

Abstract
Bone morphogenetic protein-7 (BMP-7) improves outcome in animal models of fibrotic renal disease by opposing transforming growth factor β1 (TGF-β)-dependent fibrosis. However, the underlying mechanisms remain obscure. Here, we studied the effect of BMP-7 on response to TGF-β in the proximal tubular cell line HK-2 (PTC). BMP-7 specifically limited Smad3 but not Smad2 signaling. BMP-7 did not inhibit Smad3 phosphorylation or nuclear accumulation, nor did BMP-7 alter phosphorylated Smad3 dephosphorylation or degradation. However, BMP-7 treatment reduced Smad3 DNA binding to a consensus Smad binding element probe, and chromatin immunoprecipitation showed reduced Smad3 binding to the plasminogen activator inhibitor-1 promoter in PTCs treated with BMP-7 and TGF-β compared with TGF-β alone. Degradation of the transcriptional repressor SnoN has recently been shown to be necessary for Smad3 (but not Smad2) signaling. SnoN expression was transiently lost in PTCs after TGF-β stimulation, but BMP-7 prevented this. Furthermore, BMP-7 had no effect on Smad3 signaling after siRNA-mediated SnoN knockdown, whereas prevention of SnoN degradation with the proteasome inhibitor MG132 reproduced the inhibitory action of BMP-7 on Smad3 signaling. We conclude that BMP-7 prevents TGF-β-mediated loss of the transcriptional repressor SnoN and hence specifically limits Smad3 DNA binding, altering the balance of transcriptional responses to TGF-β in PTCs. These results provide an important mechanistic insight into a key regulator of TGF-β signaling.
Transforming growth factor β1 (TGF-β) is a key profibrotic cytokine in the kidney and other solid organs.1 However, TGF-β induces numerous cellular responses and, in addition to its profibrotic role, acts as a central orchestrator of development, wound healing, and cancer and a suppressor of inflammation and immune responses. The factors governing how cells read TGF-β signals are thus central to understanding pathology in many contexts.2
Bone morphogenetic protein-7 (BMP-7) has emerged as a key antifibrotic cytokine in the kidney. BMP-7 prevents fibrosis and antagonizes the effects of TGF-β in animal models including unilateral ureteric obstruction,3 nephrotoxic serum nephritis,4 collagen IVα3-deficient mice (Alport's syndrome)5 MRLlpr/lpr mice (lupus nephritis-like glomerulonephritis),5 and nephropathy associated with streptozotocin-induced diabetes.6 Accordingly, BMP-7 has attracted substantial interest as a potential therapy for chronic kidney disease.
Surprisingly, there are few data on mechanisms by which BMP7 opposes the profibrotic effects of TGF-β. In mesangial cells, BMP-7 inhibits nuclear accumulation of the key signaling molecule regulated by TGF-β, Smad3, after TGF-β stimulation.7 We have shown previously that BMP7 ameliorates proinflammatory cellular interactions in chronic kidney disease8 and inhibits monocyte-stimulated proximal tubular cell TGF-β generation.9 However, these effects do not fully explain the inhibition of the profibrotic effects of TGF-β seen in response to BMP-7 in vivo. In particular, reversal of TGF-β-dependent tubular cell epithelial to mesenchymal transition may be central to the action of BMP-7 in renal fibrosis,4,10 but the underlying mechanisms remain undefined. Cytokines from the TGF-β family elicit their biological functions by binding to a series of structurally related type I and type II receptors. The activated receptor complex recruits and serine-phosphorylates receptor-regulated Smad proteins (R-Smads, the intracellular signaling molecules of the TGF-β family). Phosphorylated R-Smads form complexes with the co-Smad (Smad4), accumulate in the nucleus, and alter gene transcription. Combinatorial interactions of type II and type I receptors occur, but the receptors and R-Smads phosphorylated by them can be functionally grouped into “TGF-β-like” and “BMP-like” (reviewed by Ross and Hill.11). The R-Smads for TGF-β and related ligands are Smads 2 and 3, and for BMP-type ligands the R-Smads are Smads 1,5, and 8.
The aim of the current study was to systematically examine the effect of BMP-7 on the signaling response to TGF-β. The data show that BMP7 specifically inhibits Smad3 but not Smad2 signaling in proximal tubular cells, leading to inhibition of profibrotic transcriptional responses elicited by TGF-β. The inhibition of the Smad3 transcriptional responses is due to reduction of Smad3 DNA binding, related to a failure to degrade the transcriptional repressor SnoN in BMP-7-treated cells. These results provide important mechanistic insight into the antifibrotic actions of BMP-7 in kidney disease.
Materials and Methods
Cell Culture
HK-2 cells (human renal proximal tubular epithelial cells immortalized by transduction with human papilloma virus 16 E6/E7 genes [PTCs]) were cultured as described previously.12 With the exception of the cells used for transfection, cells were growth-arrested in serum-free medium for 48 hours before use in experiments. All experiments were performed in serum-free conditions.
Quantitative RT-PCR
Quantitative (q) RT-PCR was performed as described previously13 using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA). Fold changes in expression were calculated using the formula 2 − (Ctsample 1 − Ctsample 2), where Ct is the difference between the amplification threshold for the gene of interest and normalizer. P values were calculated by t-test using Microsoft Excel.
Transient Transfection and Reporter Gene Analysis
CAGA(4) Smad3 responsive reporter construct was a gift from Arisitdis Moustakas.14 ARE and MF1 Smad2 responsive reporter constructs were a gift from Lalage Wakefield.15 pRL-CMV Renilla control plasmid was purchased from Promega (Madison, WI). Transient transfection and reporter gene analysis using HK-2 cells were performed as described previously.12 For Smad3 experiments, 0.9 μg of the Smad3/4-specific reporter SBE-Luc was transfected with 0.1 μg of pRL-CMV Renilla to control for transfection efficiency. For Smad2-responsive experiments, 0.45 μg of the Smad2/4-specific promoter ARE-Luc was transfected together with 0.45 μg of its co-plasmid MF1 and 0.1 μg of pRL-CMV Renilla. After lysis of the cells in Reporter Lysis Buffer (Promega), firefly and Renilla luciferase content was quantified using the Dual-Glo Assay (Promega).
Immunoblotting
Cell extracts were prepared in SDS sample buffer and boiled for 5 minutes at 95°C before 10% SDS-polyacrylamide gel electrophoresis performed under reducing conditions, transfer to a nitrocellulose membrane (Amersham, Little Chalfont, Buckinghamshire, UK), incubation with primary antibody in PBS-0.1% (v/v) Tween-20, and then incubation with horseradish peroxidase-conjugated secondary antibody (Sigma-Aldrich, Poole, UK). Protein was visualized using enhanced chemiluminescence (Amersham) according to the manufacturer's instructions.
Immunofluorescence Microscopy
Subconfluent monolayers of cells grown in eight-well glass chamber slides were fixed in acetone/methanol (1:1, v/v) (Fisher Scientific, Pittsburgh, PA) for 10 minutes and then were washed in calcium/magnesium-free PBS, pH 7.4 (Invitrogen, Carlsbad, CA), blocked with 1% (w/v) bovine serum albumin (BSA)/HBSS, and washed in 0.1% (w/v) BSA/HBSS. The slides were incubated with primary antibody and then secondary antibody diluted in 0.1% BSA/HBSS for 2 hours at room temperature. The source and dilution of the antibodies were as follows: polyclonal mouse anti-Smad3 1:50 (sc-8332, Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and fluorescein isothiocyanate-conjugated rabbit anti-mouse IgG 1:40 (Dako North America, Inc., Carpenteria, CA). The cells were washed extensively with 0.1% BSA in HBSS, mounted in Vectashield fluorescent mountant (Vecta Laboratories, Peterborough, UK), and examined on a Leica Dialux 20 fluorescent microscope [Leica Microsystems (UK) Ltd., Milton Keynes, UK].
Electrophoretic Mobility Shift Assay
Nuclear protein extraction and electrophoretic mobility shift assays for nuclear factor-κB were performed as described previously.16 In brief, cells were harvested in ice-cold PBS (pH 7.4) and pelleted by centrifugation. Cells were resuspended in ice-cold buffer A (10 mm HEPES-KOH [pH 7.9], 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.5 mmol/L dithiothreitol, and 0.2 mmol/L phenylmethylsulfonyl fluoride) and incubated on ice for 10 minutes. The cell pellet was collected by centrifugation, resuspended in buffer B (20 mmol/L HEPES-KOH [pH 7.9], 25% glycerol, 420 mmol/L NaCl, 1.5 mmol/L MgCl2, 0.2 mmol/L EDTA, 0.3 mmol/L dithiothreitol, and 0.2 mmol/L phenylmethylsulfonyl fluoride), and incubated on ice for 20 minutes followed by a brief high-speed centrifugation (12,000 × g for 10 seconds at 4°C), and the resulting supernatants (nuclear extract) were collected. Smad Binding Element Oligonucleotide Probe (sc-2603) was bought from Santa Cruz Biotechnology, Inc. and annealed for use in the electrophoretic mobility shift assay. These double-stranded fragments were labeled with [α32P]dTTP (Amersham-Pharmacia Biotech, Little Chalfont, Buckinghamshire, UK) using the Klenow fragment of DNA polymerase I. The composition of protein/DNA complexes was determined by supershift assays using rabbit polyclonal antibodies specific for Smad1 (9512, New England Biolabs, Ipswich, MA), Smad2 (3122, New England Biolabs), Smad3 (51-1500, Invitrogen), Smad4 (sc-7955, Santa Cruz Biotechnology. Inc.), and Smad 5 (9517, New England Biolabs).
Chromatin Immunoprecipitation
Cells were cross-linked with 1% formaldehyde at room temperature for 20 minutes and then were resuspended in lysis buffer (50 mmol/L HEPES-KOH. pH 7.5, 140 mmol/L NaCl, 1 mmol/L EDTA, pH 8, 1% Triton X-100, 0.1% sodium deoxycholate, and 0.1% SDS) before sonication to yield DNA fragments ranging in size from 200 to 500 bp (Bioruptor, Diagenode SA, Liège, Belgium). Approximately 25 μg of protein of the cross-linked, sheared chromatin solution was used for immunoprecipitation. A small portion of each immunoprecipitation mixture was saved as input DNA (5%). Supernatants were diluted 10-fold in radioimmunoprecipitation assay buffer (50 mmol/L Tris-HCl, pH 8, 150 mmol/L NaCl, 2 mmol/L EDTA, pH 8, 1% Nonidet P-40, and 0.5% sodium deoxycholate) and immunoprecipitated for 4 hours at 4°C with 5 μg of rabbit anti-Smad3 with constant mixing. Control immunoprecipitations were performed at the same time with preimmune IgG. After addition of 60 μl of protein A-Sepharose containing 1.5 μg of sheared salmon sperm DNA, samples were incubated with constant mixing overnight at 4°C, centrifuged for 1 minute at 13,000 rpm, and washed three times with wash buffer I (0.1% SDS, 1% Triton X-100, 2 mmol/L EDTA, pH 8, 150 mmol/L NaCl, and 20 mmol/L Tris-HCl, pH 8) and once with wash buffer II (0.1% SDS, 1% Triton X-100, 2 mmol/L EDTA, pH 8, 500 mmol/L NaCl, and 20 mmol/L Tris-HCl, pH 8). DNA was eluted in 1% SDS, 100 mmol/L NaHCO3 for 1 hour. After proteinase K digestion, DNA was extracted with phenol: chloroform and ethanol-precipitated in the presence of 10 μl of glycogen (5 mg/ml). DNA was amplified by real-time PCR using SYBR Green Master Mix. Primers were designed using Primer 3 software for the second and third Smad binding elements in the plasminogen activator inhibitor-1 (PAI-1) promoter, previously shown to cooperatively regulate the transcriptional response of PAI-1 to TGF-β.17 Primer sequences were as follows: second Smad binding element, −580CAGA: forward 5′-GGGAGTCAGCCGTGTATCAT-3′ and reverse 5′-ACCTCCATCAAAACGTGGAA-3′; third Smad binding element −280CAGA, forward 5′-AGTCAACCTGGCAGGACATC-3′ and reverse 5′-ACACCTCCCTCTCTGGGACT-3′.
Small Interfering RNA
Transfection of HK-2 cells with small interfering RNA (siRNA) was performed as described previously.13 siRNAs were purchased from Ambion (Austin, TX).
Statistical Analysis
Unless otherwise specified, data are presented for a representative experiment of at least three separate experiments giving consistent results. Statistical analysis between groups was performed using an unpaired Student's t-test and for multiple groups using analysis of variance, with a value of P < 0.05 considered to represent a significant difference. The data are presented as means of n biological replicates as indicated in the figure legends. Error bars indicate the SEM.
Results
Inhibition of TGF-β-Dependent Responses by BMP-7
TGF-β-induced fibrosis is Smad3-dependent, with key downstream targets including connective tissue growth factor, PAI-1, and TGF-β itself (reviewed by Roberts et al18). Initial experiments were performed to investigate inhibition of TGF-β-dependent transcriptional responses by BMP-7. HK-2 cells were incubated with 1 ng/ml TGF-β or control medium plus 50 ng/ml BMP-7 for 24 hours. Subsequent quantitative RT-PCR demonstrated increased expression of connective tissue growth factor, PAI-1, and TGF-β in response to TGF-β (Figure 1, A–C). BMP-7 alone did not significantly alter expression of connective tissue growth factor, PAI-1, or TGF-β, but BMP-7 significantly attenuated responses to TGF-β (Figure 1, A–C). PAI-1 is a powerful fibrogenic molecule that is not expressed in the normal kidney (reviewed by Eddy and Fogo19) and has a well characterized sustained transcriptional response to TGF-β.20 Additional experiments demonstrated inhibition of PAI-1 expression when BMP-7 was added to TGF-β-treated cells at time points up to 8 days (Figure 1D).
Figure 1.
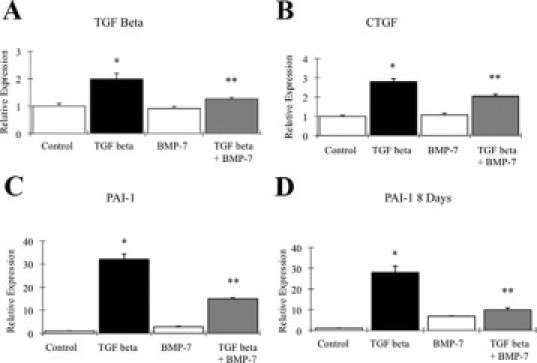
Regulation of profibrotic TGF-β transcriptional targets by BMP-7. A–C: qRT-PCR analysis shows expression of profibrotic TGF-β-inducible genes in PTCs after 24 hours with or without1 ng/ml TGF-β incubation with or without 50 ng/ml BMP-7. A: TGF-β. B: Connective tissue growth factor (CTGF). C: PAI-1. D: PAI-1 expression after 8 days of culture with or without 1 ng/ml TGF-β with or without 50 ng/ml BMP-7. n = 3, mean of duplicate determinations. Error bars indicate +SEM. Representative data from one of three experiments giving similar results. *P < 0.01 versus control. **P < 0.05 versus TGF-β.
To further investigate alterations in TGF-β response caused by BMP7, transient transfection of reporter genes sensitive to Smad3 (CAGA) and Smad2 (ARE/MF1) was undertaken. BMP-7 inhibited subsequent Smad3 signaling response to TGF-β after 12 or more hours (Figure 2A) and at concentrations of 50 ng/ml or greater (Figure 2B). There was little difference in response to BMP-7 at doses above 50 ng/ml. In contrast, BMP-7 did not significantly alter Smad2 signaling at any of the doses or times examined (Figure 2C and data not shown).
Figure 2.
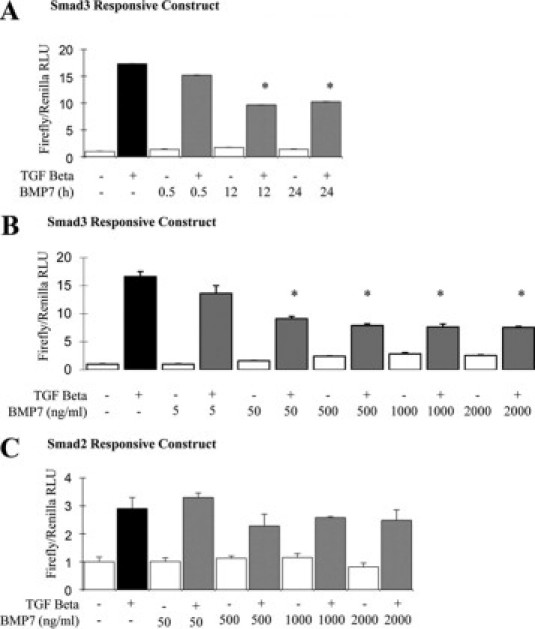
Regulation of Smad signaling response to TGF-β by BMP-7. HK-2 cells were transfected with Smad3-responsive (A and B) or Smad2-responsive (C) plasmids and then were incubated with BMP-7 for various times or doses, before incubation with 1 ng/ml TGF-β (black bar, control medium followed by TGF-β; gray bars, BMP-7 followed by TGF-β) or control medium (white bars) for 6 hours. A: Effect on Smad3 signaling of time course of 50 ng/ml BMP-7 incubation. B: Effect on Smad3 signaling of 24 hours preincubation with a BMP-7 dose range of 0 to 2000 ng/ml. C: Effect on Smad2 signaling of a 24-hour preincubation with BMP-7 at a dose range of 0 to 2000 ng/ml. *P < 0.05 versus TGF-β. RLU, relative light units.
BMP-7 Does Not Alter Smad3 Phosphorylation, Nuclear Accumulation, or Dephosphorylation
Inhibition of Smad3 but not Smad2 signaling suggested that BMP-7 acts beyond the point of TGF-β ligand-receptor interaction. Accordingly, the time course of Smad1, 2, and 3 phosphorylation in PTCs exposed to TGF-β and BMP-7 was examined, alone and in combination. Preliminary experiments showed that Smad2 and 3 phosphorylation was detectable for 24 hours after TGF-β treatment, with a maximal response by 1 hour (not shown). Although Smad1 phosphorylation was readily detected after BMP-7 treatment, confirming the signaling response to BMP-7, BMP-7 had no effect on Smad2 or Smad3 phosphorylation when cells were coincubated with TGF-β and BMP-7 (Figure 3A). No change in Smad3 phosphorylation was seen in cells preincubated with BMP-7 and then exposed to TGF-β (data not shown). Transient Smad1 phosphorylation was also evident after TGF-β treatment (Figure 3A), in keeping with recent reports of transient Smad1/5 phosphorylation in epithelial cells in response to TGF-β.21
Figure 3.
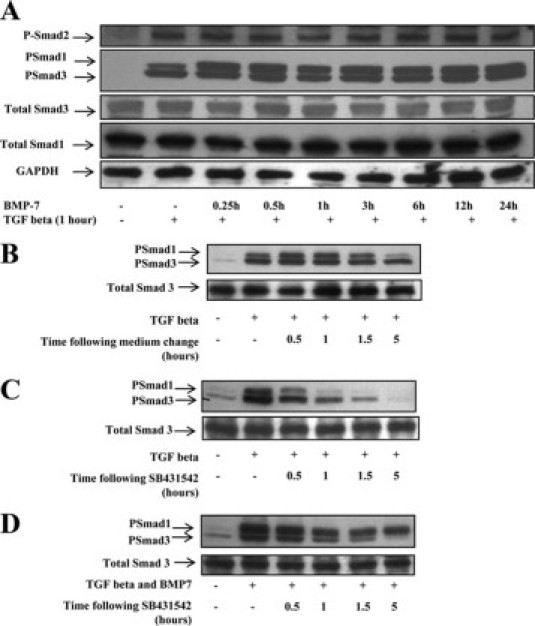
Time course of Smad phosphorylation and dephosphorylation/degradation in response to TGF-β and BMP-7. A: HK-2 cells were incubated with 50 ng/ml BMP-7 for 0 to 24 hours before incubation with 1 ng/ml TGF-β for 1 hour. Whole-cell lysates were immunoblotted with antibodies against Phospho-Smads (PSmad) 1, 2, 3, and total Smads 1 and 3. Stripping and reprobing for glyceraldehyde-3-phosphate dehydrogenase (GADPH) was used to confirm approximately equal loading. B–D: Time course of Smad1/3 dephosphorylation/degradation. HK-2 cells were incubated with control medium (B and C) or 50 ng/ml BMP-7 (D) for 24 hours before incubation with 1 ng/ml TGF-β for 30 minutes. Cells were washed extensively and incubated in cytokine-free control medium (B) or medium containing the Alk5 kinase inhibitor SB431542 (C and D) for time points up to 5 hours. Residual Phospho-Smad3 activity was detected by immunoblot before stripping and reprobing for total Smad3.
Nuclear import and export of Smad3, together with its dephosphorylation, ubiquitinylation, and degradation, are highly regulated (reviewed by Shi and Massague22). Increased Smad3 degradation has been implicated in altered renal responsiveness to TGF-β in the kidney in a cell phenotype-specific fashion,23 and in mesangial cells, nuclear accumulation of Smad3 is inhibited by BMP-7.7 To study possible Smad3 dephosphorylation or degradation in response to BMP-7, we examined the half-life of phosphorylated (Phospho)-Smad3. HK-2 cells were incubated with BMP-7 or control medium, and then Smad3 phosphorylation was stimulated with TGF-β. After 30 minutes, further Smad3 phosphorylation was inhibited using the Alk5 kinase inhibitor SB431542, and the rate of decline of Phospho-Smad3 was followed with immunoblotting. A similar rate of decline in Phospho-Smad3 was observed in control cells and cells treated with BMP-7 (Figure 3, B–D), suggesting that Phospho-Smad3 dephosphorylation and degradation were not altered in response to BMP-7. Subsequently, nuclear accumulation of Phospho-Smad3 was assessed by immunoblotting and by fluorescence microscopy. Immunoblots of nuclear protein fractions suggested that BMP-7 did not alter nuclear accumulation of Smad3 (Figure 4A). Similarly, immunocytochemical analysis did not detect differences in TGF-β-dependent nuclear accumulation of Smad3 after BMP-7 treatment (Figure 4B).
Figure 4.
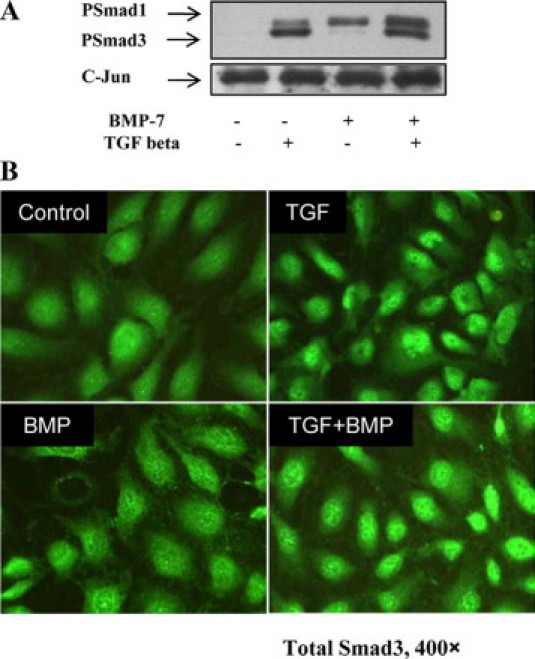
Nuclear accumulation of Smad3. HK-2 cells were incubated with 50 ng/ml BMP-7 or control medium for 24 hours before incubation with 1 ng/ml TGF-β or control medium for 1 hour. A: Immunoblotting of nuclear extracts for Phospho-Smad (PSmad) 1/3 and subsequent reprobing for c-Jun to confirm approximately equal loading. B: Immunofluorescent localization of Smad3. HK-2 cells were incubated with 50 ng/ml BMP-7 or control medium for 24 hours in eight-well chamber slides before incubation with 1 ng/ml TGF-β or control medium for 1 hour and detection of Smad3 by immunofluorescence microscopy.
BMP-7 Inhibits Smad3 DNA Binding
The above data suggest that BMP-7 predominantly limits Smad3 transcriptional responses to TGF-β, without altering TGF-β-dependent Smad phosphorylation and nuclear accumulation or the subsequent Phospho-Smad3 decay rate. Therefore, potential alterations in Smad3 DNA binding were investigated by an electrophoretic mobility shift assay. Probe retardation was readily detectable in protein extracts from TGF-β-treated cells, but this was attenuated in cells coincubated with BMP-7 (Figure 5A). To confirm the presence of Smad3 and Smad4 in the complexes formed, supershift experiments were performed with relevant antibodies (Figure 5B). Smad5 is reported to bind to consensus Smad3 elements, such that the potential for competitive inhibition of Smad3 binding by Smad5 exists.24 However, no binding of Smad 5 was seen after addition of anti-Smad5 antibody (Figure 5B).
Figure 5.
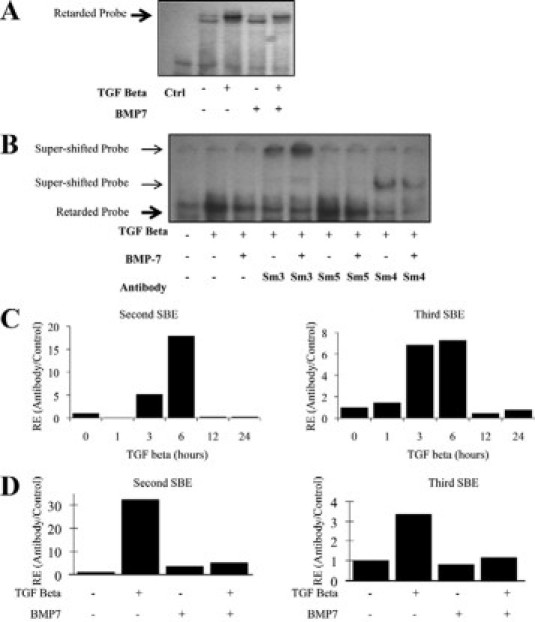
BMP-7 inhibits Smad3 DNA binding. A and B: Electrophoretic mobility shift assay with a consensus Smad binding element probe. HK-2 cells were incubated with BMP-7 or control (Ctrl) medium for 24 hours before incubation with 1 ng/ml TGF-β or control medium for 1 hour. Bold arrow, retarded probe; arrow, supershifted probe. A: Electrophoretic mobility shift assay performed with nuclear protein extract and consensus Smad binding element (SBE) probe. B: Supershift assay performed with antibodies to Smad3, 4, and 5. Sm, Smad. C and D: ChIP: Smad3 binding to the PAI-1 promoter. After chromatin immunoprecipitation with Smad3 antibody or pre-immune globulin, PAI-1 promoter Smad binding elements (SBE) were detected by qRT-PCR. Data are presented as Smad3-precipitated signal/pre-immune globulin-precipitated signal, normalized to control. C: HK-2 cells were incubated with TGF-β for time points to 24 hours before ChIP. D: HK-2 cells were incubated with BMP-7 and TGF-β as indicated for 6 hours before ChIP. RE, Relative Expression.
Subsequently, endogenous Smad3 DNA binding was evaluated by chromatin immunoprecipitation (ChIP). Induction of PAI-1 expression in the kidney is a major contributor to progressive renal fibrosis in chronic kidney disease (reviewed by Eddy and Fogo19). PAI-1 is transcriptionally activated by TGF-β with fast kinetics, and the PAI-1 promoter element contains several Smad binding elements, of which two cooperatively regulate the transcriptional response of PAI-1 to TGF-β.17,20 ChIP showed increased Smad3 binding to these two PAI-1 promoter Smad-responsive elements, with maximal Smad3 binding detected 6 hours after TGF-β stimulation (Figure 5C). BMP-7 alone did not significantly alter Smad3 binding to PAI-1 promoter sites, but inhibition of binding was seen when cells were coincubated with TGF-β and BMP-7 (Figure 5D).
BMP-7 Inhibits Smad3 DNA Binding by Preventing SnoN Degradation
Ski and SnoN are related proteins that bind to Smad proteins and to Smad binding elements in DNA and repress Smad-dependent transcriptional activation. Whereas Ski has broad TGF-β family repressor activity, with inhibitory action on BMP-dependent as well as TGF-β-dependent Smad signals, SnoN seems to be restricted to TGF-β-dependent Smad inhibition.25 Recent work demonstrated that SnoN degradation is required for Smad3- but not Smad2-dependent transcription.26 Accordingly, Ski and SnoN expression were examined in PTCs after TGF-β stimulation. Immunoblot studies showed reduced SnoN expression from 15 minutes to 6 hours after TGF-β stimulation, but from 12 or more hours after TGF-β stimulation, re-accumulation of SnoN was evident (Figure 6A). No changes in Ski expression were seen in response to TGF-β over the time course examined (Figure 6A). Subsequently, SnoN expression was evaluated in cells treated with TGF-β and BMP7, alone or in combination. BMP-7 alone did not alter SnoN expression (Figure 6B), but addition of BMP-7 prevented TGF-β-mediated SnoN degradation (Figure 6C). In additional experiments, cells were incubated for 24 hours with 50 ng/ml BMP-7 or control medium, followed by addition of 1 ng/ml TGF-β for 30 minutes. Preincubation with BMP-7 prevented loss of SnoN expression seen with TGF-β alone (Figure 6, D and E).
Figure 6.
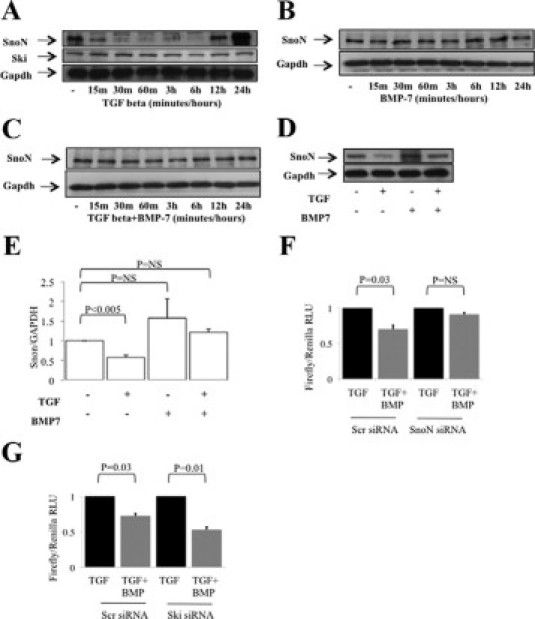
BMP-7 prevents TGF-β-mediated down-regulation of the repressor of Smad3 transcription, SnoN. A–C: HK-2 cells were incubated with 1 ng/ml TGF-β, 50 ng/ml BMP-7, or both, for time points to 24 hours before immunoblotting of whole-cell extracts. Subsequently, blots were reprobed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to confirm approximately equal loading. A: SnoN and Ski expression after TGF-β. B: SnoN expression after BMP-7. C: SnoN expression after TGF-β and BMP-7. D and E: HK-2 cells were incubated with 50 ng/ml BMP-7 or control medium for 24 hours before incubation with 1 ng/ml TGF-β or control medium for 30 minutes and immunoblotting of whole-cell extracts for SnoN. Subsequently, blots were reprobed for GAPDH to confirm approximately equal loading. D: Representative immunoblot of four experiments giving similar results. E: Densitometry of four independent experiments. F: HK-2 cells were transfected with SnoN siRNA together with Smad3 responsive (Scr) (CAGA luciferase) and control (Renilla) vectors for 48 hours. Subsequently, cells were incubated with BMP-7 or control medium for 24 hours and then incubated with TGF-β for 6 hours. Data are presented as firefly luciferase/Renilla luciferase, normalized to the TGF-β control. G: HK-2 cells were transfected with Ski siRNA together with Smad3-responsive (Scr) (CAGA luciferase) and control (Renilla) vectors for 48 hours. Subsequently, cells were incubated with BMP-7 or control medium for 24 hours and then incubated with TGF-β for 6 hours. Data are presented as firefly luciferase/Renilla luciferase, normalized to TGF-β control. RLU, relative light units.
The above data are consistent with a reduction in Smad3 DNA binding due to a failure to degrade SnoN in BMP-7-treated cells. Accordingly, the effect of SnoN and Ski knockdown on modification of TGF-β signaling by BMP-7 was examined. Preliminary immunoblot and qRT-PCR studies confirmed >80% reduction in the expression of SnoN or Ski in PTCs transfected with the respective siRNA after 48 hours (not shown). In subsequent experiments, PTCs were co-transfected with siRNA and with the Smad3 reporter vector CAGA.4 Basal Smad3 reporter activity was not altered by SnoN knockdown. BMP-7 inhibited reporter response in control transfected cells but not in cells transfected with SnoN siRNA (Figure 6F). In contrast, Ski knockdown did not prevent inhibition of reporter activation by BMP-7 (Figure 6G).
Degradation of SnoN may be prevented by the proteasome and calpain inhibitor MG132.27 The effect of MG132 on SnoN expression and Smad3 signaling in PTCs was studied. Immunoblotting showed that MG132 did not alter basal SnoN expression but that TGF-β-induced SnoN degradation was prevented by prior treatment with MG132 (Figure 7A). MG132 treatment of cells transfected with CAGA4 reporter did not alter reporter activity. However, reporter activity in response to TGF-β was diminished in cells pretreated with MG132 (Figure 7B). Subsequently, induction of PAI-1 by TGF-β was studied in MG132-treated PTCs by qRT-PCR. PAI-1 induction by TGF-β was diminished by MG132 (Figure 7, C and D). Taken together, these data show that prevention of SnoN degradation reproduces the effect of BMP-7 on Smad3 signaling, whereas SnoN knockdown prevents it, and links preservation of SnoN expression in response to BMP-7 mechanistically to inhibition of Smad3 signaling by BMP-7.
Figure 7.
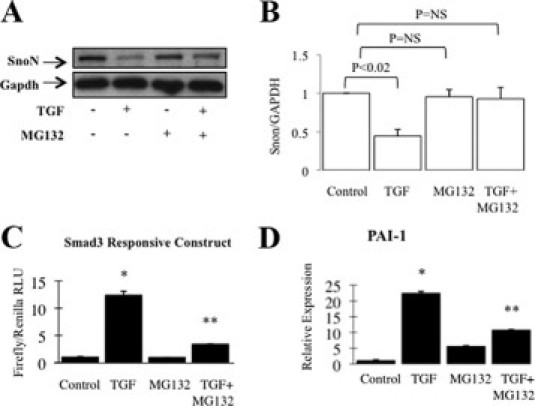
Effect of proteasome inhibitor MG132 on SnoN expression, Smad3 signaling, and TGF-β-dependent PAI-1 expression. A and B: HK-2 cells were incubated with control medium or 1 ng/ml TGF-β for 30 minutes with or without 10 μmol/L MG132, and immunoblots of whole-cell extracts for SnoN are shown. Subsequently, blots were reprobed for GAPDH to confirm approximately equal loading. A: Representative immunoblot of four experiments giving similar results. B: Densitometry of four independent experiments. C: HK-2 cells were transfected with Smad3 and Renilla control reporter constructs for 48 hours, before incubation with control medium or 1 ng/ml TGF-β for 6 hours with or without 10 μmol/L MG132. Data are presented as firefly/Renilla luciferase activity, normalized to control. D: HK-2 cells were incubated with control medium or 1 ng/ml TGF-β for 24 hours with or without 10 μmol/L MG132 before detection of PAI-1 mRNA by qRT-PCR. *P < 0.001 versus control; **P < 0.01 versus TGF-β. RLU, relative light units.
Discussion
BMP-7 is widely expressed during embryonic life and plays a crucial role in renal development,28 during which podocyte-derived BMP-7 is required for normal proximal tubular development.29 Renal tubular BMP-7 expression is diminished in animal models of diabetic nephropathy.30 PTCs express abundant BMP receptors,31 and recombinant BMP-7 improves renal fibrosis and accelerates return of renal function after relief of ureteric obstruction.3 BMP-7 counteracts the TGF-β-induced renal epithelial to mesenchymal transition and reverses chronic renal injury.4 Intriguingly, BMP-7 also opposes TGF-β-induced endocardial to mesenchymal transition in cardiac fibrosis,32 suggesting that BMP-7 may be of general importance in maintenance of the cellular phenotype in the face of excessive TGF-β stimulation. However, the mechanisms underlying these in vivo observations have remained largely obscure. Thus, the aim of this study was to discover the mechanisms by which BMP-7 inhibits TGF-β-dependent profibrotic changes in phenotype in PTCs.
TGF-β is a ubiquitous cytokine that functions in an autocrine or paracrine manner to elicit a multiplicity of effects, principally related to cell growth and extracellular matrix accumulation, which has been implicated widely in the pathogenesis of tissue fibrosis.33 In renal disease, it has been implicated in directing fibrosis in both glomerular and interstitial compartments34–37 and is postulated to be a major biological signal that regulates the switch in tubular cell activities toward a profibrogenic phenotype.38 The activated TGF-β-receptor complex phosphorylates Smad2 and 3, which transduce the TGF-β signal to the nucleus. Deletion of Smad3 prevents the TGF-β-induced epithelial to mesenchymal transition and attenuates fibrotic sequelae,18 suggesting that Smad3 signaling is the dominant pathway in terms of the profibrotic effects of TGF-β.
In the current study, we have found that BMP-7 specifically limits Smad3-dependent transcriptional activation in PTCs, via prevention of SnoN degradation, which is required for Smad3- but not Smad2-dependent transcriptional events (Figure 8, A–C).26 Thus, we have uncovered a major pathway by which BMP-7 prevents profibrotic events in the kidney. It is interesting to compare our data with the action of BMP-7 in mesangial cells, in which induction of the inhibitory Smad6 by BMP-7 leads to loss of TGF-β signaling and failure of nuclear Smad3 accumulation.7 This finding suggests that BMP-7 inhibits TGF-β signaling via multiple mechanisms. However, our data demonstrating a specific effect on Smad3, which is reversed by SnoN knockdown, suggest that in PTCs the dominant mechanism occurs via SnoN.
Figure 8.
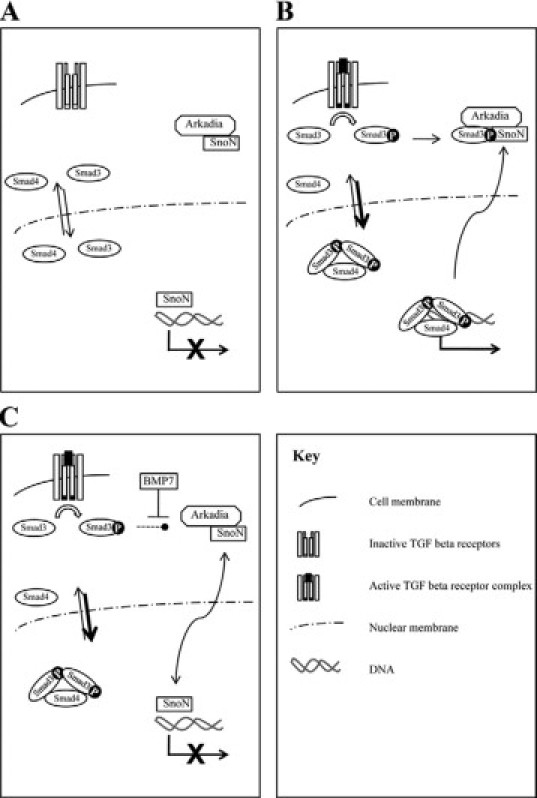
Proposed mechanism of regulation of Smad3 signaling by BMP7. A: In the absence of TGF-β, Smads shuttle into and out of the nucleus. SnoN binds to Smad binding elements and prevents R-Smad binding. B: After ligand binding, active TGF-β receptor complex leads to R-Smad phosphorylation. R-Smad-Arkadia-SnoN complexes lead to SnoN degradation. R-Smad-Smad4 complexes accumulate in the nucleus and bind to DNA. C: BMP7 prevents loss of SnoN expression. R-Smad-Smad4 complexes accumulate in the nucleus, but DNA binding to consensus Smad binding elements is prevented by SnoN. Arkadia-dependent SnoN degradation appears necessary for Smad3 (or Smad2Δ-exon3)-dependent responses, but not Smad1/Smad4-dependent responses or responses driven by Smad2-Smad4-FoxH1 complexes (see Levy et al26).
SnoN is a member of the ski family of oncoproteins that bind to the consensus Smad3 binding element (GTCTAGAC) and repress transcription.39 SnoN has a specific inhibitory action on Smad3 DNA binding, and SnoN degradation is required for transduction of the Smad3 but not Smad2 signal.26 SnoN plays a role in maintaining the repressed state of TGF-β-responsive genes in the absence of ligand and also in negative feedback regulation of TGF-β signaling.40 SnoN expression is progressively reduced in the kidney after unilateral ureteric obstruction in mice, and loss of SnoN expression sensitizes tubular epithelial cells to the epithelial to mesenchymal transition.41 SnoN may be degraded by recruitment to the proteasome by Smad3,27 and loss of SnoN expression in animal models of progressive renal fibrosis occurs via enhanced ubiquitin-mediated degradation.42,43 Interestingly, hepatocyte growth factor exerts an antifibrotic effect in the kidney by enhancing SnoN expression in PTCs.44,45 Taken together with our current data demonstrating that preservation of SnoN expression underlies the antifibrotic actions of BMP-7, it is possible to conclude that loss of SnoN expression is a key element of progressive renal fibrosis.
Footnotes
Supported by a Clinician Scientist Fellowship from the Welsh Office of Research Development (to D.F.).
References
- 1.Wynn T. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Massague J. How cells read TGF-β signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 3.Morrissey J, Hruska K, Guo G, Wang S, Chen Q, Klahr S. Bone morphogenetic protein-7 improves renal fibrosis and accelerates the return of renal function. J Am Soc Nephrol. 2002;13(Suppl 1):S14–S21. [PubMed] [Google Scholar]
- 4.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 5.Zeisberg M, Bottiglio C, Kumar N, Maeshima Y, Strutz F, Muller GA, Kalluri R. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am J Physiol Renal Physiol. 2003;285:F1060–F1067. doi: 10.1152/ajprenal.00191.2002. [DOI] [PubMed] [Google Scholar]
- 6.Sugimoto H, Grahovac G, Zeisberg M, Kalluri R. Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein-7 and advanced glycation end product inhibitors. Diabetes. 2007;56:1825–1833. doi: 10.2337/db06-1226. [DOI] [PubMed] [Google Scholar]
- 7.Wang S, Hirschberg R. Bone morphogenetic protein-7 signals opposing transforming growth factor β in mesangial cells. J Biol Chem. 2004;279:23200–23206. doi: 10.1074/jbc.M311998200. [DOI] [PubMed] [Google Scholar]
- 8.Selbi W, de la Motte C, Hascall V, Phillips A. BMP-7 modulates hyaluronan-mediated proximal tubular cell-monocyte interaction. J Am Soc Nephrol. 2004;15:1199–1211. doi: 10.1097/01.asn.0000125619.27422.8e. [DOI] [PubMed] [Google Scholar]
- 9.Zhang XL, Selbi W, de la Motte C, Hascall V, Phillips AO. Bone morphogenic protein-7 inhibits monocyte-stimulated TGF-β1 generation in renal proximal tubular epithelial cells. J Am Soc Nephrol. 2005;16:79–89. doi: 10.1681/ASN.2004050395. [DOI] [PubMed] [Google Scholar]
- 10.Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF-β and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell. 2005;16(4):1987–2002. doi: 10.1091/mbc.E04-08-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ross S, Hill CS. How the Smads regulate transcription. Int J Biochem Cell Biol. 2008;40:383–408. doi: 10.1016/j.biocel.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Fraser D, Brunskill N, Ito T, Phillips A. Long-term exposure of proximal tubular epithelial cells to glucose induces transforming growth factor-β1 synthesis via an autocrine PDGF loop. Am J Pathol. 2003;163:2565–2574. doi: 10.1016/s0002-9440(10)63611-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang M, Fraser D, Phillips A. ERK, p38, and Smad signaling pathways differentially regulate transforming growth factor-β1 autoinduction in proximal tubular epithelial cells. Am J Pathol. 2006;169:1282–1293. doi: 10.2353/ajpath.2006.050921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piek E, Moustakas A, Kurisaki A, Heldin CH, ten Dijke P. TGF-β type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci. 1999;112:4557–4568. doi: 10.1242/jcs.112.24.4557. [DOI] [PubMed] [Google Scholar]
- 15.Felici A, Wurthner JU, Parks WT, Giam LR-y, Reiss M, Karpova TS, McNally JG, Roberts AB. TLP, a novel modulator of TGF-β signaling, has opposite effects on Smad2- and Smad3-dependent signaling. EMBO J. 2003;22:4465–4477. doi: 10.1093/emboj/cdg428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McLoughlin RM, Witowski J, Robson RL, Wilkinson TS, Hurst SM, Williams AS, Williams JD, Rose-John S, Jones SA, Topley N. Interplay between IFN-γ and IL-6 signaling governs neutrophil trafficking and apoptosis during acute inflammation. J Clin Invest. 2003;112:598–607. doi: 10.1172/JCI17129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim BC, Song CY, Hong HK, Lee HS. Role of CAGA boxes in the plasminogen activator inhibitor-1 promoter in mediating oxidized low-density lipoprotein-induced transcriptional activation in mesangial cells. Transl Res. 2007;150:180–188. doi: 10.1016/j.trsl.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 18.Roberts AB, Tian F, Byfield SD, Stuelten C, Ooshima A, Saika S, Flanders KC. Smad3 is key to TGF-β-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev. 2006;17:19–27. doi: 10.1016/j.cytogfr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 19.Eddy AA, Fogo AB. Plasminogen activator inhibitor-1 in chronic kidney disease: evidence and mechanisms of action. J Am Soc Nephrol. 2006;17:2999–3012. doi: 10.1681/ASN.2006050503. [DOI] [PubMed] [Google Scholar]
- 20.Stroschein SL, Wang W, Luo K. Cooperative binding of Smad proteins to two adjacent DNA elements in the plasminogen activator inhibitor-1 promoter mediates transforming growth factor β-induced smad-dependent transcriptional activation. J Biol Chem. 1999;274:9431–9441. doi: 10.1074/jbc.274.14.9431. [DOI] [PubMed] [Google Scholar]
- 21.Daly AC, Randall RA, Hill CS. Transforming growth factor β-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol Cell Biol. 2008;28:6889–6902. doi: 10.1128/MCB.01192-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 23.Poncelet A-C, Schnaper HW, Tan R, Liu Y, Runyan CE. Cell phenotype-specific down-regulation of Smad3 involves decreased gene activation as well as protein degradation. J Biol Chem. 2007;282:15534–15540. doi: 10.1074/jbc.M701991200. [DOI] [PubMed] [Google Scholar]
- 24.Li W, Chen F, Nagarajan RP, Liu X, Chen Y. Characterization of the DNA-binding property of Smad5. Biochem Biophys Res Commun. 2001;286:1163–1169. doi: 10.1006/bbrc.2001.5529. [DOI] [PubMed] [Google Scholar]
- 25.Luo K. Ski and SnoN: negative regulators of TGF-β signaling. Curr Opin Genet Dev. 2004;14:65–70. doi: 10.1016/j.gde.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 26.Levy L, Howell M, Das D, Harkin S, Episkopou V, Hill CS. Arkadia activates Smad3/Smad4-dependent transcription by triggering signal-induced SnoN degradation. Mol Cell Biol. 2007;27:6068–6083. doi: 10.1128/MCB.00664-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stroschein SL, Bonni S, Wrana JL, Luo K. Smad3 recruits the anaphase-promoting complex for ubiquitination and degradation of SnoN. Genes Dev. 2001;15:2822–2836. doi: 10.1101/gad.912901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Godin RE, Robertson EJ, Dudley AT. Role of BMP family members during kidney development. Int J Dev Biol. 1999;43:405–411. [PubMed] [Google Scholar]
- 29.Kazama I, Mahoney Z, Miner JH, Graf D, Economides AN, Kreidberg JA. Podocyte-derived BMP7 is critical for nephron development. J Am Soc Nephrol. 2008;19:2181–2191. doi: 10.1681/ASN.2007111212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang SN, Lapage J, Hirschberg R. Loss of tubular bone morphogenetic protein-7 in diabetic nephropathy. J Am Soc Nephrol. 2001;12:2392–2399. doi: 10.1681/ASN.V12112392. [DOI] [PubMed] [Google Scholar]
- 31.Gould SE, Day M, Jones SS, Dorai H. BMP-7 regulates chemokine, cytokine, and hemodynamic gene expression in proximal tubule cells. Kidney Int. 2002;61:51–60. doi: 10.1046/j.1523-1755.2002.00103.x. [DOI] [PubMed] [Google Scholar]
- 32.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 33.Border WA, Noble NA. Transforming growth factor β in tissue fibrosis. N Engl J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 34.Coimbra T, Wiggins R, Noh JW, Merritt S, Phan SH. Transforming growth factor-β production in anti-glomerular basement membrane disease in the rabbit. Am J Pathol. 1991;138:223–234. [PMC free article] [PubMed] [Google Scholar]
- 35.Nakamura T, Miller D, Ruoslahti E, Border WA. Production of extracellular matrix by glomerular epithelial cells is regulated by transforming growth factor-β1. Kidney Int. 1992;41:1213–1221. doi: 10.1038/ki.1992.183. [DOI] [PubMed] [Google Scholar]
- 36.Border WA, Okuda S, Languino LR, Sporn MB, Ruoslahti E. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor β1. Nature. 1990;346:371–374. doi: 10.1038/346371a0. [DOI] [PubMed] [Google Scholar]
- 37.Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-β by anti-TGF-β antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes. 1996;45:522–530. doi: 10.2337/diab.45.4.522. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int. 2006;69:213–217. doi: 10.1038/sj.ki.5000054. [DOI] [PubMed] [Google Scholar]
- 39.Cohen SB, Nicol R, Stavnezer E. A domain necessary for the transforming activity of SnoN is required for specific DNA binding, transcriptional repression and interaction with TAFII110. Oncogene. 1998;17:2505–2513. doi: 10.1038/sj.onc.1202177. [DOI] [PubMed] [Google Scholar]
- 40.Stroschein SL, Wang W, Zhou S, Zhou Q, Luo K. Negative feedback regulation of TGF-β signaling by the SnoN oncoprotein. Science. 1999;286:771–774. doi: 10.1126/science.286.5440.771. [DOI] [PubMed] [Google Scholar]
- 41.Yang J, Zhang X, Li Y, Liu Y. Downregulation of Smad transcriptional corepressors SnoN and Ski in the fibrotic kidney: an amplification mechanism for TGF-β1 signaling. J Am Soc Nephrol. 2003;14:3167–3177. doi: 10.1097/01.asn.0000099373.33259.b2. [DOI] [PubMed] [Google Scholar]
- 42.Tan R, Zhang J, Tan X, Zhang X, Yang J, Liu Y. Downregulation of SnoN expression in obstructive nephropathy is mediated by an enhanced ubiquitin-dependent degradation. J Am Soc Nephrol. 2006;17:2781–2791. doi: 10.1681/ASN.2005101055. [DOI] [PubMed] [Google Scholar]
- 43.Fukasawa H, Yamamoto T, Togawa A, Ohashi N, Fujigaki Y, Oda T, Uchida C, Kitagawa K, Hattori T, Suzuki S, Kitagawa M, Hishida A. Ubiquitin-dependent degradation of SnoN and Ski is increased in renal fibrosis induced by obstructive injury. Kidney Int. 2006;69:1733–1740. doi: 10.1038/sj.ki.5000261. [DOI] [PubMed] [Google Scholar]
- 44.Yang J, Dai C, Liu Y. A novel mechanism by which hepatocyte growth factor blocks tubular epithelial to mesenchymal transition. J Am Soc Nephrol. 2005;16:68–78. doi: 10.1681/ASN.2003090795. [DOI] [PubMed] [Google Scholar]
- 45.Tan R, Zhang X, Yang J, Li Y, Liu Y. Molecular basis for the cell type specific induction of SnoN expression by hepatocyte growth factor. J Am Soc Nephrol. 2007;18:2340–2349. doi: 10.1681/ASN.2007010128. [DOI] [PubMed] [Google Scholar]