

Abstract
Pulmonary immunity depends on the ability of leukocytes to neutralize potentially harmful and frequent insults to the lung, and appropriate regulation of leukocyte migration and adhesion is integral to this process. Arhgef1 is a hematopoietic-restricted signaling molecule that regulates leukocyte migration and integrin-mediated adhesion. To explore a possible regulatory role for Arhgef1 in pulmonary immunity we examined the lung and its leukocytes in wild-type and Arhgef1-deficient animals. Here we report that the lungs of Arhgef1−/− mice harbored significantly more leukocytes, increased expression and activity of matrix metalloproteinases (MMPs), airspace enlargement, and decreased lung elastance compared with wild-type lungs. Transfer of Arhgef1−/− lung leukocytes to wild-type mice led to airspace enlargement and impaired lung function, indicating that loss of Arhgef1 in leukocytes was sufficient to induce pulmonary pathology. Furthermore, we showed that Arhgef1-deficient peritoneal macrophages when either injected into the lungs of wild-type mice or cultured on fibronectin significantly increased expression and activity of MMPs relative to control macrophages, and the in vitro fibronectin induction was dependent on the α5β1 integrin pair. Together these data demonstrate that Arhgef1 regulates α5β1-mediated MMP expression by macrophages and that loss of Arhgef1 by leukocytes leads to pulmonary pathology.
Leukocytes are resident in the lungs of healthy individuals and are necessary for the innate and adaptive immune response toward potentially harmful foreign antigens that are exposed to the lung on a constant basis. Protection provided by innate lung immunity is controlled in part through the action of sentinel alveolar macrophages (AMs)1 and alveolar epithelial cells.2 Integrin-mediated signaling is a critical component of macrophage function and has been shown to be important for pulmonary immunity.3,4 Furthermore, expression of extracellular matrix proteins are increased in several types of immune-related lung disease including chronic obstructive pulmonary disease (COPD).5
Inappropriate regulation of pulmonary immune function has significant consequences and contributes to a number of lung diseases. In particular, it has been shown that the severity of COPD in humans correlates with the extent of inflammatory cell infiltrate comprising macrophages, neutrophils, CD4, CD8, and B lymphocytes.6 COPD has also been characterized by lung tissue damage, elevated production of matrix metalloproteinases (MMPs) by pulmonary leukocytes, and impaired lung function.7–9 Precisely how, if at all, the increased leukocyte presence leads to lung pathology is not yet established.
Leukocytes migrate in response to chemoattractants that signal via G-protein–coupled receptors to polarize the cell.10 Cell polarization is accomplished by the localized activation within the cell of Rho GTPase family members where Cdc42 and Rac are activated at the migrating cell leading edge and Rho at the trailing edge.10 Arhgef1 (Human Genome Organization nomenclature, formerly known as Lsc in mouse and p115RhoGEF in humans) is an intracellular protein restricted in expression to hematopoietic cells that regulates signaling from select G-protein–coupled receptors as well as RhoA activation.11,12 Characterization of Arhgef1−/− mice by our lab, and similar mouse mutants by others, have demonstrated a role for Arhgef1 in regulating leukocyte migration and adhesion.13–16
We have previously demonstrated that T lymphocytes depend on Arhgef1 to mount an adaptive secondary immune response to airway challenge.17 In the course of those studies, we found that naïve Arhgef1-deficient lungs reproducibly harbor an increased number of leukocytes compared with wild-type controls.17 To determine whether this increased presence of pulmonary leukocytes has functional consequences, we examined mutant pulmonary leukocytes and lung function in Arhgef1−/− mice in the absence of experimental challenge. In this report, we show that loss of Arhgef1 expression by pulmonary leukocytes leads to airspace enlargement and decreased lung elastance and that elevated integrin-mediated expression of MMPs by mutant macrophages likely contributes to lung pathology.
Materials and Methods
Animals
Arhgef1-deficient15 and wild-type C57BL/6 mice (obtained from the Jackson Laboratory, Bar Harbor, ME) were maintained in our animal colony. All experiments with animals were approved by the Institutional Animal Care and use Committee.
Histological Analysis
For histological examination lungs were inflated via a cannula to 25 cm of pressure with 4% paraformaldehyde, immersed for 24 hours, imbedded in paraffin, and stained with hematoxylin and eosin.
Bronchoalveolar Lavage
Lungs were lavaged with Hanks’ balanced salt solution with 5 mmol/L EDTA and counted on a Z2 particle count and size analyzer (Beckman Coulter, Fullerton, CA). Subsets of bronchoalveolar lavage (BAL) samples were cytocentrifuged (Cytospin 2; Shandon Ltd, Runcorn, Cheshire, UK) and stained with Leukostat (Fisher Diagnostics, Pittsburgh, PA).
Lung Digest
Leukocytes were isolated from perfused lungs after treatment with collagenase types II and IV (Sigma-Aldrich, St. Louis, MO) and dispase II (Roche, Basel, Switzerland), as previously described.17 An aliquot of resuspended cells was enumerated as described above for BAL leukocyte analysis.
Flow Cytometry
After isolation of leukocytes, cells were stained using standard methods and the following antibodies. Leukocytes were identified using a pan-CD45 antibody (30-F11; eBiosciences, San Diego, CA) and Gr-1 (17–5931-81 eBiosciences), B220 or CD3 for identification of neutrophils, B cells, and T cells respectively, and back-gated during analysis to confirm appropriate forward and 90° light scatter. CD4 and CD8 staining further differentiated T cell subsets. Monocytes and macrophages were determined by F4/80+ (MCA497PE, Serotec, Raleigh, NC) staining and either small or large forward scatter respectively. Data were collected with a FACSCalibur (BD Pharmingen, San Diego, CA) and analyzed with FlowJo 4.3 software (Tree Star, Inc., Ashland, OR).
Lung Mechanics
Lung mechanics were assessed as previously described18 using the Flexivent (Scireq, Montreal, Canada) small animal ventilator. For animals at 3 months of age and older, a stepwise inflation up to 1.2 ml of air was applied to the lungs. For 3-week-old animals, 0.6 ml of air was applied to the lungs. Pressure-volumes graphs were generated with the expiratory phase using the pressure and volume values obtained after a one-second pause in piston movement.
Lung Morphometry
After measurement of lung mechanics, lung tissue was processed as follows. Lungs were inflated to 25 cm of pressure using a tower filled with 4% paraformaldehyde connected to cannula. The trachea was then tied off below the cannula, and then lungs were removed and immersed in 4% formaldehyde for 24 hours. Lung were then imbedded in paraffin and cut into 2- to 3-μm-thick slices at a random orientation and stained with hematoxylin and eosin. Next we used a digital image analysis approach originally described by Tschanz and Burri19 and subsequently adapted into a macro for ImagePro 4.5 (Media, Cybernetics). The number of intercepts between the skeletonized image of the alveolar septum and the line probes were tallied automatically and reported to a spreadsheet. Mean linear intercept was calculated by the formula:
To ensure that measuring lung mechanics did not affect lung morphometry we measured mean linear intercept as described above in mice that had not been subjected to lung mechanics measurements as well as mice that had been subjected to lung mechanics measurements. We found no difference in the mean linear intercept between these two groups (data not shown).
Adoptive Transfer Experiments
Transfer of BAL Cells
BAL cells were harvested from either wild-type or Arhgef1-deficient mice as described above using HBSS + 5 mmol/L EDTA. Lavage was pooled according to genotype, and cells were pelleted and resuspended in 40 μl of HBSS per donor animal. 40 μl of resuspended cells were instilled into 8-week-old wild-type recipient as previously described for delivery of bleomycin.18 The number and composition of cells recovered for the transfer from wild-type and Arhgef1-deficient mice are given in the Results. This procedure was performed on four occasions one week apart. One week after the last instillation recipient mice were assessed for lung mechanics and histological analysis as previously described.
Transfer of Peritoneal Macrophages
Naïve resident peritoneal macrophages were harvested by peritoneal lavage with 5 to 10 ml of ice-cold HBSS with 5 mmol/L EDTA as previously described.20 Macrophages were counted using a Z2 particle count and size analyzer (Beckman Coulter, Fullerton, CA). Next, macrophages from either wild-type or Arhgef1-deficient mice were instilled in the lungs of 8-week-old wild-type recipient mice as described for BAL leukocyte transfer. An equal number of macrophages 5 × 106 from either wild-type or Arhgef1-deficient mice was transferred into recipient mice. Forty-eight hours after transfer BAL cells and BAL supernatants were collected and analyzed for MMP expression and activity.
Real-Time RT-PCR
Total RNA was purified using TRIzol (Invitrogen, Carlsbad CA) according to the manufacturer's instructions, and differences in gene expression were determined using Real-Time RT-PCR as previously described.21 Differences in expression between samples was determined using the comparative threshold cycle (ΔΔCT) method as suggested by the manufacturer (Applied Biosystems, Foster City, CA), normalizing each sample to 18s rRNA levels (cat No. 4310893E). MMP2 (cat# Mm 00439508_m1), MMP9 (cat# Mm00442991_m1), and MMP12 (cat# Mm00500554_m1) gene expression was determined using primers and probes sets purchased from Assays on Demand (Applied Biosystems).
Gelatin Zymography
Zymography was performed using Novex (Invitrogen) Zymogram 10% gelatin gels per manufacturer's instructions on BAL supernatant or conditioned media. All gels were incubated at 37°C for 48 hours in developing buffer with fresh developing buffer added after 24 hours. Protease activity was quantified by densitometric analysis using Image Quant 5.1 (GE Healthcare, United Kingdom).
MMP Adsorption Assay
Tissue-culture 96-well plates (Costar, high binding cat#3590, Corning Incorporated, Corning, NY) were coated with10 μg/ml of either anti-MMP2 (R&D Systems cat#AF1488), anti-MMP9 (R&D Systems, Minneapolis, MN, cat#AF909), or BSA (1%). 100 μl of BAL supernatant was incubated for two hours at room temperature. Sample was transferred to a fresh well every ten minutes to maximize adsorption. After incubation sample was mixed with zymogram loading buffer and gelatin zymogram was performed as previously described.
Peritoneal Elicited Macrophages and Bone Marrow Neutrophils
Peritoneal macrophages were obtained by peritoneal lavage with 5 to 10 ml of ice-cold HBSS with 5 mmol/L EDTA from mice 5 days after intraperitoneal injection of 1 ml of sterile thioglycollate. Neutrophils were isolated from bone marrow as previously described.22
Static Adhesion Assay
Adhesion was assayed in the following manner. Briefly, tissue-culture 96-well plates (Costar, high binding cat#3590) were coated with 3 μg/ml of Fc-ICAM-1 (R&D Systems, cat# 796IC), 3 μg/ml of Fc-VCAM-1 (R&D Systems, cat#643VM), 10 μg/ml of fibronectin (Molecular Innovations, Nori, MN, cat#MFBN), 20 μg/ml of laminin (Invitrogen, cat # phe0033), heat denatured BSA (1%) in DPBS without Ca++ or Mg++ (Mediatech, Inc., Manassas, VA) for 2 hours at room temperature followed by blocking with heat denatured fatty-acid-free BSA in DPBS. Macrophages once harvested were labeled with CellTracker Green CMFDA (Invitrogen) per manufacturers instructions. Cells were resuspended in fresh DMEM without fetal calf serum at a density of 1 × 106 cells/ml. 100 μl of the resuspended cells were added to each well of the precoated 96-well plates. Plates were then centrifuged at 40g for 1 minute and examined by light microscopy to confirm 40% to 60% confluence. Plates were incubated for either 1 hour or 48 hours at 37°C with 10% CO2. Next, media was removed and DPBS was added followed by recording of fluorescence 485 nm/535 nm for 1 second per well using a Wallac VICTOR2 plate reader. The fluorescence at this point was used as the number of cells plated. The media and nonadherent cells were subsequently removed by inverting the plate and gently flicking off the media. Next, the plate was submerged in warm DPBS and the solution removed by inverting the plate and gentle flicking. 100 μl of fresh warm DPBS was added and the fluorescence measured again, and this value was plotted against the cells plated value to determine the percentage of cells adherent. Conditions were performed in triplicate wells, and the average of the triplicate was used for each condition and genotype.
Cultures of Peritoneal-Elicited Macrophages on Integrin Ligands
Tissue-culture 96-well plates (Costar high-binding) were coated and peritoneal-elicited macrophages (PEMs) harvested as described for static adhesion assays. Cells were resuspended in DMEM with 5% heat inactivated fetal calf serum at a concentration of either 1.25 × 105 cells/ml for 48 hours cultures or 2.0 × 106 cells/ml for 24 hours cultures, and 100 μl of cells were added to each well. Plates were then centrifuged at 40g for 1 minute and then incubated for either 24 or 48 hours at 37°C with 10% CO2. Next, cells were treated with TRIzol for RNA purification as previously described.21 For antibody treatment experiments cells were preincubated for 15 minutes with 50 μg/ml of anti-CD49E (eBiosciences, clone eBioHMa5-1) or 2 μg/ml of anti-CD29 (eBiosciences, clone eBioHMb1-1) before plating on fibronectin for 24 hours. For zymographic analysis of conditioned media the media was replaced after the first 24 hours of culture with fresh DMEM without fetal calf serum for an additional 24 hours before collection.
Statistical Analysis
All statistical analysis data are presented as mean ± SE Statistical significance for data points was determined using Student two-tailed t test. All statistical analysis was performed with JMP IN software package (SAS, Cary, NC).
Results
Loss of Arhgef1 Leads to an Increased Presence of Pulmonary Leukocytes
To assess whether Arhgef1 participates in lung immunity, histological comparisons of lungs from Arhgef1−/− and wild-type mice from three weeks to one year of age were performed. These analyses revealed reproducible discrete leukocyte aggregates at all ages in Arhgef1-deficient lung that was not typically observed in the respective controls (Figure 1A and data not shown). Higher magnification of these accumulations indicated that these aggregates contain lymphocytes, macrophages, and neutrophils (Figure 1A) and was supported by immunofluorescent histological staining with antibodies recognizing CD3, CD11b, and Gr-1–expressing cells, respectively (data not shown).
Figure 1.
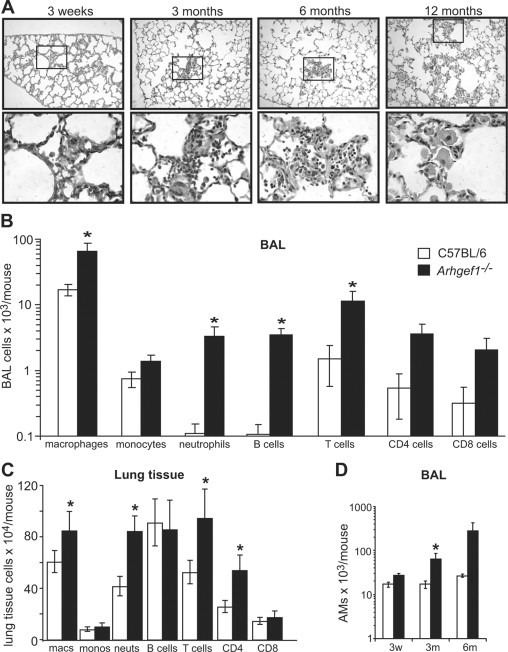
Arhgef1-deficient animals harbor increased numbers of lung leukocytes. A: Hematoxylin and eosin (H&E) stained lung from Arhgef1-deficient mice at the indicated ages. Top row is taken at ×10 magnification. The boxed area in the top row is shown at ×60 magnification below the respective sample. B: Leukocyte populations recovered from bronchoalveolar lavage (BAL) of 3-month-old mice. Wild-type (open bars) or Arhgef1−/− (solid bars) mice were lavaged with 3 × 1 ml aliquots of HBSS with 5 mmol/L EDTA. Cells were enumerated with a BD Coulter counter followed by flow cytometric analysis as described in methods. Data represent mean ± SE; n = 16 for C57BL/6; n = 15 for Arhgef1−/−. C: Leukocytes recovered from lavaged, perfused, and enzymatically digested wild-type (open bars) and mutant (solid bars) lung tissue. Cells were enumerated and defined as in B. Data represent mean ± SE; n = 8 for both Arhgef1−/− and C57BL/6 mice. D: Number of macrophages recovered from BAL at 3 weeks, 3 months, and 6 months in wild-type (open bars, n = 4, 16 and 3, respectively) and mutant (solid bars, n = 4, 15 and 5, respectively) animals. AM indicates alveolar macrophages. *P < 0.05 Student two-tailed t test compared with C57BL/6 samples.
To confirm this apparent increased leukocyte presence and to begin to identify the pulmonary compartment in which these cells were located, we performed BAL followed by flow cytometric analysis to identify and quantitate leukocyte populations. Consistent with the histological examination, Arhgef1−/− animals harbored more macrophages, monocytes, neutrophils, and lymphocytes (B cells and CD4 and CD8 cells) that were for all cell types except monocytes significantly increased in number compared with wild-type controls (Figure 1B). These data demonstrate that loss of Arhgef1 leads to a significant increase in the number of airspace leukocytes compared with wild-type.
Additionally, we extended our pulmonary leukocyte analysis by enzymatic digestion of 3-month-old perfused and lavaged lung tissue again followed by flow cytometric analysis. These experiments revealed that the increase in leukocyte cell types in the Arhgef1-deficient lung tissue was more restricted and modest compared with the BAL. Specifically, the numbers of tissue macrophages, neutrophils, and CD4+ T cells were significantly increased relative to wild-type animals but the B cell and CD8 T cell numbers were equivocal (Figure 1C). Our histological analysis (Figure 1A) indicated accumulation of leukocytes could be detected as early as 3 weeks of age. Thus, to assess whether Arhgef1−/− leukocytes accumulate with age we enumerated BAL macrophages in both Arhgef1-deficient and control mice at three weeks, three months, and six months of age (Figure 1D). These data showed that although the number of alveolar macrophages in the wild-type remained relatively constant at these different ages, the number of these cells in Arhgef1-deficient mice increased with age and were significantly different from controls at three months of age. Considered together, we conclude from these data that loss of Arhgef1 leads to chronic elevation in the number of resident pulmonary leukocytes.
Arhgef1−/− Animals Display Pulmonary Pathology and Impaired Respiratory Function
Our histological examination of Arhgef1−/− lungs suggested a loss of alveolar septum or alveolar simplification (Figure 1A), which is characteristic of emphysema and a hallmark of COPD.8,9 Furthermore, loss of alveolar structure is reflected in both airspace enlargement and loss of elastic recoil of the lungs and we evaluated both properties in mutant and wild-type animals throughout development.
To evaluate lung elastic properties, we performed quasi-static pressure-volume loops on mechanically ventilated mice. These data revealed that the lungs of wild-type animals display loss of elastic recoil (Figure 2A) with age as previously demonstrated in C57BL/6 mice23 and similar to humans.24 Importantly, at all ages tested, from 1.5 weeks to 12 months, Arhgef1−/− mice exhibited a greater impairment in lung function compared with wild-type animals.
Figure 2.
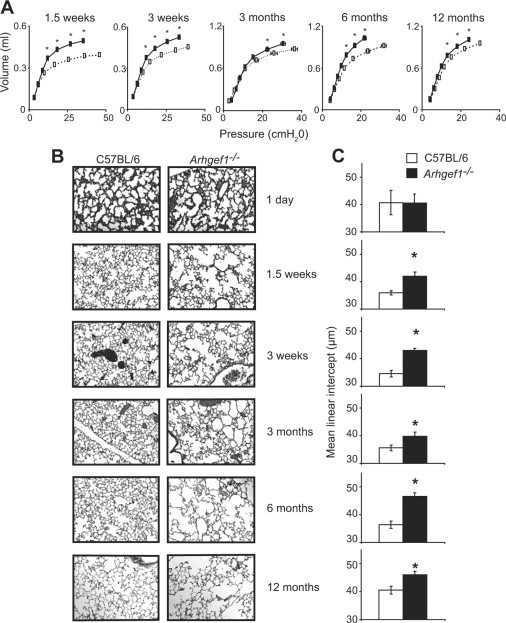
Impaired lung mechanics and airspace enlargement in the absence of Arhgef1. A: Quasi-static pressure-volume loops performed on wild-type (open boxes, dotted lines) and Arhgef1−/− mice (solid boxes, solid lines) from 1.5 weeks to 12 months of age. Data represent mean ± SE C57BL/6 mice n = 4, 4, 11, 4, and 6; Arhgef1−/−n = 4, 4, 12, 4, and 8 for 1.5 weeks, 3 weeks, 3 months, 6 months, and 1 year of age, respectively.B: Representative H&E stained sections of lung from C57BL/6 (left column) and Arhgef1−/− (right column) mice from 1 day old to 1 year of age. C: Mean linear intercept (μm) of alveolar septae as measured on H & E–stained wild-type (open bars) and mutant (solid bars) inflated lungs. Data represent mean ± SE For C57BL/6 mice n = 4, 4, 7, 7, 7, and 9 for Arhgef1−/− mice n = 3, 4, 7, 9, 7, and 18 for 1 day, 1.5 weeks, 3 weeks, 3 months, 6 months, and 1 year of age, respectively. *P < 0.05 for Student two-tailed t test compared with C57BL/6 mice.
Loss of elastic recoil often results from destruction of alveolar tissue, so we next addressed whether the impaired lung function reflected differences in lung tissue damage as measured by changes to the lung airspace. To determine this we used a morphometric analysis of lung sections stained with hematoxylin and eosin (Figure 2B). Sections were micrographed at low-power magnification and analyzed with a digital image analysis approach19 to provide a quantitative measurement of airspace as the mean linear intercept. The mean linear intercept represents the average distance between intercepts of alveolar septa and a line of a given length. These analyses revealed that the lungs from Arhgef1−/− animals from 1.5 weeks to 12 months of age exhibited significantly increased airspace enlargement compared with wild-type mice, whereas no differences were detected in newborn animals (Figure 2C). We interpret these data to indicate that Arhgef1 function is not required for normal lung structure formation in utero, but after birth loss of Arhgef1 function, presumably by hematopoietic cells, leads to impaired lung function and damaged alveolar tissue.
Increased MMP Expression and Activity by Arghef1-Deficient Leukocytes
In humans with COPD, and mice exposed to cigarette smoke, alveolar damage is considered to result, at least in part, from enhanced production of MMPs. The expression and activity of a number of MMPs, including MMP2, MMP9, and MMP12, are elevated in patients with emphysema and also in several different mutant mice that develop airspace enlargement.7,25 Thus, we questioned whether aberrant expression of MMPs might account for the airspace enlargement observed in Arhgef1−/− lungs. In initial experiments we relied on real-time RT-PCR (qPCR) to measure the expression of MMP2, MMP9, and MMP12 in various control and mutant tissues and cells. These results demonstrated that MMP2, MMP9, and MMP12 expression in 1-day-old lung, 3-month-old liver, bone marrow neutrophils, and PEMs was comparable between wild-type and mutant animals (Figure 3A). In contrast, Arhgef1−/− BAL leukocytes from 3-month-old mice displayed significantly increased (≈100-fold) expression of MMP2, MMP9, and MMP12 compared with BAL cells from age-matched wild-type animals (Figure 3A). It is important to note that this MMP expression as measured by qPCR is normalized to 18S RNA for each sample and therefore is assumed to reflect relative expression per cell. Moreover, the observed increase of MMP expression in mutant BAL leukocytes is much higher than one would expect if the difference was merely attributable to the increased number of leukocytes present in the Arhgef1−/− samples (Figure 1B).
Figure 3.
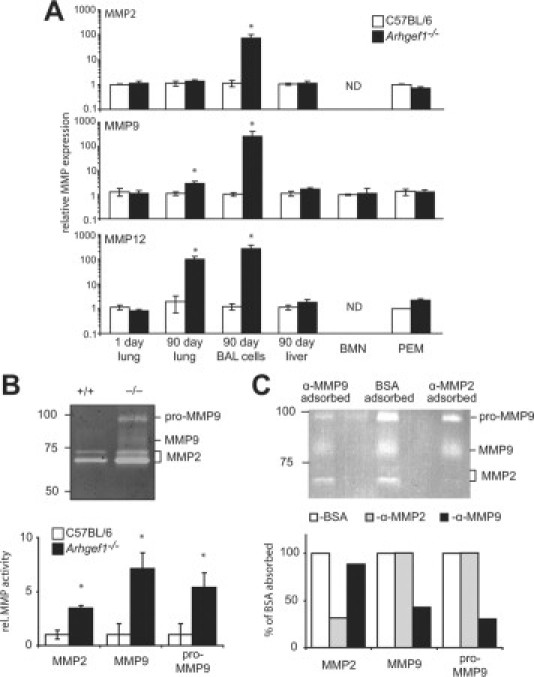
Loss of Arhgef1 leads to increased metalloproteinase expression and activity in the lung. A: MMP2 (top), MMP9 (middle), and MMP12 (bottom) expression measured by qPCR in whole lung at day 1, adult lavaged lung tissue, adult BAL leukocytes, adult liver, bone marrow neutrophils (BMN), or peritoneal elicited macrophages (PEM). Arhgef1−/− (solid bars, n = 8) MMP expression is shown as fold relative to control samples (open bars n = 8) with all samples normalized to 18s RNA. B: Gelatin zymography of BAL supernatant from wild-type (+/+) and homozygous (−/−) mutants. Molecular weight standards and respective enzymatic activity of MMPs are shown. Shown below zymogram is the quantitation of MMP activity as determined by densitometric analysis. MMP activity is shown as fold relative to control samples wild-type (open bars) n = 4 and Arhgef1−/− (solid bars) n = 6 from two independent experiments. C: Gelatin zymography of BAL supernatant from Arhgef1−/− mice adsorbed with anti-MMP2 or anti-MMP9. Molecular weight standards and respective enzymatic activity of MMPs are noted. Shown below zymogram is the quantitation of MMP as determined by densitometric analysis. MMP activity is shown as fold relative to BSA adsorbed. Open bars represent BSA adsorbed sample, gray bars represent anti-MMP2 adsorbed sample, and solid bars represent anti-MMP9 adsorbed samples. Data represent mean ± SE. *P < 0.05 Student two-tailed t test compared with C57BL/6 samples. ND indicates not detected.
Because MMP mRNA expression may not accurately reflect protease activity, we also performed gelatin zymography on BAL supernatant from control and mutant animals. In these analyses we consistently observed increased gelatinase activities in Arhgef1-deficient samples migrating at a molecular weights consistent with MMP2 and MMP926 and as shown in Figure 3B. To confirm the identity of the protease activity elevated in Arhgef1−/− BAL, we adsorbed BAL supernatant from Arhgef1-deficient mice with antibodies specific for MMP2 or MMP9 (Figure 3C). Incubation of BAL supernatant with plate-bound anti-MMP9 substantially decreased (>50%) the gelatinase activity at approximately 95 kDa and 85 kDa, whereas the gelatinase activity below 75 kDa was only decreased ≈10% (Figure 3C). Conversely, incubation with antibodies directed against MMP2 decreased the gelatinase activity below 75 kDa to ≈35% of the BSA adsorbed sample but had no measurable effect on the gelatinase activities at 95 kDa and 85 kDa (Figure 3C). Based on the ability of these anti-MMP antibodies to deplete specific gelatinase activities, we conclude that the 95-kDa and 85-kDa proteases are likely pro-MMP9 and MMP9 respectively, whereas the activities below 75 kDa are derived from MMP2. Based on these identities, densitometric analysis of Figure 3B revealed Arhgef1−/− BAL supernatant harbors significantly more MMP2 and MMP9 activities compared with wild-type (Figure 3B). Thus, loss of Arhgef1-function leads to exaggerated MMP expression in the pulmonary compartment but not in other adult tissues.
Pulmonary Pathophysiology Is Transferred with Arhgef1−/− BAL Leukocytes
Arhgef1 expression is hematopoietic-specific (http://biogps.gnf.org/#goto=genereport&id=9138)27 suggesting that the pathological and functional differences between wild-type and Arhgef1−/− mice results from the increased presence of leukocytes that lack Arhgef1 function. However, Arhgef1−/− lung pathology is observed as early as 1.5 weeks after birth, possibly indicating a direct role for Arhgef1 role in lung development. To directly test whether an Arhgef1 deficiency in leukocytes could result in lung pathology, BAL cells were collected from either naïve wild-type or Arhgef1-deficient mice and transferred into adult wild-type recipient mice by intratracheal instillation. Instillations were performed once a week for four weeks followed by an evaluation of pulmonary function and measurement of airspace one week after the last challenge. The results from these experiments revealed that instillation of Arhgef1-deficient BAL cells promotes a significant decrease in pulmonary elastic recoil and a significant increase in mean linear intercept of wild-type recipient lungs compared with instillation of wild-type BAL cells (Figure 4, A and B). These data suggest that Arhgef1-deficient BAL cells are sufficient to cause pathology in an adult wild-type lung and suggests that the lung pathology observed in Arhgef1−/− animals results from loss of Arhgef1 function in pulmonary leukocytes. However, Arhgef1 mutant BAL harbors an elevated number of leukocytes and more diverse cell types relative to wild-type (Figure 1B) obscuring whether lung pathology resulted from a particular leukocyte population(s) or increased numbers of transferred cells.
Figure 4.
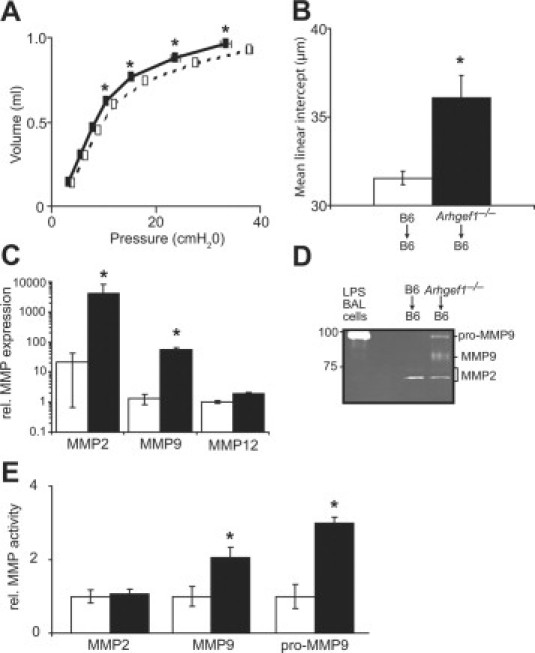
Transfer of Arhgef1-deficient BAL cells causes pathology in adult lungs. Eight-week-old wild-type mice received intratracheal instillations of BAL cells recovered from either C57BL/6 mice or Arhgef1-deficient mice once a week for 4 weeks. A: Quasi-static pressure-volume loops performed on wild-type mice that received either wild-type BAL cells (open boxes, dotted lines, n = 5) or Arhgef1−/− BAL cells (solid boxes, solid lines, n = 9). Data represent mean ± SE. B: Mean linear intercept (μm) of alveolar septae as measured on H & E–stained inflated lungs from wild-type mice that received either wild-type BAL cells (open bar, n = 5) or Arhgef1−/− BAL cells (solid bar, n = 9). C: MMP2, MMP9, and MMP12 expression measured by qPCR in BAL cells recovered 48 hours after the transfer of peritoneal macrophages from wild-type (n = 4) and Arhgef1−/− (n = 7) into wild-type recipient mice. D: Representative gelatin zymography of BAL supernatant recovered from mice described in C. Whole cell lysate of BAL cells from LPS-treated wild-type mouse is included as positive control for pro-MMP9. E: MMP activity in BAL supernatant from mice described in C as determined by densitometric analysis. MMP activity is shown as fold relative to control samples. Data represent mean ± SE. *P < 0.05 for Student two-tailed t test comparing mice that received wild-type cells to mice that received Arhgef1−/− cells.
Under inflammatory conditions macrophages have been shown to express all three of the MMPs25,28,29 that are elevated in Arhgef1−/− BAL leukocytes, and macrophages also represent more than 60% of the BAL cells recovered from Arhgef1-deficient mice. Thus, we considered that Arhgef1−/− alveolar macrophages were the cell type responsible for lung tissue damage. To test this hypothesis we transferred equal numbers (5 × 106) of naïve resident peritoneal macrophages from wild-type and Arhgef1-deficient mice into the lungs of wild-type recipient mice via intratracheal instillation. Importantly, unlike BAL leukocytes, resident and thioglycollate-elicited peritoneal macrophages from Arhgef1-deficient mice exhibit similar expression of MMP2, MMP9, and MMP12 compared with control cells (Figure 3A and data not shown). Approximately 48 hours after transfer, BAL fluid and cells were collected from recipient mice and analyzed for MMP expression by qPCR and activity by gelatin zymography (Figure 4, C–E). The results from these experiments demonstrated that the transfer of Arhgef1−/− resident peritoneal macrophages resulted in an approximate 100-fold increase in the expression of MMP2 and MMP9 by BAL cells compared with wild-type peritoneal macrophages (Figure 4C). However, although both MMP2 and MMP9 expression was elevated in the BAL cells after the transfer of Arhgef1-deficient macrophages, only MMP9 activity was elevated in the BAL supernatant (Figure 4, D and E). Thus, the transfer of Arhgef1-deficient macrophages into the lung environment results in exaggerated MMP expression and activity.
Arhgef1-Deficient Macrophages Display Exaggerated MMP Expression when Cultured on the Fibronectin α5β1 Integrin Ligand
We have previously shown that Arhgef1-deficient B lymphocytes inefficiently resolve integrin-mediated adhesive events,15 and integrin engagement has been reported to induce the expression of MMPs in fibroblasts, lymphocytes, and macrophages.30–32 Thus, we evaluated the ability of Arhgef1−/− macrophages to adhere to integrin ligands and the influence of these interactions on MMP expression. For these experiments we used peritoneal macrophages isolated five days after thioglycollate treatment of wild-type and mutant animals and measured adhesion to cell-bound (ICAM-1 and VCAM-1) and extracellular matrix (laminin and fibronectin) integrin ligands. The results from these experiments revealed that both Arhgef1−/− and wild-type macrophages exhibited similar adhesion to all integrin ligands tested after 48 hours although to different levels (Figure 5A). Specifically, both wild-type and mutant macrophage populations displayed considerable adhesion to VCAM-1 (≈70% of cells) and fibronectin (≈85% of cells) whereas adhesion to ICAM-1 was similar to BSA-coated control wells and adhesion to laminin was negligible (Figure 5A). These results show that loss of Arhgef1 in peritoneal-elicited macrophages does not appear to significantly alter their adhesion to cell-bound or extracellular matrix integrin ligands.
Figure 5.
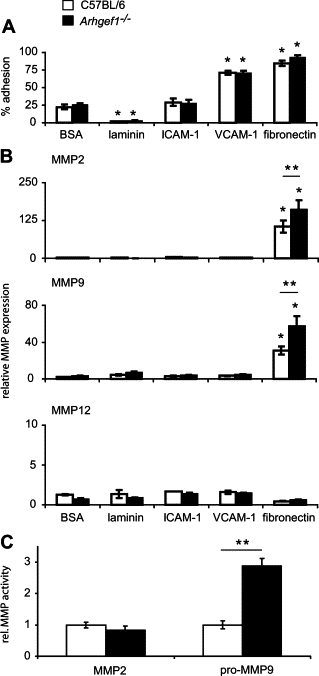
Arhgef1-deficient peritoneal elicited macrophages exhibit increased integrin-mediated MMP expression. A: Adhesion of wild-type (open bars) and Arhgef1−/− (solid bars) PEMs to BSA, laminin, ICAM-1, VCAM-1, and fibronectin after 48 hours of incubation. Wild-type (n = 6) and Arhgef1−/− (n = 6) from 3 independent experiments. B: MMP2 (top), MMP9 (middle), and MMP12 (bottom) expression measured by qPCR of wild-type (open bars) and Arhgef1−/− (solid bars) PEMs cultured for 48 hours on the indicated substrates. MMP expression is shown as fold relative to wild-type samples cultured on BSA with all samples normalized to 18s RNA. Wild-type (n = 8) and Arhgef1−/− (n = 12) from 4 independent experiments. C: MMP activity as quantified by densitometric analysis of conditioned media from macrophages cultured on fibronectin, wild-type (open bars; n = 6), and Arhgef1−/− (solid bars; n = 6). Data represent mean ± SE. *P < 0.05 Student two-tailed t test compared with cells on BSA. **P < 0.05 Student two-tailed t test comparing wild-type to Arhgef1−/− cells.
We next measured the expression of MMP2, MMP9, and MMP12 induced by the culture of macrophages on these various integrin ligands and whether loss of Arhgef1 would influence this expression. These experiments (Figure 5B) demonstrated that wild-type macrophages cultured only on fibronectin induced the expression of MMP2 and MMP9 and as previously shown.32,33 Expression of MMP2 and MMP9 was again significantly increased by Arhgef1−/− macrophages under the same conditions, reflecting our previous in vivo findings. Specifically, on fibronectin wild-type macrophages increased MMP2 and MMP9 expression approximately 100- and 25-fold, respectively, whereas this induction was elevated in mutant macrophages to 150-fold and 60-fold, respectively. In contrast, induction of MMP12 expression by wild-type or mutant macrophages was not observed on any integrin ligand tested. Thus, although both macrophage cell types revealed comparable and considerable adhesion to VCAM-1 and fibronectin, only incubation on fibronectin led to an induction of MMP expression and the absence of Arhgef1 further enhanced this expression.
We also measured MMP2 and MMP9 activities in the conditioned media of wild-type and mutant macrophages and observed fibronectin was the only integrin ligand that induced an increase in gelatinase activity. However, although MMP2 expression was present in both cell types when cultured on fibronectin, neither cell type displayed a significant increase in MMP2 activity relative to other integrin ligands. In contrast, Arhgef1-deficient macrophages elaborated significant increased pro–MMP-9 activity on fibronectin compared with wild-type (Figure 5C).
Fibronectin can serve as a ligand for a diverse number of integrins, and many of these integrins can be expressed by monocytes/macrophages.32–34 Of interest, the Arg-Gly-Asp (RGD) tripeptide sequence is recognized by a subset of these fibronectin-binding integrins with the α5β1 and αv-containing integrins most prominent in this region.31,32,35,36 Thus, to identify the integrin(s) responsible for signaling MMP expression by wild-type and mutant macrophages we pretreated macrophages with monoclonal antibodies previously shown both in vitro and in vivo to block integrin mediated signaling.37,38 Preincubation of macrophages with antibodies directed against either integrin α5 (anti-CD49E) or β1 (anti-CD29) significantly inhibited the induction of both MMP2 and MMP9 expression (Figure 6, A and B) by both mutant and control macrophages and consistent with previous reports of this integrin in inducing these MMPs.31–32,39 From these data we conclude that both wild-type and Arhgef1−/− macrophages signal MMP expression via α5β1 integrin interaction with its ligand on fibronectin. Furthermore, loss of Arhgef1 leads to the exaggerated expression and activity of MMPs as a consequence of this integrin–ligand interaction.
Figure 6.
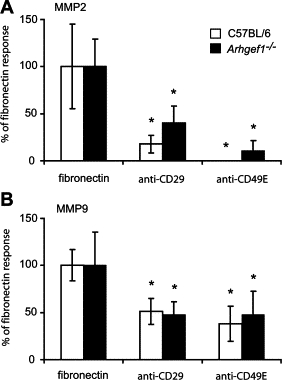
Induction of MMPs on fibronectin is dependent on α5β1 integrin signaling. MMP2 expression (A) and MMP9 expression (B) as measured by qPCR of wild-type (open bars, n = 6) and Arhgef1−/− (solid bars, n = 6) PEMs cultured for 24 hours on fibronectin with either anti-CD29 (2 μg/ml) or anti-CD49E (50 μg/ml) antibodies. MMP expression is shown as percentage of untreated fibronectin plated PEMs. Data represent mean ± SE from two independent experiments. *P < 0.05 Student two-tailed t test comparing untreated samples to respective antibody treatments.
Discussion
Our results demonstrate that naïve Arhgef1−/− mice chronically harbor increased numbers of pulmonary leukocytes that promote lung tissue destruction and lead to impaired lung function. We further identify that Arhgef1-deficient leukocytes express increased levels of MMPs in vivo and is recapitulated in vitro when mutant macrophages are cultured on fibronectin and as a consequence of α5β1 integrin signaling. Fibronectin is a major extracellular matrix protein and is expressed by diverse cell types in the lung and particularly during inflammation. Therefore, these findings suggest that Arhgef1-deficient macrophage adhesion to this integrin ligand would elaborate exaggerated amounts of MMPs that account, at least in part, for the observed lung pathophysiology displayed by Arhgef1−/− mice. Thus, a major finding presented by these data are that Arhgef1 function in pulmonary macrophages is critical for homeostatic lung function.
At day 1 after birth, both lung airspace and pulmonary MMP expression is similar between mutant and wild-type neonates. However, by day 21 and onward, Arhgef1−/− lungs display impaired respiratory function as measured by increased compliance (decreased elastance) and airspace enlargement. This pulmonary pathophysiology is accompanied by an increased number of Arhgef1-deficient BAL macrophages at 3 weeks of age, the earliest time point quantified. Because Arhgef1 expression is restricted to hematopoietic cells we considered that mutant leukocytes were responsible for the observed pulmonary pathology and impaired respiratory function. Howevever, alveolarization of murine lungs is extensive after birth,40 and airspace enlargement might also be expected in Arhgef1−/− mice if Arhgef1 participated in alveolarization during lung development. Therefore, to clearly distinguish between these possibilities, wild-type and mutant pulmonary leukocytes were transferred intratracheally to adult wild-type mice. The transfer of Arhgef1−/− leukocytes conferred pulmonary pathophysiology, leading us to conclude that Arhgef1-deficient leukocytes are sufficient for the lung tissue destruction and associated decreased lung elastance in Arhgef1−/− mice.
Our findings suggest that elevated MMP expression and activity by Arhgef1−/− alveolar macrophages is associated with the lung pathology and impaired lung function observed with Arhgef1-deficient mice. This conclusion is supported by several lines of evidence that include the finding that expression of all of these MMPs are elevated in Arhgef1−/− BAL leukocytes and mutant BAL contained elevated MMP2 and MMP9 protease activity. Furthermore, wild-type BAL harbored significantly elevated MMP expression and activity after instillation of Arhgef1−/− macrophages when compared with instillation of wild-type cells. Indeed, macrophages are the predominant leukocyte population in the airspace of naïve Arhgef1−/− animals, and alveolar macrophages have been shown to express each of these MMPs under various inflammatory conditions.25 Thus, considered together these data provide strong support for the notion that Arhgef1−/− macrophages through the elaboration of MMPs facilitate lung pathophysiology.
Importantly, our data do not formally demonstrate that elevated MMP activity is responsible for the pathology observed in Arhgef1 mouse mutants. However, we note that in both humans and mouse models, lung tissue destruction leading to increased airspace has been associated with an imbalance in the production of proteases and anti-proteases41,42 and, accordingly, alterations in the expression of the MMP2, MMP9, and MMP12 proteases have been implicated in tissue damage.7,43 It is important to note, however, that elevated MMP expression as a consequence of an Arhgef1-deficiency appears restricted to the lung because neither Arhgef1−/− liver nor peritoneal macrophages exhibit elevated MMP expression. How tissue environment may influence MMP production in the absence of Arhgef1 is not clear, but our results clearly show that the interaction of Arhgef1−/− macrophages with fibronectin leads to exaggerated MMP production. This suggests the possibility that the unique extracellular matrix composition in the lung may provide integrin ligands that lead to increased MMP production in the absence of Arhgef1. Along these lines it is also noteworthy that aberrant fibronectin expression is not only a feature of inflammation44 but also of inflammatory diseases of the lung, such as COPD,5 and is also elevated in Arhgef1-deficient lungs (data not shown).
Although both mutant and wild-type macrophages displayed considerable adhesion to both VCAM-1 and fibronectin, only fibronectin induced MMP2 and MMP9 expression. Integrin-mediated signaling has long been recognized to induce MMP expression,30,31,45 and fibronectin-induced MMP expression by monocytes/macrophages has also previously been reported.32,46 Our results confirm the previous findings that the α5β1 integrin pair is responsible for the induction of MMP2 and MMP9 expression in macrophages cultured on fibronectin.32,33 These data extend those findings to demonstrate that Arhgef1 regulates α5β1 integrin signaling that promotes MMP2 and MMP9 expression and is supported by a recent report demonstrating that Arhgef1 is activated on integrin-mediated adhesion.47
MMP activity is controlled at transcriptional, posttranscriptional, and translational levels,48 and our results underscore the complexity of this regulation. Despite that we document Arhgef1−/− leukocytes display significantly increased MMP2 expression and activity in vivo (Figure 3), adoptive transfer of Arhgef1−/− peritoneal macrophages leads to elevated MMP2 expression but not activity (Figure 4) and is paralleled with in vitro cultures of macrophages on fibronectin that only exhibit elevated MMP2 expression (Figure 5). Similarly, whereas both the ≈95-kDa and ≈85-kDa MMP9 activities were enhanced in the BAL supernatant of Arhgef1−/− mice and wild-type that had received Arhgef1-deficient macrophages, in the in vitro macrophage cultures on fibronectin only the 95-kDa MMP9 was detected in either wild-type or Arhgef1-deficient samples. Moreover, MMP12 expression was significantly elevated in mutant lung, although MMP12 activity was not reproducibly observed in Arhgef1−/− BAL. Finally, we did not observe induction of MMP12 expression in either wild-type or Arhgef1-deficient macrophage when cultured on any of the integrin ligands tested.
Thus, based on these in vivo findings and our in vitro experiments of integrin-mediated MMP expression, we propose that under homeostatic noninflammatory conditions, Arhgef1−/− leukocytes traffic to the lungs and adhere to integrin ligands including fibronectin. In the absence of Arhgef1, pulmonary leukocyte integrin-fibronectin adhesion leads to enhanced integrin-mediated MMP production and activity that ultimately degrades lung tissue and results in airspace enlargement and eventual impaired lung function. Furthermore, although our data indicate that Arhgef1−/− macrophages display normal integrin adhesion, Arhgef1−/− marginal zone B cells display sustained integrin adhesion15 and thus increased integrin signaling by Arhgef1−/− pulmonary leukocytes may similarly be sustained and contribute to elevated MMP production. Finally, the increase in MMP activity in the airspace may facilitate the cleavage of either extracellular matrix proteins or chemokines, and these products, in turn, may further recruit additional leukocytes leading to additional inflammatory damage.49,50
These studies were initiated from the observation made in a previous report from our laboratory documenting the lack of an inflammatory response and consequent airway hyperreactivity in the Arhgef1-defcient mice using a mouse model of asthma.17 In the previous study Arhgef1-deficient mice fail to develop an adaptive inflammatory response in the lung that is attributable to impaired Arhgef1−/− T-cell function, whereas the current study documents the spontaneous presence of an innate inflammatory process in the lung compartment. Recent debate51 has centered on whether human asthma and COPD are different expressions of one disease entity (Dutch hypothesis) or distinct entities elaborated by different mechanisms (British hypothesis). Thus, Arhgef1-deficient mice are resistant to the development of asthma-like symptoms while simultaneously displaying pulmonary characteristics similar to patients with COPD. If ARHGEF1 has a similar influence in human subjects it would seemingly provide insight into the differences and possibly the mechanisms that may predispose individuals to the development of one disease over the other.
In sum, we present data showing that Arhgef1 regulates α5β1 integrin-mediated signaling of MMP expression and is required more generally by pulmonary leukocytes for appropriate homeostatic lung immunity. In its absence, and without intentional challenge, Arhgef1-deficient lungs exhibit a number of pulmonary features similar to COPD in humans that include a stable increase in the number of pulmonary leukocytes, elevated MMP expression and activity, lung tissue damage resulting in airspace enlargement, and impaired lung function. Furthermore, lung tissue destruction can be accounted for by exaggerated in vivo expression of MMPs and likely mediated via Arhgef1−/− alveolar macrophage association with fibronectin. Thus, these data not only present the Arhgef1−/− mouse mutant as a possible animal model of human emphysema, but also suggest the aberrant regulation of integrin signaling may underscore the molecular basis for this disease in humans.
Acknowledgements
We thank Pamela Strauch for technical assistance, and we particularly acknowledge Dr. Peter Henson for his advice on experiments and comments on this manuscript.
Footnotes
Supported by the Sandler Program in Asthma Research (R.M.T.), the National Jewish Health COPD Translational Research Initiative (R.M.T.), a postdoctoral NIAID training grant AI07045-16 (J.M.H.), and a FAMRI YCSA award (J.M.H.).
References
- 1.Fels AO, Cohn ZA. The alveolar macrophage. J Appl Physiol. 1986;60:353–369. doi: 10.1152/jappl.1986.60.2.353. [DOI] [PubMed] [Google Scholar]
- 2.Diamond G, Legarda D, Ryan LK. The innate immune response of the respiratory epithelium. Immunol Rev. 2000;173:27–38. doi: 10.1034/j.1600-065x.2000.917304.x. [DOI] [PubMed] [Google Scholar]
- 3.Henderson WR, Jr, Chi EY, Albert RK, Chu SJ, Lamm WJ, Rochon Y, Jonas M, Christie PE, Harlan JM. Blockade of CD49d (alpha4 integrin) on intrapulmonary but not circulating leukocytes inhibits airway inflammation and hyperresponsiveness in a mouse model of asthma. J Clin Invest. 1997;100:3083–3092. doi: 10.1172/JCI119863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paine R, 3rd, Morris SB, Jin H, Baleeiro CE, Wilcoxen SE. ICAM-1 facilitates alveolar macrophage phagocytic activity through effects on migration over the AEC surface. Am J Physiol. 2002;283:L180–L187. doi: 10.1152/ajplung.00430.2001. [DOI] [PubMed] [Google Scholar]
- 5.Kranenburg AR, Willems-Widyastuti A, Moori WJ, Sterk PJ, Alagappan VK, de Boer WI, Sharma HS. Enhanced bronchial expression of extracellular matrix proteins in chronic obstructive pulmonary disease. Am J Clin Pathol. 2006;126:725–735. doi: 10.1309/jc477fael1ykv54w. [DOI] [PubMed] [Google Scholar]
- 6.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Pare PD. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 7.Hogg JC, Senior RM. Chronic obstructive pulmonary disease - part 2: pathology and biochemistry of emphysema. Thorax. 2002;57:830–834. doi: 10.1136/thorax.57.9.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22:672–688. doi: 10.1183/09031936.03.00040703. [DOI] [PubMed] [Google Scholar]
- 9.Saetta M, Kim WD, Izquierdo JL, Ghezzo H, Cosio MG. Extent of centrilobular and panacinar emphysema in smokers' lungs: pathological and mechanical implications. Eur Respir J. 1994;7:664–671. doi: 10.1183/09031936.94.07040664. [DOI] [PubMed] [Google Scholar]
- 10.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 11.Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science. 1998;280:2109–2111. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- 12.Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Galpha13. Science. 1998;280:2112–2114. doi: 10.1126/science.280.5372.2112. [DOI] [PubMed] [Google Scholar]
- 13.Hu J, Strauch P, Rubtsov A, Donovan EE, Pelanda R, Torres RM. Lsc activity is controlled by oligomerization and regulates integrin adhesion. Mol Immunol. 2008;45:1825–1836. doi: 10.1016/j.molimm.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francis SA, Shen X, Young JB, Kaul P, Lerner DJ. Rho GEF Lsc is required for normal polarization, migration, and adhesion of formyl-peptide-stimulated neutrophils. Blood. 2006;107:1627–1635. doi: 10.1182/blood-2005-03-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubtsov A, Strauch P, Digiacomo A, Hu J, Pelanda R, Torres RM. Lsc regulates marginal-zone B cell migration and adhesion and is required for the IgM T-dependent antibody response. Immunity. 2005;23:527–538. doi: 10.1016/j.immuni.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 16.Girkontaite I, Missy K, Sakk V, Harenberg A, Tedford K, Potzel T, Pfeffer K, Fischer KD. Lsc is required for marginal zone B cells, regulation of lymphocyte motility and immune responses. Nat Immunol. 2001;2:855–862. doi: 10.1038/ni0901-855. [DOI] [PubMed] [Google Scholar]
- 17.Brown JP, Taube C, Miyahara N, Koya T, Pelanda R, Gelfand EW, Torres RM. Arhgef1 is required by T cells for the development of airway hyperreactivity and inflammation. Am J Respir Crit Care Med. 2007;176:10–19. doi: 10.1164/rccm.200702-270OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lovgren AK, Jania LA, Hartney JM, Parsons KK, Audoly LP, Fitzgerald GA, Tilley SL, Koller BH. COX-2-derived prostacyclin protects against bleomycin-induced pulmonary fibrosis. Am J Physiol. 2006;291:L144–L156. doi: 10.1152/ajplung.00492.2005. [DOI] [PubMed] [Google Scholar]
- 19.Tschanz SA, Burri PH. A new approach to detect structural differences in lung parenchyma using digital image analysis. Exper Lung Res. 2002;28:457–471. doi: 10.1080/01902140290096719. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008 doi: 10.1002/0471142735.im1401s83. Chapter 14:Unit 14.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hartney JM, Coggins KG, Tilley SL, Jania LA, Lovgren AK, Audoly LP, Koller BH. Prostaglandin E2 protects lower airways against bronchoconstriction. Am J Physiol. 2006;290:L105–L113. doi: 10.1152/ajplung.00221.2005. [DOI] [PubMed] [Google Scholar]
- 22.Suratt BT, Petty JM, Young SK, Malcolm KC, Lieber JG, Nick JA, Gonzalo JA, Henson PM, Worthen GS. Role of the CXCR4/SDF-1 chemokine axis in circulating neutrophil homeostasis. Blood. 2004;104:565–571. doi: 10.1182/blood-2003-10-3638. [DOI] [PubMed] [Google Scholar]
- 23.Huang K, Rabold R, Schofield B, Mitzner W, Tankersley CG. Age-dependent changes of airway and lung parenchyma in C57BL/6J mice. J Appl Physiol. 2007;102:200–206. doi: 10.1152/japplphysiol.00400.2006. [DOI] [PubMed] [Google Scholar]
- 24.Turner JM, Mead J, Wohl ME. Elasticity of human lungs in relation to age. J Appl Physiol. 1968;25:664–671. doi: 10.1152/jappl.1968.25.6.664. [DOI] [PubMed] [Google Scholar]
- 25.Wert SE, Yoshida M, LeVine AM, Ikegami M, Jones T, Ross GF, Fisher JH, Korfhagen TR, Whitsett JA. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. Proc Natl Acad Sci U S A. 2000;97:5972–5977. doi: 10.1073/pnas.100448997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoon HK, Cho HY, Kleeberger SR. Protective role of matrix metalloproteinase-9 in ozone-induced airway inflammation. Environ Health Perspect. 2007;115:1557–1563. doi: 10.1289/ehp.10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aasheim HC, Pedeutour F, Smeland EB. Characterization, expression and chromosomal localization of a human gene homologous to the mouse Lsc oncogene, with strongest expression in hematopoetic tissues. Oncogene. 1997;14:1747–1752. doi: 10.1038/sj.onc.1200994. [DOI] [PubMed] [Google Scholar]
- 28.Demedts IK, Brusselle GG, Bracke KR, Vermaelen KY, Pauwels RA. Matrix metalloproteinases in asthma and COPD. Curr Opin Pharmacol. 2005;5:257–263. doi: 10.1016/j.coph.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 29.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huhtala P, Humphries MJ, McCarthy JB, Tremble PM, Werb Z, Damsky CH. Cooperative signaling by alpha 5 beta 1 and alpha 4 beta 1 integrins regulates metalloproteinase gene expression in fibroblasts adhering to fibronectin. J Cell Biol. 1995;129:867–879. doi: 10.1083/jcb.129.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esparza J, Vilardell C, Calvo J, Juan M, Vives J, Urbano-Marquez A, Yague J, Cid MC. Fibronectin upregulates gelatinase B (MMP-9) and induces coordinated expression of Gelatinase a (MMP-2) and its activator MT1-MMP (MMP-14) by human T lymphocyte cell lines. a process repressed through RAS/MAP kinase signaling pathways. Blood. 1999;94:2754–2766. [PubMed] [Google Scholar]
- 32.Xie B, Laouar A, Huberman E. Fibronectin-mediated cell adhesion is required for induction of 92-kDa type IV collagenase/gelatinase (MMP-9) gene expression during macrophage differentiation. The signaling role of protein kinase C-beta. J Biol Chem. 1998;273:11576–11582. doi: 10.1074/jbc.273.19.11576. [DOI] [PubMed] [Google Scholar]
- 33.Sudhakaran PR, Radhika A, Jacob SS. Monocyte macrophage differentiation in vitro: Fibronectin-dependent upregulation of certain macrophage-specific activities. Glycoconj J. 2007;24:49–55. doi: 10.1007/s10719-006-9011-2. [DOI] [PubMed] [Google Scholar]
- 34.Berton G, Lowell CA. Integrin signalling in neutrophils and macrophages. Cell Signal. 1999;11:621–635. doi: 10.1016/s0898-6568(99)00003-0. [DOI] [PubMed] [Google Scholar]
- 35.Mould AP, Askari JA, Aota S, Yamada KM, Irie A, Takada Y, Mardon HJ, Humphries MJ. Defining the topology of integrin alpha5beta1-fibronectin interactions using inhibitory anti-alpha5 and anti-beta1 monoclonal antibodies. Evidence that the synergy sequence of fibronectin is recognized by the amino-terminal repeats of the alpha5 subunit. J Biol Chem. 1997;272:17283–17292. doi: 10.1074/jbc.272.28.17283. [DOI] [PubMed] [Google Scholar]
- 36.Ruoslahti E, Pierschbacher M. Arg-Gly-Asp: a versatile cell recognition signal. Cell. 1986;44:517–518. doi: 10.1016/0092-8674(86)90259-x. [DOI] [PubMed] [Google Scholar]
- 37.Noto K, Kato K, Okumura K, Yagita H. Identification and functional characterization of mouse CD29 with a mAb. Int Immunol. 1995;7:835–842. doi: 10.1093/intimm/7.5.835. [DOI] [PubMed] [Google Scholar]
- 38.Ridger VC, Wagner BE, Wallace WA, Hellewell PG. Differential effects of CD18. CD29, and CD49 integrin subunit inhibition on neutrophil migration in pulmonary inflammation. J Immunol. 2001;166:3484–3490. doi: 10.4049/jimmunol.166.5.3484. [DOI] [PubMed] [Google Scholar]
- 39.Das S, Banerji A, Frei E, Chatterjee A. Rapid expression and activation of MMP-2 and MMP-9 upon exposure of human breast cancer cells (MCF-7) to fibronectin in serum free medium. Life Sci. 2008;82:467–476. doi: 10.1016/j.lfs.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 40.Amy RW, Bowes D, Burri PH, Haines J, Thurlbeck WM. Postnatal growth of the mouse lung. J Anat. 1977;124:131–151. [PMC free article] [PubMed] [Google Scholar]
- 41.Elkington PT, Friedland JS. Matrix metalloproteinases in destructive pulmonary pathology. Thorax. 2006;61:259–266. doi: 10.1136/thx.2005.051979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mercer PF, Shute JK, Bhowmik A, Donaldson GC, Wedzicha JA, Warner JA. MMP-9. TIMP-1 and inflammatory cells in sputum from COPD patients during exacerbation. Respir Res. 2005;6:151. doi: 10.1186/1465-9921-6-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Demedts IK, Morel-Montero A, Lebecque S, Pacheco Y, Cataldo D, Joos GF, Pauwels RA, Brusselle GG. Elevated MMP-12 protein levels in induced sputum from patients with COPD. Thorax. 2006;61:196–201. doi: 10.1136/thx.2005.042432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roman J. Extracellular matrix and lung inflammation. Immunol Res. 1996;15:163–178. doi: 10.1007/BF02918505. [DOI] [PubMed] [Google Scholar]
- 45.Werb Z, Tremble PM, Behrendtsen O, Crowley E, Damsky CH. Signal transduction through the fibronectin receptor induces collagenase and stromelysin gene expression. J Cell Biol. 1989;109:877–889. doi: 10.1083/jcb.109.2.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shapiro SD, Kobayashi DK, Pentland AP, Welgus HG. Induction of macrophage metalloproteinases by extracellular matrix. Evidence for enzyme- and substrate-specific responses involving prostaglandin-dependent mechanisms. J Biol Chem. 1993;268:8170–8175. [PubMed] [Google Scholar]
- 47.Dubash AD, Wennerberg K, Garcia-Mata R, Menold MM, Arthur WT, Burridge K. A novel role for Lsc/p115 RhoGEF and LARG in regulating RhoA activity downstream of adhesion to fibronectin. J Cell Sci. 2007;120:3989–3998. doi: 10.1242/jcs.003806. [DOI] [PubMed] [Google Scholar]
- 48.Yan C, Boyd DD. Regulation of matrix metalloproteinase gene expression. J Cell Physiol. 2007;211:19–26. doi: 10.1002/jcp.20948. [DOI] [PubMed] [Google Scholar]
- 49.Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III. PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. 2000;96:2673–2681. [PubMed] [Google Scholar]
- 50.Henson PM, Vandivier RW. The matrix degrades, neutrophils invade. Nat Med. 2006;12:280–281. doi: 10.1038/nm0306-280. [DOI] [PubMed] [Google Scholar]
- 51.Kraft M. Asthma and chronic obstructive pulmonary disease exhibit common origins in any country! Am J Respir Crit Care Med. 2006;174:238–240; discussion 243–234. doi: 10.1164/rccm.2604007. [DOI] [PubMed] [Google Scholar]