulmonary arterial hypertension (IPAH)1 and somatic and germline mutations in the transforming growth factor β superfamily, particularly of bone morphogenetic protein receptor 2,2,3 led to the concept that this abnormal vascular cell proliferation process resembles that seen in neoplastic processes.4 The hypoxia-driven activation of hypoxia inducible factor (HIF), which contributes to several of the key features present in neoplastic processes,5 also plays a role in the pathogenesis of experimental pulmonary hypertension.6,7
HIF, a heterodimer of the HIF-1α or -2α, and HIF-1β, mediates adaptive molecular responses to low oxygen availability,8 leading to transcriptional activation of genes that regulate energy metabolism, erythropoiesis, vasomotor tone, and angiogenesis.9 HIF-1α therefore plays a pathological role in tumor angiogenesis, neoplastic invasion,9 and in the metabolic shift of cancer cells toward glycolysis, which underlies the Warburg phenomena.10 HIF-2α coordinates fetal lung development11 and adaptive lung responses to chronic hypoxia, which include the control of expression of genes involved in pulmonary vascular cell proliferation and angiogenesis.12 Identification of vascular endothelial growth factor, HIF-1α, and HIF1β expression within endothelial plexiform lesions13 and in pulmonary artery medial smooth muscle cells14 suggests that HIF-dependent signaling may contribute to the proliferative vasculopathy of IPAH. The potential pathogenic role of HIF-1α in PH was supported by the findings that HIF-1α6 or HIF-2α7 heterozygous mice have decreased hypoxic PH, when compared with littermates, and abnormalities in HIF-1α underlie the mitochondria pathology and PH in the Fawn-Hooded rat model of spontaneous disease.14 The role of abnormal HIF-dependent signaling in human IPAH has not been addressed.
Our prior work indicated that cultured ECs derived from human IPAH lungs (IPAH-ECs) exhibit an abnormal metabolic phenotype that is characterized by low numbers of mitochondria and decreased oxygen consumption, significantly higher glycolytic rate,15 apoptosis-resistance, and increased cell proliferation.16 Moreover, pulmonary and total body nitric oxide (NO) are lower in IPAH patients, as compared with healthy controls.17–21 Here, we hypothesize that IPAH-ECs have altered hypoxia sensing, with increased expression of HIF-1α, which accounts for the decreased mitochondria in IPAH-ECs.15 Moreover, we postulate that the oxidative stress seen in IPAH lungs,22–25 caused by decreased expression of manganese superoxide dismutase and nitric oxide, accounts for the up-regulation of HIF-1α, and ultimately, for the decreased mitochondria numbers in IPAH-ECs. Our findings using disease-relevant cells provide the rationale for the development of therapies targeting the energetic shift and HIF-1 activation in pulmonary arterial hypertension.
Materials and Methods
Clinical Characteristics
IPAH patients were identified by the clinical classification of pulmonary hypertension,26 recently updated in the fourth World Symposium on Pulmonary Hypertension.27 Altogether, five IPAH and three control subjects were studied. Clinical characteristics among subjects were similar (age in years, IPAH 43 ± 6, control 29 ± 12; sex [female/male], IPAH 4/1, control 2/1; race [Caucasian/African American/Hispanic], IPAH 5/0/0, control 3/0/0). Pulmonary hypertension was diagnosed by right heart catheterization performed for clinical care (pulmonary artery pressures [mm Hg], IPAH, systolic 88 ± 3, diastolic 37 ± 4, mean 59 ± 3). The study was approved by the Cleveland Clinic Institutional Review Board. The immunohistochemical studies were performed in histological sections of IPAH (n = 5) or normal lungs (n = 5) (cases P1 to P5, see table in28). These five patients were treated with intravenous prostacyclin before lung transplant. The study was approved by the Western Institutional Review Board (in lieu of the Johns Hopkins Institutional Review Board).
Cell Culture
Human pulmonary artery ECs were dissociated and cultured as described before16 in endothelial cell growth medium (EGM-2, Cambrex, Walkersville, MD) on plates precoated with fibronectin 1 μg/cm2 at 37°C for 1 hour. Cells were passaged at 70% to 80% confluence by dissociation from plates with 0.25% trypsin-EDTA (Invitrogen Corporation, Carlsbad, CA). Primary cultures of passages 5 to 8 were used in experiments. HeLa cells were maintained in Dulbecco's Modified Eagle Medium (Invitrogen Corporation) with 10% heat-inactivated fetal calf serum, and 1% penicillin/streptomycin. Human umbilical vein ECs (HUVECs) (Lonza, Walkersville, MD) were cultured in endothelial cell growth medium (EGM-2, Cambrex).
For nitric oxide (NO) donor treatment, cells were incubated with DETA NONOate (detaNO) (AXXORA LLC, San Diego, CA) as the time indicated. For hypoxia treatment, cells were placed directly in a 5% CO2 and 95% air incubator (21% O2) or exposed to hypoxia in a sealed chamber or a hypoxic incubator, and cultured at 37°C.
Western Blot
Cytosolic and nuclear extracts of pulmonary artery ECs were prepared according to manufacturer's protocol (Panomics Inc, Fremont, CA). Whole cell lysates were prepared as previously described21 or in radioimmunoprecipitation assay buffer (Santa Cruz Biotechnology, Santa Cruz, CA). Protein was separated by electrophoresis on a 4% to 15% Tris-HCl precast gel (Bio-Rad Lab, Hercules, CA), and then transferred onto polyvinylidene difluoride membranes (Millipore Corporation, Bedford, MA). Alternatively, XT Precast Gels (Bio-Rad) and 3-(N-morpholino) propanesulfonic acid buffer were used for SDS polyacrylamide gel electrophoresis. Antibodies used for Western analyses included mouse monoclonal anti-HIF-1α antibody (Ab) (BD Biosciences, Mississauga, ON, Canada), anti-catalase Ab (Sigma-Aldrich, St. Louis, MO), anti-Complex III-2 Ab (Molecular Probes, Inc., Eugene, OR), anti-cytochrome c Ab (Santa Cruz Biotechnology) rabbit polyclonal anti-manganese superoxide dismutase (MnSOD) Ab, anti-CuZnSOD Ab (Santa Cruz Biotechnology), anti-von Hippel-Lindau (VHL) Ab, anti-prolyl hydroxylase 2 Ab (Novus Biologicals, Littleton, CO), or anti-EC nitric oxide synthase (eNOS) Ab (Affinity Bioreagents, Golden, CO), followed by a secondary anti-rabbit or anti-mouse Ab (Amersham, Arlington Heights, IL, or Cell Signaling Technology, Beverly, MA). Goat polyclonal anti-lamin B Ab, rabbit polyclonal anti-enolase Ab, or mouse monoclonal anti-α-tubulin Ab (Santa Cruz), anti-glyceraldehyde phosphate dehydrogenase Ab (Research Diagnostics, Inc., Concord MA), and anti-tubulin Ab (Santa Cruz Biotechnology), were used for protein loading.
VHL Gene Promoter Methylation Assay
DNA was isolated and purified from IPAH-ECs and IPAH whole lung tissue specimens. The DNA was then bisulfite modified with Methyl Detector Bisulfite Modification Kit (Active Motif, Carlsbad CA). The positive control consisted of p16Kip1 methylation assessed by nested PCR, using primers included in the kit. Methylation status of the VHL promoter was assessed using the following primers29: unmethylated: forward (−185), 5′-GTTGGAGGATTTTTTTGTGTATGT-3′, reverse (−20), 5′-CCCAAACCAAACACCACAAA-3′, with an expected amplicon of 165 bp; and methylated: forward (−183), 5′-TGGAGGATTTTTTTGCGTACGC-3′, reverse (−25), 5′-GAACCGAACGCCGCGAA-3′, with expected amplicon of 158 bp. Methylated DNA and unmethylated DNA from Chemicon were used as positive and negative controls, respectively, in reactions run in parallel with sample DNA. The PCR protocol were as follows: 95°C × 5 minutes, (95°C × 45 seconds, 59°C × 45 seconds, 72°C × 60 seconds) × 35, 72°C × 10 minutes, followed by maintenance at 4°C.
Transient Transfection and Luciferase Assay
Wild-type hypoxia response element (HRE)-luciferase reporter construct and mutant HRE-luciferase reporter construct were generous gifts from Dr. M.C. Simon.30 Both plasmids have three copies of HRE in tandem and cloned upstream of the Firefly luciferase gene in the pGL2 vector. According to the manufacturer protocol of Lipofectamine 2000 Reagent (Invitrogen Corp), IPAH-ECs, or control human pulmonary artery ECs were transiently transfected with wild-type or mutant constructs for 5 hours and cotransfected with Renilla luciferase construct for normalization of transfection efficiency. Alternatively, cells were transfected using Fugene6 (Roche Applied Science, Indianapolis, IN) or Targeting Systems (El Cajon, CA) instead of lipofectamine. In parallel experiments, enhanced green fluorescence protein was used to determine percentage of cells transfected. After transfection, cells were exposed to hypoxia with 2.5% O2, treated with 125 μmol/L CoCl2, or left untreated under normoxia (21% O2); 48-hour after transfection, cells were assayed for Western blot, luciferase activity using a Dual-Luciferase Reporter Assay System (Promega Corp, Madison, WI), and for protein content by Bradford method (BioRad Laboratories, Inc., Hercules, CA). Wild-type HRE-luciferase activity was determined by values of Firefly luciferase normalized by the Renilla luciferase activity, protein content, and mutant HRE-luciferase reporter construct activity. Fold-induction of HRE binding activation was the ratio of normalized HRE-luciferase activity under treatment with CoCl2 or hypoxia over the relative activity in cells exposed to normoxia.
Immunohistochemistry
Immunohistochemical detection of HIF-1α was performed with the mouse anti-human HIF-1α Ab (Novus Biol.) at 1:8000 dilution for 30 minutes at 37C using the Dako Cytomation CSA system for mouse antibodies (Dako). Histological sections of formalin-fixed, paraffin-embedded lung tissues were dehydrated followed by antigen retrieval with a preheated citrate buffer (Dako). The sections were then blocked with avidin-biotin and then treated with 3% H2O2 for 7 minutes. Following incubation with primary Ab or negative control (mouse IgG), the slides were incubated with the link Ab for 15 minutes, then with streptavidin-biotin for 15 minutes, and the biotinylated tyramide amplification reagent diluted 1:10 in 0.10 M Tris.HCl, 0.15 M NaCl, 0.5% BMP blocking buffer. The secondary streptavidin-horseradish peroxidase was incubated for 15 minutes followed by development with diaminobenzidine. Carbonic anhydrase IX (CAIX) expression was detected with a rabbit anti-human CAIX (Novus Biol) at 1:500 dilution for 1 hour at room temperature. Histological sections of formalin-fixed, paraffin-embedded lung tissues were dehydrated and blocked with serum block for 30 minutes, followed by incubation with primary Ab or rabbit serum. The slides were then developed by the avidin-biotin system with diaminobenzidine as chromogen (Vector).
MnSOD Small Interfering RNA
MnSOD small interfering (si)RNA was synthesized by Ambion (Austin, TX). The sense and antisense MnSOD siRNA were 5′-GGAACAACAGGCCUUAUUCtt-3′ (sense) and 5′-GAAUAAGGCCUGUUGUUCCtt-3′ (antisense). SilencerTM Negative control #1 siRNA (siRNA control) (Ambion, Austin TX) was used as a negative control.31
SOD Activity
SOD activity determined by the rate of reduction of cytochrome c with one unit (U) of SOD activity is defined as the amount of SOD required to inhibit the rate of cytochrome c reduction by 50%.
HIF-1α Micro-RNA
HIF-1α micro (mi)RNA (AF304431) served as the target sequence. BLOCK-iT RNAi Designer from Invitrogen was used to design two single-stranded DNA oligonucleotides encoding the target pre-miRNA. The top and bottom strands were annealed to generate a double-stranded oligonucleotide suitable for cloning into BLOCK-iT Pol II miR RNAi Expression Vectors (pcDNA6.2-GW/EmGFP-miR). This vector contains enhanced green fluorescent protein coding sequence that allows visual delivery assessment. Overall 12 different constructs with double-stranded oligos were tested and pcDNA6.2-GW/EmGFP-miR-neg control plasmid (Invitrogen) which is predicted not to target any known vertebrate gene was used as a negative control. The most effective blocking sequence was selected by Dual-Luciferase Reporter Assay (Promega), based on the inhibition of HRE-luciferase activity. Percent inhibition was calculated according to the formula: (A − B)/A × 100%, where A = relative (fold) HRE-luciferase activity induced by CoCl2 or hypoxia in control vector transfected cells, B = relative (fold) HRE-luciferase activity induced by CoCl2 or hypoxia in HIF-1α miRNA-transfected cells. The most potent silencing sequence used for further experiments targeted the sequence AGGATCAGACACCTAGTCCT (1483 to 1503) within HIF-1α mRNA, which had 54% to 75% inhibition of HRE-luciferase activity as determined for each cell line separately.
Mitochondria Staining and Confocal Microscopy
IPAH-ECs and control cells in chamber slides were transfected with HIF-1α miRNA or control miRNA. Seventy-two hours following transfection, mitochondria were stained with Mitotracker Red 580 (Invitrogen). The slides were mounted with Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (Vector Labs, Burlingame, CA), sealed, and analyzed by confocal laser-scanning microscopy (TCS-40; Leica Microsystems, Cambridge, UK).
Southern Analysis
As previously described,15 total DNA extracted from IPAH-EC was digested with restriction enzyme PvuII, electrophoresed through a 0.8% agarose gel, and transferred to Duralon-UV membranes (Stratagene, Cedar Creek, TX). The filters were hybridized with the 32P-labeled PCR generated mitochondrial DNA (mtDNA) probe and visualized with autoradiography.
Statistical Analyses
Data were shown as mean ± SE. All statistical comparisons were performed using the Student's t-test, paired t-test, analysis of variance, or Wilcoxon nonparametric analyses as appropriate. The level of significance set at or below 0.05. All data were analyzed by the JMP 7 software (SAS Institute, Cary, NC).
Results
Greater HIF-1α Expression and Transcriptional Activity Found in IPAH-ECs and IPAH Lungs
As HIF-1α contributes to proliferative properties of neoplastic cells,5 is expressed in plexiform lesions in IPAH,13 and has been recently implicated in experimental pulmonary hypertension,14 we evaluated HIF-1α expression in the highly proliferative IPAH-ECs. HIF-1α was expressed in cultured IPAH-ECs (n = 3) but not control ECs (n = 3) under normoxia (control: 1.00 ± 0.14 vs. IPAH: 1.49 ± 0.27, normalized relative expression levels, n = 10 replicate experiments, Student's t-test, P < 0.05). IPAH-ECs had approximately twofold higher HIF-1α expression under hypoxia as well (Figure 1, A–B). Hypoxia dose response showed that HIF-1α in IPAH-ECs expression was greater than controls at any oxygen tension (paired t-test, P = 0.02). Consistent with greater HIF-1α expression, IPAH-ECs (n = 3) had greater HRE-luciferase reporter activation than control cells (n = 3) under normoxia (control: 1.00 ± 0.18 vs. IPAH: 1.95 ± 0.35, normalized fold induction, n ≥8 replicate experiments, Wilcoxon 1-way test, P < 0.05), under 2.5% O2 (controls: 1.24 ± 0.22 vs. IPAH: 2.48 ± 0.26, normalized fold induction, t-test, P = 0.01) or hypoxia-mimic CoCl2 (controls: 1.67 ± 0.24 vs. IPAH: 3.02 ± 0.37, normalized fold induction, n = 8 replicate experiments, t-test, P = 0.009) (Figure 1C). These results were consistent with the finding of increased expression of HIF-1α in intimal cells of remodeled large muscular arteries and in cells lining vascular slits in plexiform lesions (previously shown to represent endothelial cells32) in IPAH lungs, when compared with pulmonary artery intima of normal lungs (Figures 2, A–C and Supplemental Figure S1 at http://ajp.amjpathol.org). Consistent with the finding of increased expression and activity of HIF-1α in IPAH pulmonary arteries (which have also been shown to co-express its co-activator partner, HIF-1β13), we detected expression of the HIF-1α target gene CAIX33 in intimal cells lining IPAH pulmonary arteries (Figures 2, D–F and Supplemental Figure S1 at http://ajp.amjpathol.org).
Figure 1.
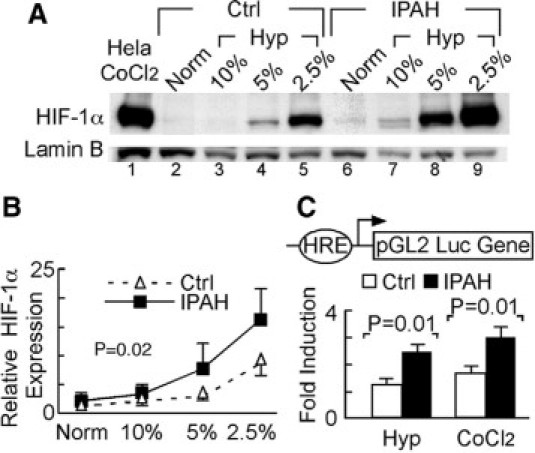
Enhanced HIF-1α expression and transcriptional activity in IPAH-ECs. A: Representative Western blot analysis showing HIF-1α expression in nuclear extracts of IPAH-ECs (n = 3) and control cells (Ctrl, n = 3) under normoxia (Norm) or under a range of hypoxia (Hyp) for 4 hours. B: Quantitative densitometric analysis of Western blots for HIF-1α expression in nuclear extracts of IPAH-ECs (n = 3) and control ECs (n = 3). C: HIF-1α-luciferase reporter activity in cells, transiently transfected with wild-type or mutant HRE-luciferase reporter and cotransfected with renilla construct 24 hours after being exposed to 2.5% O2 hypoxia, treated with 125 μmol/L CoCl2, or left untreated under normoxia. Data shown as fold induction over normoxia expression levels (n ≥4 replicate experiments).
Figure 2.
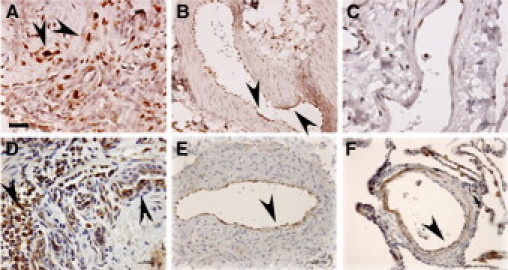
Expression of HIF-1α and its transcriptional target CAIX in IPAH plexiform lesions and remodeled pulmonary arteries. Note increased expression of HIF-1α in cells forming the vascular slits in IPAH plexiform lesions (A, arrowheads) and intimal cells lining the remodeled pulmonary arteries (B, arrowheads), when compared with intima of a normal pulmonary artery (C). This pattern of HIF-1α expression correlated with expression of its transcriptional target CAIX in an IPAH plexiform lesion (D, arrowheads) and pulmonary artery intima (E, arrowhead) when compared with a normal pulmonary artery (F, arrowhead). Scale bars: 25 μm (A, D, E); 50 μm (B, C, F). Images representative of overall five IPAH and five normal lungs tested.
The greater expression of HIF-1α in IPAH-ECs was not due to different expression of prolyl hydroxylase-2 (Figure 3A) or VHL tumor suppressor protein (Supplemental Figure S2 at http://ajp.amjpathol.org). Consistent with these results, VHL gene promoter was not methylated in IPAH-ECs as compared with control ECs when assayed by bisulfite DNA modification followed by PCR with methylation-specific primers (data not shown).
Figure 3.
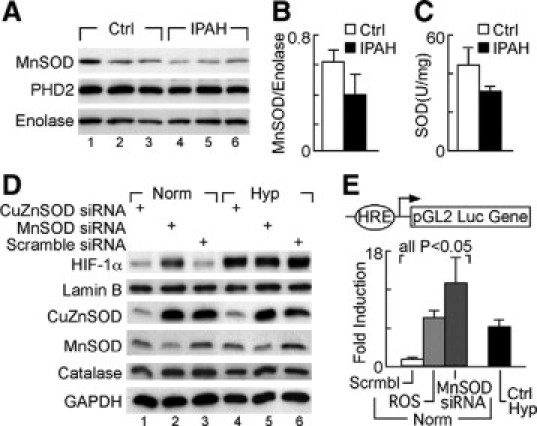
Reduction of MnSOD and SOD activity in IPAH-ECs and regulation of HIF-1α expression by MnSOD in HUVECs. A−B: Western blot analysis of MnSOD and prolyl hydroxylase 2 protein expression in whole cell extracts of IPAH (n = 5) and control pulmonary artery ECs (n = 3), normalized by enolase expression (one-way analysis of variance, P = 0.0374). C: SOD activity (U/mg) is reduced in pulmonary arterial hypertension (n = 5), as compared with control pulmonary artery ECs (n = 3) (one way analysis of variance, P = 0.0275). D: HIF-1α expression measured by Western blot of nuclear extracts of HUVECs transiently transfected with MnSOD siRNA, CuZnSOD, or scramble siRNA exposed to 2.5% O2 (Hyp) or normoxia for 4 hours, normalized for nuclear lamin B. MnSOD, CuZnSOD, and catalase protein expression were evaluated in cytosolic fractions, and normalized for cytosolic glyceraldehyde phosphate dehydrogenase. E: HRE activity in HUVECs transiently transfected with wild-type HRE-luciferase reporter and renilla, and either cotransfected with MnSOD siRNA or scramble siRNA (Scrmbl), or treated with pyrogallol (ROS) for 24 hours under normoxia (Norm). Positive controls consisted of HUVECs exposed to 2.5% O2 for 24 hours (Ctrl Hyp).
Lower SOD Expression and Activity Is Related to Higher HIF Activation in IPAH-ECs
Previous studies identified enhanced oxidative stress,22 increased perivascular inflammation,32,34 and low SOD activity in IPAH lungs.22,35 Consistent with these findings, we observed that IPAH-ECs had decreased MnSOD protein expression and overall SOD activity (Figure 3, A–C). Since MnSOD is both necessary and sufficient to suppress activation of HIF-1α by hypoxia,36,37 we addressed the mechanistic link between decreased MnSOD and increased HIF-1α levels. MnSOD knockdown by siRNA, but not CuZnSOD siRNA or scrambled siRNA, resulted in increased HIF-1α expression in normoxic HUVEC. Under hypoxia, HIF-1α accumulation in HUVEC was not influenced by CuZnSOD knockdown (Figure 3D). Similarly, HRE-luciferase activation in normal pulmonary artery ECs was increased by MnSOD knockdown, as well as by treatment with pyrogallol treatment, which generates intracellular reactive oxygen species (ROS) (all comparisons P < 0.05) (Figure 3E).
HIF-1α Expression in IPAH-ECs Is Modulated by Nitric Oxide
ROS and NO may influence HIF-1α stability.14,38,39 Pulmonary and total body NO are lower in IPAH patients, as compared with healthy controls17–21 and IPAH-ECs produce lower total amounts of NO products than control cells in vitro.15,21 Since SODs modulate consumption of NO by superoxide and control the bioavailability of free NO,35 we postulated that lower MnSOD expression may consequently lead to low bioavailable NO in IPAH-ECs. Indeed, HUVECs in which the SODs were knocked down by RNA interference had reduced NO production measured as nitrite in media overlying cells (NO2− [nM]: scramble siRNA, 108 ± 4; MnSOD siRNA 72 ± 12; CuZnSOD siRNA 47 ± 1; one-way analysis of variance, P = 0.02). Although abrogation of both MnSOD and CuZnSOD reduced NO production, only MnSOD knockdown increased HIF-1α expression in normal cells (Figure 3E). These results suggest that the mitochondrial MnSOD, which influences levels of mitochondrial ROS and/or NO bioavailability, might be mechanistically implicated in the enhanced HIF-1α expression in IPAH-ECs.
To test if NO affects HIF-1α expression in IPAH-ECs, HIF-1α expression was analyzed in cells after treatment with a range of different concentrations of the NO donor detaNO. HIF-1α induction by high-levels of detaNO under normoxia was also greater in IPAH-ECs than in control cells (Figure 4, A–B). As previously reported with non-EC cultured cells,40 low levels of NO (ie, 10 μmol/L – 100 μmol/L detaNO) reduced normoxic and hypoxia-induced HIF-1α accumulation in IPAH-ECs (Figure 4A). These findings suggest that loss of NO production (as by decreased EC NO synthesis21) might result in loss of inhibition of HIF-1α under normoxia.
Figure 4.
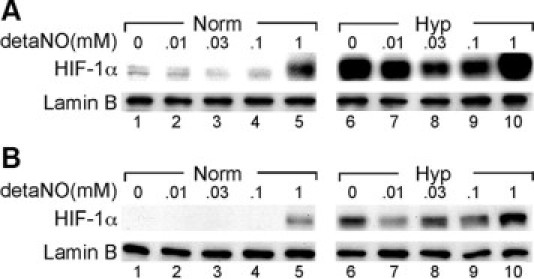
Nitric oxide modulates HIF-1α expression in IPAH-ECs. IPAH (A) and control (B) pulmonary artery ECs were treated with the NO donor detaNO (10 μmol/L, 30 μmol/L, 100 μmol/L, and 1 mmol/L) or vehicle under normoxia (Norm) or 2.5% O2 (Hyp). HIF-1α expression was determined in nuclear extracts, normalized for expression of lamin-B.
Knockdown of HIF-1α Increases Mitochondria Numbers in IPAH-ECs in Vitro
The proliferative phenotype of ECs in IPAH has been further substantiated by the findings that IPAH-ECs have decreased cellular respiration and lower numbers of mitochondria than control cells, which can be rescued by increase in NO bioavailability.15 We tested whether HIF-1α expression lies downstream of NO and therefore contributes to the shift to anaerobic metabolism using an anti-HIF-1α miRNA construct, which resulted in ∼75% inhibition of HRE-luciferase activity in cells exposed to cobalt or hypoxia. Using Mitotracker Red 580 and confocal laser-scanning microscopy to evaluate changes in mitochondria, we found that IPAH-ECs have approximately half the number of mitochondria as control cells (relative fluorescence: IPAH, 0.53 ± 0.29 vs. Ctrl 1.00 ± 0.21; t-test, P = 0.01). These findings were also supported by the assessment of mitochondria DNA (Figure 5C) and expression of mitochondria complex III-2 and cytochrome c (Figure 5D) in representative IPAH-ECs, when compared with control ECs. These results are in line with our prior documentation of decreased mitochondria DNA based on quantitative PCR and on electron microscopy.15 HIF-1α miRNA increased the mitochondria numbers, mtDNA and mitochondrial proteins Complex III-2, and cytochrome c in IPAH-ECs (Figure 5, A–D). We previously showed that mitochondria numbers increase with supplementation of NO donor (with 125 mmol/L detaNO).15 As shown above, a similar concentration of this NO donor also decreased HIF-1α expression in the IPAH-ECs (Figure 4A). Taken together, this suggests that the effect of NO donor on mitochondria numbers might be via HIF-1α inhibition. To rule out any potential effect of HIF-1α knockdown on NO production by the cells, we measured nitrite in supernatants. Nitrite levels in media overlying cells were similar with anti-HIF-1α miRNA or control vector transfected (t-test, P > 0.05), despite that eNOS expression in IPAH-ECs was HIF-dependent as eNOS protein levels were reduced by anti-HIF-1α miRNA41 (Figure 5D).
Figure 5.
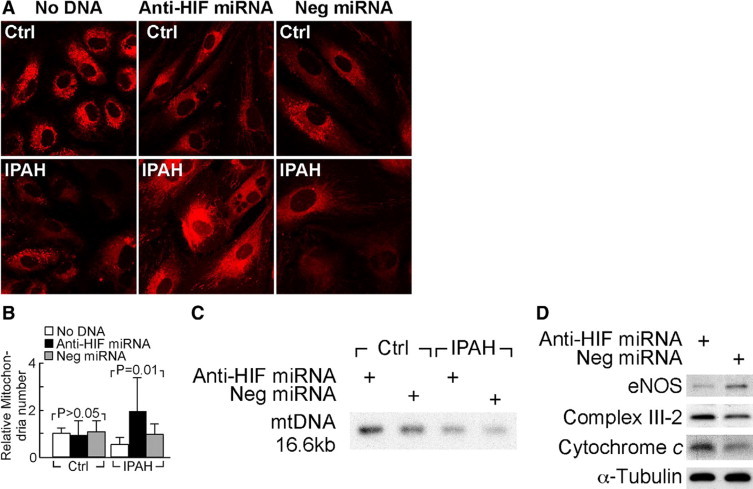
Knockdown HIF-1α by miRNA increases mitochondria in IPAH-pulmonary artery ECs. A and B: Mitochondria were quantified with Mitotracker Red 580 followed by confocal laser-scanning microscopy in IPAH and control pulmonary artery ECs transiently transfected with anti HIF-1α miRNA, negative control miRNA (Neg miRNA), or left untransfected (No DNA). C: Southern analysis of mtDNA content in representative IPAH-ECs and control endothelial cells transfected with anti-HIF-1α miRNA or scrambled miRNA. D: Expression of eNOS, mitochondrial complex III-2, and cytochrome c protein of representative IPAH-EC and control endothelial cells transfected with anti-HIF-1α miRNA or scrambled miRNA assessed by Western blot analysis.
Discussion
Because of its central role in the cancerous cell shift to a glycolytic metabolism, HIF-1α expression is one of the fundamental mechanisms in the neoplastic cell survival and proliferation. Furthermore, HIF-1α also controls energy metabolism, erythropoiesis, vasomotor tone, and angiogenesis.9 Here, we show that IPAH-ECs have greater HIF-1α accumulation and HRE activation than controls under normoxia and hypoxia, which parallel the findings that expression of HIF-1α and its transcriptional target CAIX are increased in ECs of lesional vessels, including the plexiform lesions of IPAH lungs in vivo. The up-regulation of HIF-1α in IPAH-ECs is potentially dependent on the down-regulation of MnSOD and decreased levels of NO. Our data also indicate that a decrease in MnSOD expression in normal ECs suffices to increase the expression of HIF-1α under normoxia. In aggregate, our findings therefore point to a link between oxidative stress due to down-regulation of MnSOD expression and the associated lower levels of NO and up-regulation of HIF-1α in IPAH-ECs. Excessive ROS generation and loss of MnSOD activity has been documented in human pulmonary arterial hypertension, whether idiopathic or associated with other lung diseases, and has been linked to HIF activation in model systems,14,18,22,25,35,42 therefore potentially contributing to pulmonary vascular remodeling.
The well-described deficiency of NO in patients with IPAH and pulmonary arterial hypertension associated with collagen vascular disease18–20 is therapeutically targeted by the use of the phosphodiesterase inhibitor sildenafil. We have previously shown that cultured IPAH-ECs have decreased NO levels,21 which contribute to the decrease in mitochondria numbers and glycolytic shift previously documented in these cells.15 Under normoxia, NO and/or its reaction products mimic hypoxia, leading to HIF-1α protein stabilization, HIF-1 dimer formation, and gene induction.43 Several mechanism(s) of accumulation of HIF-1α by high levels of NO under normoxia have been proposed, including (i) NO inhibition of the terminal enzyme cytochrome c oxidase (complex IV) in the mitochondria, which would lead to a state of mitochondrial ‘hypoxia’33 or (ii) NO reaction with superoxide to form ONOO− which may result in S-nitrosylation of thiol groups in the HIF-1α and stabilization of HIF-1α,39,40,44–47 and/or (iii) reactive species inhibition of enzymes responsible for HIF-1α hydroxylation.47–50 In contrast, low levels of NO reduce hypoxia-induced HIF-1α accumulation and HIF-1 gene induction.51 Under hypoxia, the blockade of cellular respiration by NO enables higher overall intracellular O2 availability for increased degradation of HIF-1α.50,52 Here, we found that HIF-1α induction was greater in IPAH than control ECs by supplementation with high levels of NO. On the other hand, low levels of the NO donor reduced hypoxia-induced HIF-1α accumulation and also reduced the HIF-1α expressed under normoxia in IPAH-ECs. Thus, while decreased NO synthesis has been shown to account for decreased mitochondria numbers and metabolic shift,15 we postulated that the downstream mechanisms mediating these effects may involve HIF-1α.
The mitochondrial MnSOD plays a central role in enzymatic scavenging of superoxide. Mitochondria are the largest producer of ROS through the process of cellular respiration in which mitochondria catalyze one-electron reduction of oxygen to superoxide radical followed by formation of hydrogen peroxide, in addition to the four-electron reduction of oxygen to water.53 We confirmed that the lower SOD activity in IPAH-ECs as compared with control cells was probably related to lower MnSOD protein. The lower MnSOD expression could be due to the lower numbers of mitochondria in IPAH cells15 or down-regulation of MnSOD gene due to AKT-dependent phosphorylation of FOXO transcription factors.13,54 We further documented that HIF-1α protein accumulated in normoxic HUVECs treated with MnSOD siRNA and that HRE-luciferase activation increased with MnSOD siRNA or pyrogallol treatment (which generates intracellular ROS). Notwithstanding the complexity of the mechanisms underlying pathological NO deficiency,15,18,21 our data supports that lower MnSOD controls the accumulation of HIF-1α protein and may be consequential to low NO production by IPAH cells.
There is growing evidence of the shared features between severe pulmonary hypertension and neoplastic processes. Genetic instability, a characteristic hallmark of cancer, has also been documented in IPAH lungs.55 We have evidence of significant somatic gene losses in pulmonary hypertensive cells (Aldred et al, submitted). The link between genetic instability and HIF-1α activation was recently documented in an animal model of Kaposi's sarcoma, also a vascular proliferative process sharing many of the features abnormal angiogenesis seen in PH lungs.56,57
Taken together, accumulating evidence from our group and others identifies that abnormalities of NO production and reduced cellular respiration/mitochondria function coupled to increased glycolysis are universal hallmarks of PH across species, including murine, rodent, avian, and human.14,15,58,59 Here, increased HIF-1α expression is mechanistically linked to both the alterations of NO and the ensuing metabolic changes present in pulmonary arterial hypertension.
Our studies are clinically relevant as they provide the translational framework to understand the recent successes of pharmacological strategies that target cellular utilization of mitochondria respiration vis-a-vis glycolysis, including imatinib60 (an inhibitor of PDGF, c-kit receptors, and also hexokinases61), the nonspecific inhibitors of HIF-1α 2-metoxyestradiol62 or cyclosporine-A,63 and the pyruvate kinase inhibitor dichloroacetate.64 Novel therapies aimed at glycolytic pathways and/or HIF-1 signaling may control vascular cell growth programs shared by endothelial and smooth muscle cells that contribute to occlusive vascular lesions in this untreatable disease.
Note Added In Proof
Since acceptance of this manuscript, we have been able to detect HIF-1α by immunohistochemistry and the endothelial cell marker Factor VIII-related antigen by immunofluorescence in the lung sections of two patients with IPAH. Our approach allowed the acquisition of sequential images, one in bright field showing HIF-α expression by bright field (as shown in Figure 2) and another depicting expression of Factor VIII-related antigen by immunofluorescence in the same vascular lesions (Supplemental Figure S3, available online at http://ajp.amjpathol.org). These findings confirm our initial assertion that endothelial cells express HIF-1α in IPAH lungs.
Acknowledgements
We thank Dr. M. Celeste Simon for the generous gifts of wild-type HRE-luciferase reporter construct and mutant HRE-luciferase reporter construct and Dr. Gregg L. Semenza for the detailed methods for nuclear extract preparation.
Footnotes
Supported by the NHLBI grants RO1HL60917 (to S.C.E.) and RC1HL100849 (to R.M.T.), American Heart Association (to R.M.T.), and the Pulmonary Hypertension Breakthrough Initiative of the Cardiovascular Medical Research Education Fund (to S.C.E. and R.M.T.).
I.F. and W.X. contributed similarly to this work; S.C.E. and R.M.T. are co-corresponding authors.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
References
- 1.Lee SD, Shroyer KR, Markham NE, Cool CD, Voelkel NF, Tuder RM. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Invest. 1998;101:927–934. doi: 10.1172/JCI1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The International PPH Consortium, Lane KB, Machado RD, Pauciulo. M.W., Thompson JR, Philips JA, III, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2 encoding a TGF-B receptor cause familiar pulmonary hypertension. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 4.Voelkel NF, Cool CD, Lee SD, Wright L, Geraci MW, Tuder RM. Primary pulmonary hypertension between inflammation and cancer. Chest. 1999;114:225S–230S. doi: 10.1378/chest.114.3_supplement.225s. [DOI] [PubMed] [Google Scholar]
- 5.Rai PR, Cool CD, King JAC, Stevens T, Burns N, Winn RA, Kasper M, Voelkel NF. The cancer paradigm of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:558–564. doi: 10.1164/rccm.200709-1369PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1 alpha. J Clin Invest. 1999;103:691–696. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P. Heterozygous deficiency of hypoxia-inducible factor-2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest. 2003;111:1519–1527. doi: 10.1172/JCI15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Semenza GL. Life with oxygen. Science. 2007;318:62–64. doi: 10.1126/science.1147949. [DOI] [PubMed] [Google Scholar]
- 9.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 10.Warburg O, Posener K, Negelein E. Ueber den Stoffwechsel der Carcinomzelle. Biochem. 1924;Z152:319–344. [Google Scholar]
- 11.Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E, Lupu F, Nemery B, Dewerchin M, Van Veldhoven P, Plate K, Moons L, Collen D, Carmeliet P. Loss of HIF-2[alpha] and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med. 2002;8:702–710. doi: 10.1038/nm721. [DOI] [PubMed] [Google Scholar]
- 12.Tuder RM, Flook BE, Voelkel NF. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. J Clin Invest. 1995;95:1798–1807. doi: 10.1172/JCI117858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuder RM, Chacon M, Alger LA, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, Bishop AE, Geraci MW, Semenza GL, Yacoub M, Polak JM, Voelkel NF. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol. 2001;195:367–374. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 14.Bonnet S, Michelakis ED, Porter CJ, ndrade-Navarro MA, Thebaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry S, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1 alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats—similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 15.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, Dweik RA, Tuder RM, Stuehr DJ, Erzurum SC. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA. 2007;104:1342–1347. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, Drazba J, nand-Apte B, Erzurum SC. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L548–L554. doi: 10.1152/ajplung.00428.2006. [DOI] [PubMed] [Google Scholar]
- 17.Girgis RE, Champion HC, Diette GB, Johns RA, Permutt S, Sylvester JT. Decreased exhaled nitric oxide in pulmonary arterial hypertension: response to bosentan therapy. Am J Respir Crit Care Med. 2005;172:352–357. doi: 10.1164/rccm.200412-1684OC. [DOI] [PubMed] [Google Scholar]
- 18.Kaneko FT, Arroliga AC, Dweik RA, Comhair SA, Laskowski D, Oppedisano R, Thomassen MJ, Erzurum SC. Biochemical reaction products of nitric oxide as quantitative markers of primary pulmonary hypertension. Am J Respir Crit Care Med. 1998;158:917–923. doi: 10.1164/ajrccm.158.3.9802066. [DOI] [PubMed] [Google Scholar]
- 19.Machado RF, Londhe Nerkar MV, Dweik RA, Hammel J, Janocha A, Pyle J, Laskowski D, Jennings C, Arroliga AC, Erzurum SC. Nitric oxide and pulmonary arterial pressures in pulmonary hypertension. Free Radic Biol Med. 2004;37:1010–1017. doi: 10.1016/j.freeradbiomed.2004.06.039. [DOI] [PubMed] [Google Scholar]
- 20.Ozkan M, Dweik RA, Laskowski D, Arroliga AC, Erzurum SC. High levels of nitric oxide in individuals with pulmonary hypertension receiving epoprostenol therapy. Lung. 2001;179:233–243. doi: 10.1007/s004080000064. [DOI] [PubMed] [Google Scholar]
- 21.Xu W, Kaneko FT, Zheng S, Comhair SA, Janocha AJ, Goggans T, Thunnissen FB, Farver C, Hazen SL, Jennings C, Dweik RA, Arroliga AC, Erzurum SC. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004;18:1746–1748. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- 22.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med. 2004;169:764–769. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 23.Grobe AC, Wells SM, Benavidez E, Oishi P, Azakie A, Fineman JR, Black SM. Increased oxidative stress in lambs with increased pulmonary blood flow and pulmonary hypertension: role of NADPH oxidase and endothelial NO synthase. Am J Physiol−Lung Cell MPH. 2006;290:L1069–L1077. doi: 10.1152/ajplung.00408.2005. [DOI] [PubMed] [Google Scholar]
- 24.Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox) Am J Physiol Lung Cell Mol Physiol. 2006;290:L2–L10. doi: 10.1152/ajplung.00135.2005. [DOI] [PubMed] [Google Scholar]
- 25.Wedgwood S, Black SM. Role of reactive oxygen species in vascular remodeling associated with pulmonary hypertension. Antioxid Redox Signal. 2003;5:759–769. doi: 10.1089/152308603770380061. [DOI] [PubMed] [Google Scholar]
- 26.Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, Lebrec D, Speich R, Beghetti M, Rich S, Fishman A. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:5S–12S. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 27.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, Gaine SP, Gladwin MT, Jing ZC, Krowka MJ, Langleben D, Nakanishi N, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–S54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 28.Richter A, Yeager ME, Zaiman A, Cool CD, Voelkel NF, Tuder RM. Impaired transforming growth Factor β signaling in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;170:1340–1348. doi: 10.1164/rccm.200311-1602OC. [DOI] [PubMed] [Google Scholar]
- 29.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arsham AM, Plas DR, Thompson CB, Simon MC. Phosphatidylinositol 3-kinase/Akt signaling is neither required for hypoxic stabilization of HIF-1 alpha nor sufficient for HIF-1-dependent target gene transcription. J Biol Chem. 2002;277:15162–15170. doi: 10.1074/jbc.M111162200. [DOI] [PubMed] [Google Scholar]
- 31.Comhair SAA, Xu W, Ghosh S, Thunnissen FBJM, Almasan A, Calhoun WJ, Janocha AJ, Zheng L, Hazen SL, Erzurum SC. Superoxide dismutase inactivation in pathophysiology of asthmatic airway remodeling and reactivity. Am J Pathol. 2005;166:663–674. doi: 10.1016/S0002-9440(10)62288-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tuder RM, Groves BM, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275–285. [PMC free article] [PubMed] [Google Scholar]
- 33.Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A, Wilson GD, Turley H, Talks KL, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60:7075–7083. [PubMed] [Google Scholar]
- 34.Dorfmuller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358–363. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 35.Masri FA, Comhair SA, Dostanik-Larson I, Kaneko FT, Dweik RA, Arroliga AC, Erzurum SC. Deficiency of lung antioxidants in idiopathic pulmonary arterial hypertension. Clin Transl Sci. 2008;1:99–106. doi: 10.1111/j.1752-8062.2008.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaewpila S, Venkataraman S, Buettner GR, Oberley LW. Manganese superoxide dismutase modulates hypoxia-inducible factor-1 alpha induction via superoxide. Cancer Res. 2008;68:2781–2788. doi: 10.1158/0008-5472.CAN-07-2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang M, Kirk JS, Venkataraman S, Domann FE, Zhang HJ, Schafer FQ, Flanagan SW, Weydert CJ, Spitz DR, Buettner GR, Oberley LW. Manganese superoxide dismutase suppresses hypoxic induction of hypoxia-inducible factor-1alpha and vascular endothelial growth factor. Oncogene. 2005;24:8154–8166. doi: 10.1038/sj.onc.1208986. [DOI] [PubMed] [Google Scholar]
- 38.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 39.Mateo J, Garcia-Lecea M, Cadenas S, Hernandez C, Moncada S. Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem J. 2003;376:537–544. doi: 10.1042/BJ20031155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murphy MP. Does interplay between nitric oxide and mitochondria affect hypoxia-inducible transcription factor-1 activity? Biochem J. 2003;376:e5–e6. doi: 10.1042/BJ20031605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coulet F, Nadaud S, Agrapart M, Soubrier F. Identification of hypoxia-response element in the human endothelial nitric-oxide synthase gene promoter. J Biol Chem. 2003;278:46230–46240. doi: 10.1074/jbc.M305420200. [DOI] [PubMed] [Google Scholar]
- 42.Cracowski JL, Cracowski C, Bessard G, Pepin JL, Bessard J, Schwebel C, Stanke-Labesque F, Pison C. Increased lipid peroxidation in patients with pulmonary hypertension. Am J Respir Crit Care Med. 2001;164:1038–1042. doi: 10.1164/ajrccm.164.6.2104033. [DOI] [PubMed] [Google Scholar]
- 43.Kimura H, Weisz A, Kurashima Y, Hashimoto K, Ogura T, D'Acquisto F, Addeo R, Makuuchi M, Esumi H. Hypoxia response element of the human vascular endothelial growth factor gene mediates transcriptional regulation by nitric oxide: control of hypoxia-inducible factor-1 activity by nitric oxide. Blood. 2000;95:189–197. [PubMed] [Google Scholar]
- 44.Palmer LA, Gaston B, Johns RA. Normoxic stabilization of hypoxia-inducible factor-1 expression and activity: redox-dependent effect of nitrogen oxides. Mol Pharmacol. 2000;58:1197–1203. doi: 10.1124/mol.58.6.1197. [DOI] [PubMed] [Google Scholar]
- 45.Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell. 2003;14:3470–3481. doi: 10.1091/mbc.E02-12-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Callapina M, Zhou J, Schmid T, Kohl R, Brune B. NO restores HIF-1 alpha hydroxylation during hypoxia: role of reactive oxygen species. Free Rad Biol Med. 2005;39:925–936. doi: 10.1016/j.freeradbiomed.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 47.Agani FH, Pichiuli P, Chavez JC, LaManna JC. The role of mitochondria in the regulation of the hypoxia-inducible factor 1 expression during hypoxia. J Biol Chem. 2000;275:35863–35867. doi: 10.1074/jbc.M005643200. [DOI] [PubMed] [Google Scholar]
- 48.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schroedl C, McClintock DS, Budinger GRS, Chandel NS. Hypoxic but not anoxic stabilization of HIF-1 alpha requires mitochondrial reactive oxygen species. Am J Physiol −Lung Cell MPH. 2002;283:L922–L931. doi: 10.1152/ajplung.00014.2002. [DOI] [PubMed] [Google Scholar]
- 50.Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 51.Liu Y, Christou H, Morita T, Laughner E, Semenza GL, Kourembanas S. Carbon monoxide and nitric oxide suppress the hypoxic induction of vascular endothelial growth factor gene via the 5′ enhancer. J Biol Chem. 1998;273:15257–15262. doi: 10.1074/jbc.273.24.15257. [DOI] [PubMed] [Google Scholar]
- 52.Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci USA. 2006;103:5379–5384. doi: 10.1073/pnas.0601026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vinogradov AD, Grivennikova VG. Generation of superoxide-radical by the NADH:ubiquinone oxidoreductase of heart mitochondria. Biochemistry (Mosc) 2005;70:120–127. doi: 10.1007/s10541-005-0090-7. [DOI] [PubMed] [Google Scholar]
- 54.Li M, Chiu JF, Mossman BT, Fukagawa NK. Down-regulation of manganese-superoxide dismutase through phosphorylation of FOXO3a by Akt in explanted vascular smooth muscle cells from old rats. J Biol Chem. 2006;281:40429–40439. doi: 10.1074/jbc.M606596200. [DOI] [PubMed] [Google Scholar]
- 55.Yeager ME, Halley GR, Golpon HA, Voelkel NF, Tuder RM. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ Res. 2001;88:e8–e11. doi: 10.1161/01.res.88.1.e2. [DOI] [PubMed] [Google Scholar]
- 56.Cool CD, Rai MD, Yeager ME, Hernandez-Saavedra D, Serls AE, Bull TM, Geraci MW, Brown KK, Routes JM, Tuder RM, Voelkel NF. Expression of human herpesvirus 8 in primary pulmonary hypertension. N Engl J Med. 2003;349:1113–1122. doi: 10.1056/NEJMoa035115. [DOI] [PubMed] [Google Scholar]
- 57.Ma Q, Cavallin LE, Yan B, Zhu S, Duran EM, Wang H, Hale LP, Dong C, Cesarman E, Mesri EA, Goldschmidt-Clermont PJ. Antitumorigenesis of antioxidants in a transgenic Rac1 model of Kaposi's sarcoma. Proc Natl Acad Sci USA. 2009;106:8683–8688. doi: 10.1073/pnas.0812688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang Z, Iqbal M, Cawthon D, Bottje WG. Heart and breast muscle mitochondrial dysfunction in pulmonary hypertension syndrome in broilers (Gallus domesticus) Comp Biochem Physiol A Mol Integr Physiol. 2002;132:527–540. doi: 10.1016/s1095-6433(02)00005-3. [DOI] [PubMed] [Google Scholar]
- 59.Cawthon D, Iqbal M, Brand J, McNew R, Bottje WG. Investigation of proton conductance in liver mitochondria of broilers with pulmonary hypertension syndrome. Poult Sci. 2004;83:259–265. doi: 10.1093/ps/83.2.259. [DOI] [PubMed] [Google Scholar]
- 60.Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med. 2005;353:1412–1413. doi: 10.1056/NEJMc051946. [DOI] [PubMed] [Google Scholar]
- 61.Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 62.Tofovic SP, Salah EM, Mady HH, Jackson EK, Melhem MF. Estradiol metabolites attenuate monocrotaline-induced pulmonary hypertension in rats. J Cardiovasc Pharmacol. 2005;46:430–437. doi: 10.1097/01.fjc.0000175878.32920.17. [DOI] [PubMed] [Google Scholar]
- 63.Koulmann N, Novel-Chate V, Peinnequin A, Chapot R, Serrurier B, Simler N, Richard H, Ventura-Clapier R, Bigard X. Cyclosporin A inhibits hypoxia-induced pulmonary hypertension and right ventricle hypertrophy. Am J Respir Crit Care Med. 2006;174:699–705. doi: 10.1164/rccm.200512-1976OC. [DOI] [PubMed] [Google Scholar]
- 64.Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation. 2002;105:244–250. doi: 10.1161/hc0202.101974. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.