

Abstract
Fibroblast differentiation into myofibroblasts is a key event during normal wound repair. We have previously demonstrated an age-related defect in this process associated with impaired synthesis of hyaluronan (HA) synthase (HAS) 2 but failed to prescribe its role in a mechanistic sense. Here we demonstrate that in addition to HAS2, there is loss of EGF receptor (EGF-R) in aged cells, and both are required for normal fibroblast functionality. Analysis of molecular events revealed that in young cells, transforming growth factor (TGF)-β1-dependent phenotypic activation uses two distinct but cooperating pathways that involve TGF-β receptor/Smad2 activation and EGF-mediated EGF-R/extracellular signal-regulated kinase (ERK) 1/2 signaling, and the latter is compromised with in vitro aging. Pharmacological inhibition of any of the five intermediates (TGF-β receptor, Smad2, EGF, EGF-R, and ERK1/2) attenuated TGF-β1 induction of α-smooth muscle actin. We present evidence that the HA receptor CD44 co-immunoprecipitates with EGF-R after activation by TGF-β1. This interaction is HA-dependent because disruption of HA synthesis abrogates this association and inhibits subsequent ERK1/2 signaling. In aged fibroblasts, this association is lost with resultant suppression of ERK1/2 activation. Forced overexpression of EGF-R and HAS2 in aged cells restored TGF-β1-mediated HA-CD44/EGF-R association and α-smooth muscle actin induction. Taken together, these results demonstrate that HA can serve as a signal integrator by facilitating TGF-β1-mediated CD44-EGF-R-ERK interactions and ultimately fibroblast phenotype. We propose a model to explain this novel mechanism and the functional consequence of age-dependent dysregulation.
Problems in wound and skin repair constitute a major medical problem for aging adults, and as the size of the elderly population continues to grow, this financial burden is set to increase. Chronic skin wounds have been estimated to affect 4% of the UK population older than 65, and the morbidity associated with this impaired wound healing is estimated to cost the health service in excess of £1 billion annually in the UK1 and $9 billion in the United States.2
Wound healing, regardless of the etiology of the wound, involves an overlapping sequence of events including coagulation, inflammation, epithelialization, formation of granulation tissue, and remodeling of the matrix and tissue. Fibroblasts are central to wound healing and when activated, they undergo a number of phenotypic transitions, eventually acquiring a contractile “myofibroblastic” phenotype, characterized by the expression of α-smooth muscle actin (α-SMA).3 These myofibroblasts are responsible for closure of wounds and for the formation of the collagen-rich scar. Disease-associated alterations in fibroblast behavior contribute to the pathogenesis of progressive fibrotic disorders, and our central tenant is that age-dependent impairment in wound healing may also reflect a defect in phenotypic maturation. In support of this hypothesis, it has been demonstrated previously that fibroblasts isolated from aging skin have impaired migration, premature senescence, impaired proliferative response, and defects in matrix generation compared with those from young skin.4,5 Furthermore, we have shown that aging cells are resistant to fibroblast to myofibroblast differentiation.6 Because the myofibroblast orchestrates the successful formation of granulation tissue and matrix remodeling, this is an important finding that may be central to the delayed healing seen in elderly individuals.
Transforming growth factor-β1 (TGF-β1) is the principal mediator of fibroblast to myofibroblast differentiation. In our studies, TGF-β1 addition to early passage dermal fibroblasts (young cells) induced their differentiation to α-SMA-positive myofibroblasts.7,8 Dermal fibroblasts aged in cell culture (aged cells), however, were resistant to differentiation, despite the normal activation of the TGF-β1 intracellular signaling pathways.6 We have previously demonstrated that the matrix polysaccharide hyaluronan is a key factor in the regulation of fibroblast activation.7,9 Hyaluronan (HA) is a ubiquitous connective tissue glycosaminoglycan synthesized by HA synthase (HAS) enzymes, of which three vertebrate genes have been isolated and characterized: HAS1, HAS2, and HAS3.10,11 HA has a role in maintaining matrix stability and tissue hydration. It is known to play a major role in regulating cell-cell adhesion,12 migration,13–15 differentiation,16 and proliferation17,18 and therefore plays an important role in wound healing. In addition, it is involved in mediating cellular responses to TGF-β. For example, our recent studies in epithelial cells have demonstrated that HA modulates TGF-β signaling after interaction with its receptor, CD44.19,20 Failure of TGF-β1-induced differentiation to the myofibroblast phenotype associated with aging is associated with the inability to induce HAS2, a decrease in HA synthesis, and a lack of pericellular coat formation. HA synthesis and coat assembly could be restored by forced overexpression of HAS2 followed by TGF-β1 stimulation. This did not, however, restore the myofibroblast phenotype, suggesting that other factors (in addition to TGF-β1 and HAS2) must be operative in young cells but have been lost during the aging process.
The classic signaling pathway for TGF involves the Smad family of transcriptional activators.21,22 The receptor-associated R-Smad, Smad2, and Smad3 are phosphorylated directly by the TGF-β type 1 receptor kinase, after which they hetero-oligomerize with Smad4, translocate to the nucleus, and together with their binding partners activate or repress their target genes. We have previously demonstrated the role of R-Smads in the regulation of TGF-β1-dependent fibroblast proliferation. Furthermore, this effect may be modified by HA.19,20,23 Several studies have also shown that TGF can signal in a Smad-independent fashion, recruiting pathways such as the extracellular signal-regulated kinase (ERK) pathway.24
Synergistic effects between TGF-β1 and epidermal growth factor (EGF) have been demonstrated to regulate cell phenotype and function in several systems.25–31 Previous studies have demonstrated a decrease in EGF receptor (EGF-R) expression and responsiveness in aging fibroblasts.4 We therefore postulate that a TGF-β1-dependent and an EGF-R-dependent pathway are both necessary for full myofibroblastic differentiation. In view of our data demonstrating the importance of HA in both phenotypic activation and maintenance, we also postulate that HA is the key factor in regulating the coordinated action of the TGF-β1 and EGF signaling pathways, required for acquisition of myofibroblast phenotype, such that defects in both HA synthesis and EGF signaling both contribute to age-dependent impaired wound healing.
An in vitro aging model based on cell senescence has previously been described and validated as a model of age-related alterations in human aortic smooth muscle cell function.32,33 Similarly alterations in fibroblast function in an in vitro model of aging have demonstrated the validity of this model in terms of in vivo age-related alterations in fibroblast motility and mitogenesis, which are associated with age-dependent impaired wound healing.4,5 In this study we have used this model to address the hypothesis that synergy between TGF-β1-dependent pathways and a competent EGF signaling pathway are required for myofibroblastic differentiation and that central to this process is the induction of HAS2 and synthesis of HA. Furthermore, we differentiate what contribution these pathways (Smad and non-Smad) make to age-related impairment in phenotypic activation.
Materials and Methods
Materials
Antibodies for Western blot analysis and immunofluorescence and the final working dilution were as follows: rat monoclonal anti-CD44 antibody (A020, dilution, 1:200) from Calbiochem (Nottingham, UK); polyclonal rabbit anti-EGF-R antibody (1005:sc-03, dilution, 1:1000) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); murine monoclonal anti-α-SMA clone 1A4 (dilution 1:30) from Sigma (Poole, UK); and monoclonal mouse anti-phospho-p44/42-mitogen-activated protein kinase (ERK1 and ERK2) antibody (9106, dilution, 1:1000) and rabbit polyclonal anti-phospho-Smad2 and 3 (dilution, 1:1000) from Cell Signaling Technology (Beverly, MA). All conjugated secondary antibodies (dilution, 1:10,000) were purchased from Sigma. Other reagents used were recombinant TGF-β, EGF, goat anti-human EGF antibody from R&D Systems (Oxford, UK), mouse monoclonal anti-CD44 blocking antibody from Ancell (Bayport, MN); and mitogen-activated protein kinase kinase inhibitor PD98059 and p38 kinase inhibitor SB203580 (Calbiochem). The EGF-R inhibitor AG1478 was from Invitrogen (Carlsbad, CA); the Alk5 inhibitor SB431542, bovine testicular hyaluronidase (H3884), and 4-methylumbelliferone (4MU) were from Sigma.
Cell Culture
All experiments were performed with dermal fibroblasts obtained by biopsy from consenting adults undergoing routine minor surgery. Ethical approval for the biopsies was obtained from the South East Wales Research Ethics Committee. The cells were isolated and characterized as described previously7,8,34 and cultured in Dulbecco's modified Eagle's medium supplemented with l-glutamine (2 mmol/L), 100 units/ml penicillin and 100 μg/ml streptomycin, and 10% fetal bovine serum (FBS) (Biological Industries Ltd., Cumbernauld, UK). The cultures were maintained at 37°C in a humidified incubator in an atmosphere of 5% CO2, and fresh growth medium was added to the cells every 3 to 4 days. At 90% confluence, fibroblasts were trypsinized and reseeded at the ratio of 1:3. At each passage, the total number of viable cells was determined by direct counting using a hemocytometer. The effect of aging was examined using a previously characterized and validated model of in vitro aging,32 which has been demonstrated in a fibroblast model to have applicability to in vivo aging.4 Population doubling levels (PDLs) were calculated as follows: PDL = [log10 (total cells harvested at passage − log10 (total cells reseeded)]/log10(2).35 Cumulative population doubling levels were calculated by adding the derived increase to the previous PDL, and fibroblast populations were cultured until senescence, which varied for each patient, occurring at PDL 46 to 70. In the experiments, PDL 10 to 15 and PDL 25 to 39 were used and are referred to as young and aged dermal fibroblasts, respectively. The cells were incubated in serum-free medium for 48 hours before use in experiments, and all experiments were done under serum-free conditions unless otherwise stated.
Immunocytochemistry
Cells were grown to 70% confluence in eight-well Permanox chamber slides. The culture medium was removed, and the cells washed with sterile PBS, before fixation in acetone-methanol (1:1, v/v) for 5 minutes at room temperature. After fixation, slides were blocked with 5% bovine serum albumin for 20 minutes before a further washing step with PBS. Subsequently the slides were incubated with the primary antibody diluted in 0.1% bovine serum albumin-PBS for 2 hours at room temperature. After a further washing step slides were incubated with fluorescein isothiocyanate-conjugated and/or tetramethylrhodamine isothiocyanate-conjugated secondary antibodies for 1 hour at room temperature (DAKO, Cambridgeshire, UK). Cells were then mounted and analyzed by fluorescent microscopy.
Analysis of Cell Proliferation
Fibroblast proliferation was assessed by the incorporation of d-[3H]thymidine into DNA. Cells were grown in 35-mm dishes and assessed at subconfluence (∼50% confluence). Metabolic labeling was performed by incubation with 1 μCi/ml d-[3H]thymidine for 24 hours. The medium was then discarded, and the cells were washed repeatedly with PBS containing 1 mmol/L thymidine before fixing with 500 μl of 5% trichloroacetic acid containing 1 mmol/L thymidine at 4°C for 1 hour. The cell layer was extracted by incubation with 1 ml of 0.1 M NaOH at 20°C for 24 hours and neutralized with 0.1 M HCl. Radioactivity was determined by β-counting on a Packard Tri-Carb 1900 liquid scintillation analyzer, and the results reported as disintegrations per minute.
Quantitative PCR
Young and aged dermal fibroblasts were grown to confluence in 35-mm dishes and washed with PBS before lysis with TRI reagent and RNA purification according to the manufacturer's protocol. One microgram of total RNA was retrotranscribed using High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Foster City, CA) according to the manufacturer's protocol. This uses the random primer method for initiating cDNA synthesis. As a negative control reverse transcription was performed with sterile H2O replacing the RNA sample. Quantitative PCR was performed using the 7900HT Fast Real-Time PCR System from Applied Biosystems using TaqMan Universal PCR Master Mix (Applied Biosystems) following the manufacturer's instructions. The following TaqMan gene expression assays were used: CD44 (HS00153304_ m1), HAS2 (HS0019343_m1), TSG-6 (HS00200180_m1), Smad2 (HS00183425_m1), Smad3 (HS00232222_m1), EGF-R (HS01076073_m1), EGF (HS01099999_m1), and α-SMA (HS00426835_g1). PCR was performed in a final volume of 20 μl/sample. One microliter of reverse transcription product, 1 μl of target gene primers and probe, 10 μl of TaqMan Universal PCR MasterMix, and 8 μl of sterile H2O. Amplification was performed using a cycle of 95°C for 1 second and 60°C for 20 seconds for a further 40 cycles. As a negative control, PCR was performed with sterile H2O replacing the cDNA sample. PCR was simultaneously done for ribosomal RNA (primers and probe commercially designed and purchased from Applied Biosciences) as a standard reference gene. The comparative CT method was used for relative quantification of gene expression. The CT (threshold cycle where amplification is in the linear range of the amplification curve) for the standard reference gene (ribosomal RNA) was subtracted from the target gene CT to obtain the ΔCT (dCT). The mean dCT values for similar samples were then calculated. The expression of the target gene in experimental samples relative to expression in control samples was then calculated using the formula:
where dCT(1) is the mean dCT calculated for the experimental samples and dCT(2) is the mean dCT calculated for the control samples.
Overexpression of HAS2 and EGF-R
The HAS26 and EGF-R4 open reading frame was inserted into the vector pCR3.1 using a standard ligation reaction with T4 DNA ligase (Promega, Madison, WI). Amplification of the cloned vector was performed via bacterial transformation (JM109 competent Escherichia coli, Promega). Transient transfection was performed with the aid of Nucleofector technology (Amaxa Biosystems, Gaithersburg, MD) in accordance with the manufacturer's protocol for transfection of primary mammalian fibroblasts. Fibroblasts were grown to 70% confluence in T75 flasks. The medium was removed, and the cells were harvested by trypsinization (solution containing 0.05% trypsin and 0.53 mmol/L EDTA). Once the cells detached, the resulting cell suspension was treated with an equal volume of FBS to neutralize the protease activity. Cell counting was performed using a hemocytometer, and cell numbers were adjusted to a final concentration of 0.5 × 106 cells/ml. Cells were then centrifuged (100 × g for 10 minutes), and the resulting pellet was resuspended in Basic Nucleofector Solution (Amaxa Biosciences). The cells were transfected either with HAS2-pCR3.1 and/or EGF-R-pCR3.1. The concentrations used were 100 μl of Basic Nucleofector Solution to 1 μg of DNA. The solution was then transferred to an Amaxa-certified cuvette and placed in the Nucleofector. Nucleofection was performed for 5 seconds according to the prespecified program designed for mammalian fibroblasts, and the cells were subsequently transferred to either 35-mm dishes or eight-well Permanox chamber slides containing prewarmed medium supplemented with 10% FBS for 24 hours followed by a 24 hours incubation in serum-free medium. pmaxGFP (green fluorescent protein) plasmid (Amaxa Biosystems) was used as a control (mock transfections). Two negative controls were performed: 1) 0.5 × 106 cells in 100 μl of Basic Nucleofector Solution containing 1 μg of DNA but without application of the program and 2) 0.5 × 106 cells in 100 μl of Basic Nucleofector Solution without DNA but with application of the program. The cells were incubated in medium supplemented with 10% FBS for 24 hours followed by a 24-hour incubation in serum-free medium before experimentation. pmaxGFP transfection was performed in parallel, and expression was determined by fluorescence microscopy after 48 hours to assess efficiency of electroporation.
Small Interfering RNA Transfection
Transient transfection of dermal fibroblasts was performed with specific small interfering (siRNA) nucleotides (Applied Biosystems) targeting either Smad2 (115717), Smad3 (107875), or CD44 (114068). Transfection was performed using Lipofectamine 2000 transfection reagent (Invitrogen) in accordance with the manufacturer's protocol. In brief, cells were grown to 50% confluence in antibiotic-free medium in either 12-well culture plates or 8-well Permanox chamber slides. Two microliters of transfection agent was diluted in 98 μl of Opti-MEM reduced growth medium (Gibco, Grand Island, NY) and left to incubate at room temperature for 5 minutes. Specific siRNA oligonucleotides were diluted in Opti-MEM reduced growth medium to a final concentration of 20 μmol/L in a total volume of 100 μl. The transfection agent and siRNA mixtures were then combined and incubated at room temperature for a further 10 minutes. The newly formed transfection complexes (200 μl) were subsequently added to cells and incubated at 37°C with 5% CO2 for 24 hours in medium supplemented with 10% FBS followed by a 24-hour incubation in serum-free medium before experimentation. As a control, cells were transfected with negative control siRNA (a scrambled sequence that bears no homology to the human genome) (Applied Biosystems).
Western Blot Analysis
Cells were grown to confluence in 35-mm dishes and rinsed with cold PBS. Cells were then lysed using 1% protease inhibitor cocktail, 1% phenylmethylsulfonyl fluoride and 1% sodium orthovanadate in radioimmunoprecipitation assay lysis buffer (Santa Cruz Biotechnology Inc., Heidelberg, Germany). The samples were scraped, collected, and centrifuged at 2500 × g for 10 minutes. The supernatant was collected, protein concentrations were determined by the Bradford assay, and the samples were stored at −70°C until use. Equal amounts of protein were mixed with equal volumes of reducing SDS sample buffer and boiled for 5 minutes at 95°C before loading onto 10% SDS-polyacrylamide gels. Electrophoresis was performed under reducing conditions at 150 V for 1 hour, and the separated proteins were then transferred at 150 V over 90 minutes to a nitrocellulose membrane (GE Healthcare, Little Chalfont, Buckinghamshire, UK). The membrane was blocked with Tris-buffered saline containing 5% nonfat powdered milk for 1 hour and incubated with the primary antibody at 4°C overnight. The blots were subsequently washed with Tris-buffered saline containing 1% Tween before incubation with the appropriate horseradish peroxidase-conjugated secondary antibody for 1 hour at room temperature. Proteins were visualized using enhanced chemiluminescence (GE Healthcare) according to the manufacturer's instructions.
Immunoprecipitation
Cells were grown, washed, and lysed as for Western blot analysis above. Cell protein samples (200 μg) were precleared with 25 μl of packed protein A-cross-linked 4% beaded agarose (Sigma) at 4°C overnight. The beads were removed by centrifugation (13,000 rpm, 10 minutes), and the supernatant was collected. Samples were subjected to immunoprecipitation using rat anti-CD44 antibody (2 μg/ml) and incubated at 4°C with constant mixing overnight. The immune complex was captured by the addition of packed agarose protein A beads (50 μl) overnight at 4°C. Beads were washed with radioimmunoprecipitation assay buffer (50 mmol/L Tris, 150 mmol/L NaCl, 0.5% sodium deoxycholate, 10 mmol/L MgCl2, 0.1% SDS, and 1% Triton X-100); 30 μl of sample buffer was then added before boiling for 10 minutes. Separation of the beads was achieved by centrifugation (13,000 × g for 10 minutes), and the supernatant was removed. Subsequently samples were transferred to nitrocellulose membranes and processed as described for Western blot analysis using rabbit anti-EGF-R antibody as the primary antibody. The blots were subsequently washed in Tris-buffered saline-Tween and incubated with the appropriate horseradish peroxidase-conjugated secondary antibody (Sigma) in Tris-buffered saline-Tween. Proteins were visualized using enhanced chemiluminescence (Amersham Biosciences Inc., Piscataway, NJ) according to the manufacturer's instructions. The specificity of immunoprecipitation was confirmed by negative control reactions performed with the IgG control.
Statistical Analysis
All experiments were performed at least in triplicate for individual patient donors. All values are provided as means ± SE. Comparisons were performed using an unpaired Student's t-tests, and probability values of P < 0.05 were considered to indicate a significant difference.
Results
TGF-β1-Dependent Phenotypic Activation Uses Smad2 and EGF-R-Dependent Pathways
The role of the TGF-β1 signaling intermediates Smad2 and Smad3 in TGF-β1-dependent phenotypic activation was examined by gene silencing using siRNA. Suppression of expression of Smad2, after transfection with Smad2 siRNA was confirmed by quantitative PCR (Q-PCR) (Figure 1A). Inhibition of Smad2 expression was associated with a failure of TGF-β1 to increase α-SMA in young fibroblasts (Figure 1B), confirming our earlier work suggesting the involvement of this signaling intermediate in fibroblast phenotypic activation.36 In contrast, successful knockdown of Smad3 using siRNA failed to have an effect on TGF-β1-mediated phenotypic differentiation (data not shown). Synergy between TGF-β1 and EGF in regulating cell phenotype is well recognized in other cell systems.25,29,30 The relative contribution of TGF-β receptor and EGF receptor signaling in TGF-β1-dependent fibroblast to myofibroblast phenotypic activation was examined by addition of TGF-β1 to growth-arrested young fibroblasts for 48 hours in the presence of either AG1478, a potent and selective inhibitor of EGF-R, or the TGF-β receptor/Alk5 inhibitor SB431542. Addition of either inhibitor attenuated TGF-β1-induced expression of α-SMA (Figure 1C), suggesting that in the absence of exogenous EGF, TGF-β1 uses both its own and the EGF-R to facilitate phenotypic activation.
Figure 1.
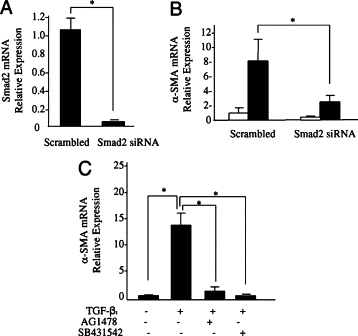
Involvement of Smad2, EGF-R, and Alk5 in phenotypic conversion. Confluent monolayers of young dermal fibroblasts were transfected with Smad2 siRNA or scramble oligonucleotide control (scramble) for 48 hours. A: Total mRNA was extracted and Smad2 mRNA expression was assessed by RT-Q-PCR. B: In parallel experiments, after transfection the medium was replaced with serum-free medium for a further 48 hours, before addition of serum-free medium alone (white bars) or serum-free medium containing 10 ng/ml TGF-β1 (black bars) for a further 72 hours, and α-SMA expression was assessed by RT-Q-PCR. C: Confluent monolayers of young dermal fibroblasts were growth-arrested in serum-free medium for 48 hours. Serum-free medium alone or serum-free medium containing 10 ng/ml TGF-β1 alone or in combination with either the EGF-R inhibitor AG1478 (10 μmol/L) or the Alk5-specific inhibitor SB431542 (10 μmol/L) was added to cells for a further 72 hours. Total mRNA was extracted, and α-SMA expression was assessed by RT-Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assessed relative to scramble (A), unstimulated-scrambled (B), and control unstimulated (C) samples. The comparative CT method was used for relative quantification of gene expression, and the results represent the mean ± SE of six individual experiments using cells isolated from two different donors. Statistical analysis was performed by Student's t-test: *P < 0.05.
Differential Effect of Aging on Smad2 and EGF-R
Given the age-dependent failure of TGF-β1-directed phenotypic activation that we have previously demonstrated,6 we sought to examine the effects of aging on Smad2 and EGF-R expression. The effect of aging on Smad2 activation was examined by Western blot analysis of TGF-β1-dependent phosphorylation of Smad2. Smad2 was rapidly phosphorylated (within 15 minutes) after addition of TGF-β1 to quiescent cells and peak activation was observed after 1 hour. This kinetic profile did not change across the range of in vitro ages extending from young (PDL 15) through to aged cells (PDL 39) (Figure 2A). For evaluation of EGF-R expression, fibroblasts were grown to confluence and serum-deprived for 48 hours before mRNA extraction and analysis of EGF-R mRNA by Q-PCR. Young fibroblasts had significantly higher (∼3-fold) EGF-R mRNA expression compared with patient-matched aged fibroblasts (Figure 2B). In parallel experiments EGF-R protein was examined by Western blot analysis (Figure 2C). Cell protein was extracted from growth-arrested patient-matched fibroblasts over a range of different in vitro ages extending from young (PDL 15) through to aged cells (PDL 39). Consistent with the Q-PCR data, in vitro aging was associated with an age-dependent decrease in EGF-R protein expression with barely detectable levels in near-senescent cells (PDL 33 to 39). Collectively these results suggest that age-dependent loss of TGF-β1-dependent phenotypic activation is associated with loss of EGF-R, whereas Smad signaling remains intact.
Figure 2.
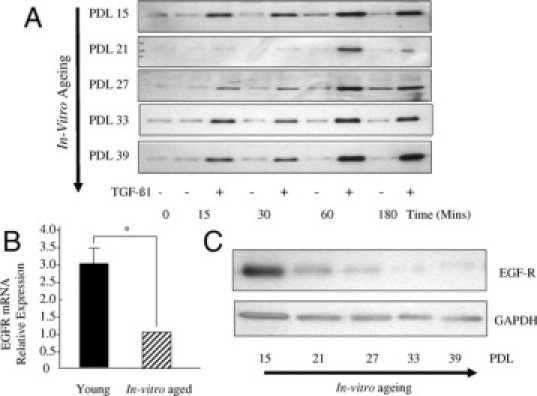
Effect of in vitro aging on EGF-R and Smad2 signaling. A: Confluent monolayers of dermal fibroblasts at the indicated PDL were growth arrested in serum-free medium for 48 hours. The medium was replaced with either serum-free medium alone or serum-free medium containing 10 ng/ml TGF-β1 for the indicated times. Cell lysates were subsequently subjected to Western blot analysis using antibodies against the phosphorylation form of Smad2. Expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was analyzed as a control to ensure equal loading (results not shown). The results shown are representative of five independent experiments from the same patient donor. B: Confluent monolayers of patient-matched young and in vitro aged dermal fibroblasts were growth-arrested in serum-free medium for 48 hours. Total mRNA was extracted and EGF-R mRNA expression was assessed by RT-Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assessed relative to in vitro aged cells. The comparative CT method was used for relative quantification of gene expression, and the results represent the mean ± SE of six individual experiments using cells isolated from two different donors. Statistical analysis was performed by Student's t-test: *P < 0.05. C: Confluent monolayers of dermal fibroblasts at the indicated PDL were growth-arrested in serum-free medium for 48 hours. Cell protein was extracted, and Western blotting for EGF-R was performed. Expression of GAPDH was analyzed as a control to ensure equal loading. The results shown are representative of five independent experiments from the same patient donor.
Restoration of EGF-R Expression in Isolation Does Not Influence Phenotypic Activation
We hypothesize that loss of EGF-R may be responsible for loss of TGF-β1 responsiveness in aged fibroblasts. Previous studies have demonstrated restoration of age-dependent EGF responsiveness by forced overexpression of EGF-R cDNA.4 To examine the role of the age-dependent decrease in EGF-R expression in TGF-β1 responsiveness, aged fibroblasts were transfected with cDNA encoding either EGF-R or GFP (as a control). Confirmation of EGF-R overexpression was demonstrated by Q-PCR (Figure 3A). TGF-β1 responsiveness of EGF-R-overexpressing cells was examined by stimulation with TGF-β1 for 72 hours after transfection. There was no difference in α-SMA expression between EGF-R-overexpressing cells and GFP-transfected cells after addition of TGF-β1 (Figure 3B), suggesting that restoration of EGF-R expression in aged cells did not restore TGF-β1 responsiveness. The functional nature of the transfected EGF-R was explored by examining the EGF-dependent proliferative response of EGF-R-overexpressing aged fibroblasts. These experiments demonstrated significantly increased proliferation, as assessed by [3H]thymidine incorporation, after addition of exogenous EGF to EGF-R-overexpressing cells compared with control GFP-transfected cells, thus confirming the functionality of the transfected receptor (Figure 3C).
Figure 3.
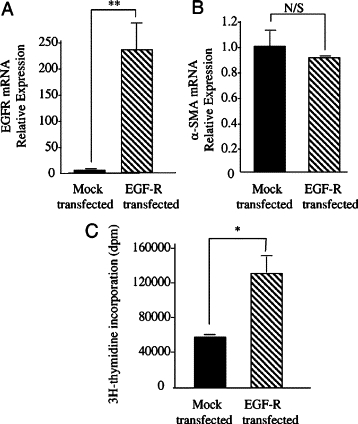
Overexpression of EGF-R in aged dermal fibroblasts. Aged dermal fibroblasts were transfected with either EGF-R-pCR3.1 (EGF-R transfected) or GFP (mock transfected). Transfection efficiency of EGF-R-pCR3.1 was 41.5%. Total mRNA was extracted and EGF-R (A) and α-SMA (B) mRNA expression was assessed by RT-Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assessed relative to mock transfected. The comparative CT method was used for relative quantification of gene expression, and the results represent the mean ± SE of nine individual experiments using cells isolated from three different donors. Statistical analysis was performed by Student's t-test: **P < 0.01. N/S, not significant. C: EGF-induced proliferation was determined by incorporation of [3H]thymidine. Expression plasmids for EGF-R or GFP (mock transfected) were introduced into aged fibroblasts using Nucleofector technology before metabolic labeling, performed by incubation with 1 μCi/ml d-[3H]thymidine after addition of either serum-free medium alone (black bars) or serum-free medium containing 10 ng/ml EGF (gray bars) for 24 hours. Data are the mean ± SE from six individual experiments, using cells isolated from two different donors. Statistical analysis was performed by Student's t-test: *P < 0.05.
TGF-β1-Mediated Phenotypic Activation Is Facilitated by EGF
Synergy between TGF-β1 and EGF in regulating cell phenotype is well recognized in other cell systems.25,29,30 The effect of in vitro aging on the expression of EGF was examined under basal conditions and after stimulation with TGF-β1 (Figure 4A). Fibroblasts were grown to confluence and serum-deprived for 48 hours before addition of either recombinant TGF-β1 or serum-free medium alone (control treatment) for a further 72 hours. Q-PCR analysis revealed that under basal conditions aged cells exhibited a small but significant attenuation in EGF mRNA. Moreover, after addition of TGF-β1, young cells responded with a significant induction (∼7-fold) in EGF mRNA, whereas, in contrast, aged cells demonstrated no change in EGF mRNA expression. These data demonstrate that in addition to the loss of expression of EGF-R, in vitro aging is associated with a loss of TGF-β1-dependent induction of EGF. This result raises the possibility that loss of autocrine EGF activity may also be involved in age-dependent loss of TGF-β1 phenotypic activation. To examine this, aged fibroblasts overexpressing EGF-R were costimulated with exogenous EGF and TGF-β1. In contrast with stimulation with TGF-β1 in isolation (Figure 3B), costimulation leads to augmented phenotypic activation as assessed by expression of α-SMA (Figure 4B). In addition, costimulation leads to a restoration of induction of the hyaladherin TSG-6 (Figure 4C) and HAS2 (Figure 4D), which did not occur after addition of TGF-β1 in isolation.
Figure 4.
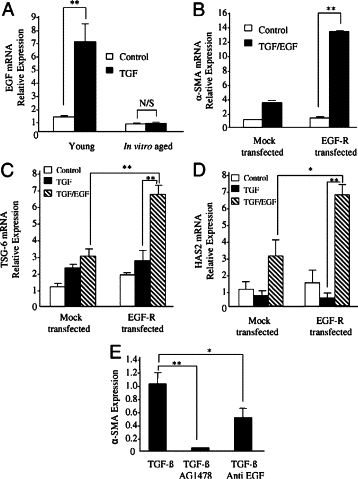
Effect of age on EGF expression (A), synergistic effects of TGF-β1 and EGF in EGF-R-overexpressed aged cells (B–D), and effect of EGF inhibition on phenotypic activation (E). A: Confluent monolayers of patient matched young and aged dermal fibroblasts were growth-arrested in serum-free medium for 48 hours, before addition of either serum-free medium alone (white bars) or serum-free medium containing 10 ng/ml TGF-β1 (black bars) for 72 hours. Total mRNA was extracted, and EGF expression was assessed by RT-Q-PCR. B–D: Aged dermal fibroblasts were transfected either with EGF-R-pCR3.1 (EGF-R transfected) or GFP (mock transfected). Transfection efficiency of cells expressing EGF-R-pCR3.1 was calculated as 42.6% (data not shown). B: 48 hours after transfection, cells were incubated in either serum-free medium alone (white bars) or serum-free medium containing 10 ng/ml TGF-β1 in combination with EGF (10 ng/ml) (black bars) for 72 hours, and α-SMA expression was assessed by RT-Q-PCR. C and D: 48 hours after transfection, cells were incubated in either serum-free medium alone (white bars), serum-free medium containing 10 ng/ml TGF-β1 (black bars), or serum-free medium containing TGF-β1 (10 ng/ml) in combination with EGF (10 ng/ml) (gray bars) for 72 hours. TSG-6 (C) and HAS2 (D) mRNA expression was assessed by RT-Q-PCR. E: confluent monolayers of dermal fibroblasts were growth-arrested in serum-free medium for 48 hours. The medium was replaced with serum-free medium containing TGF-β1 (10 ng/ml) alone or in combination with either the EGF-R inhibitor AG1478 (10 μmol/L) or anti-EGF antibody (10 μg/ml) for 72 hours, and α-SMA expression was assessed by RT-Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assessed relative to in vitro aged control cells (A), mock-transfected control cells (B–D), or untreated cells (E). The comparative CT method was used for relative quantification of gene expression, and the results represent the mean ± SE of six individual experiments using cells isolated from two different donors. Statistical analysis was performed by Student's t-test: *P < 0.05; **P < 0.01. N/S, not significant.
To investigate the participation of EGF in TGF-β1-dependent phenotypic activation young cells were treated with TGF-β1 in the presence of either an anti-EGF antibody or the EGF-R inhibitor AG1478, and the expression of α-SMA was assayed by Q-PCR (Figure 4E). Addition of neutralizing anti-EGF antibody inhibited TGF-β1-mediated α-SMA accumulation by ∼50%, whereas complete inhibition was achieved using the chemical inhibitor AG1478.
HAS2-Dependent HA Is Regulated through the EGF-R
The induction of HAS2 in aged cells after restoration of EGF signaling together with the reported up-regulation of HAS2 and HA synthesis mediated by EGF-R signaling37 led us to investigate the role of EGF signaling in regulation of HAS2 in our cell system. TGF-β1-mediated induction of HAS2 in young fibroblasts was significantly attenuated by treatment with the EGF-R inhibitor AG1478 (Figure 5A). Previous work has demonstrated that HA through CD44 may directly activate EGF-R in the context of oncogenic signaling38,39 To examine the potential of endogenous HA to activate EGF-R in the absence of exogenous EGF, aged fibroblasts overexpressing both EGF-R and HAS2 were stimulated with TGF-β1, and cell phenotype was monitored by quantitation of α-SMA. Under these experimental conditions TGF-β1-dependent phenotypic activation was restored in the absence of exogenous EGF (Figure 5B). This result suggests that in aged cells forced overexpression of HAS2-dependent HA synthesis removes the need for exogenous EGF in phenotypic activation of EGF-R-overexpressing cells. Collectively these data indicate that HAS2 participates in phenotypic activation and is downstream of TGF-β1-mediated EGF synthesis.
Figure 5.
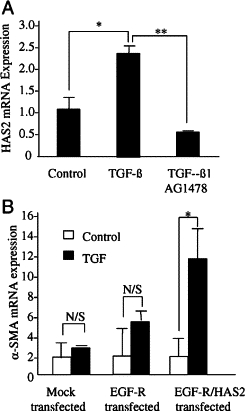
Involvement of EGF-R in TGF-β1-dependent HAS2 induction (A) and combinatory effects of EGF-R and HAS2 overexpression in aged cells (B). A: Confluent monolayers of young dermal fibroblasts were growth-arrested in serum-free medium for 48 hours, before addition of either serum-free medium alone (control) or serum-free medium containing TGF-β1 (10 ng/ml) in isolation or in combination with the EGF-R inhibitor AG1471 (10 μmol/L) for 72 hours. Total mRNA was extracted, and HAS2 expression was assessed by RT-Q-PCR. B: Aged dermal fibroblasts were transfected either with EGF-R-pCR3.1 alone (EGF-R transfected) or in combination with HAS2-pCR3.1 (EGF-R/HAS2 transfected) or GFP (mock transfected). Transfection efficiency of cells expressing EGF-R-pCR3.1 or in combination with HAS2-pCR3.1 was calculated as 44.1 and 37.8%, respectively. At 48 hours after transfection, cells were incubated in either serum-free medium alone (white bars) or serum-free medium containing 10 ng/ml TGF-β1 (black bars) for 72 hours, and α-SMA mRNA expression was assessed by RT-Q-PCR. Ribosomal RNA expression was used as an endogenous control and gene expression was assessed relative to control cells (A) or mock-transfected control cells (B). The comparative CT method was used for relative quantification of gene expression, and the results represent the mean ± SE of six individual experiments using cells isolated from two different donors. Statistical analysis was performed by Student's t-test: *P < 0.05; **P < 0.01. N/S, not significant.
TGF-β1 Activates ERK1/2, through EGF-R, in an HA-Dependent Manner and Is Impaired with Aging
Activation of EGF-R by HA associated with cytoskeletal reorganization in head and neck squamous cell carcinoma cells has been associated with downstream activation of mitogen-activated protein kinase (MAPK) (in particular ERK1 and ERK2).40 In our experimental system addition of TGF-β1 resulted in the activation of ERK1 and ERK2. Western blot analysis demonstrated that maximal phosphorylation was reached at 10 minutes after administration of TGF-β1 and rapidly returned to less than basal levels after 30 minutes (Figure 6A). Significantly, TGF-β1-mediated ERK activation was markedly attenuated in aged cells (Figure 6B), consistent with the age-dependent reduction in EGF-R expression. TGF-β1-mediated activation of ERK could be inhibited by either inhibition of the EGF-R using AG1487 or by an antibody to EGF (Figure 6C), suggesting that TGF-β1-mediated ERK activation was downstream of EGF-mediated activation of the EGF-R. In the same experiments the relationship between Smad2 and EGF-R signaling was investigated. As expected, Smad2 phosphorylation increased after TGF-β1 stimulation. In contrast to the effects of AG1487 and anti-EGF antibody on ERK phosphorylation, Smad2 phosphorylation was unaltered (Figure 6C), suggesting that Smad2 and EGF-R/ERK1/2 phosphorylation are independent.
Figure 6.
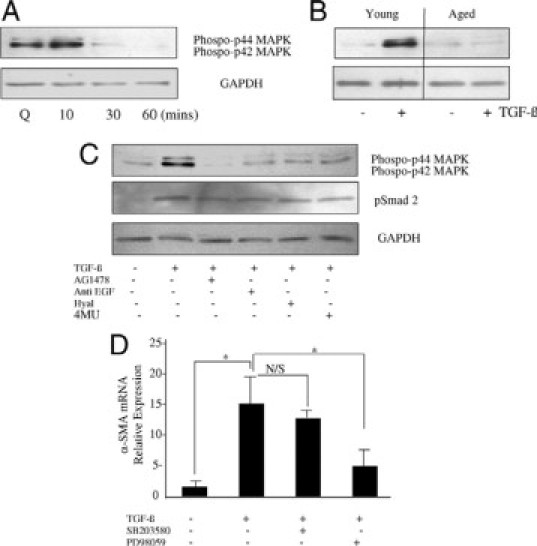
TGF-β1-dependent activation of ERK signaling (A), the effects of in vitro aging (B) involvement of HA and EGF signaling (C), and requirement for phenotypic conversion (D) on ERK activation. A: Growth-arrested confluent monolayers of young dermal fibroblasts (Q) were incubated with 10 ng/ml TGF-β1 for up to 60 minutes, and phosphorylation of ERK1/2 proteins was assessed by Western blot analysis at the time points indicated. B: 10 ng/ml TGF-β1 in serum-free medium was added to growth-arrested confluent monolayers of patient-matched young and in vitro aged dermal fibroblasts for 10 minutes. Cell lysates were prepared for Western blot with antibodies for the phosphorylated form of ERK1/2. C: The involvement of EGF and HA in activation of ERK1/2 phosphorylation and Smad 2 phosphorylation was analyzed by Western blot. Confluent monolayers of growth-arrested young dermal fibroblasts were pretreated with either 10 μmol/L EGF-R inhibitor AG1478 (lane2), 10 μg anti-EGF antibody (lane 3), 200 μg/ml hyaluronidase (Hyal) (lane 4), or 0.5 mmol/L 4MU for 30 minutes before a further 10-minute incubation with 10 ng/ml TGF-β1. Control cells received serum-free medium alone (lane 1). D: Confluent monolayers of young dermal fibroblasts were growth-arrested in serum-free medium for 48 hours before addition of either serum-free medium alone or serum-free medium containing 10 ng/ml TGF-β1 in isolation or in combination with the p38 inhibitor SB203580 (10 μmol/L) or the ERK inhibitor PD98059 (10 μmol/L) for 72 hours. Total mRNA was extracted, and α-SMA mRNA expression was assessed by RT-Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assessed relative to untreated cells. The comparative CT method was used for relative quantification of gene expression, and the results represent the mean ± SE of six individual experiments using cells isolated from two different donors. Statistical analysis was performed by Student's t-test: *P < 0.05. N/S, not significant
We next sought to determine the role of HA in TGF-β1-mediated ERK activation by either inhibition of HA synthesis by the depletion of the UDP-glucuronic acid pool using 4MU or digestion of HA using hyaluronidase. Both inhibition of HA synthesis and degradation of HA inhibited TGF-β1-mediated activation of ERK1 and ERK2 (Figure 6C). The requirement of HA for TGF-directed Smad2 phosphorylation was also investigated in the same way. Neither inhibition of HA synthesis nor degradation of HA had an effect on Smad2 activation, suggesting that the TGF-β-specific Smad pathway is HA-independent (Figure 6C).
Finally, we examined the involvement of ERK1/2 and p38MAPK in myofibroblastic differentiation by TGF-β1. ERK inhibition by the inhibitor PD98059 but not the p38MAPK inhibitor SB203580 significantly inhibited TGF-mediated α-SMA induction in young dermal fibroblasts, suggesting involvement of ERK but not p38MAPK signaling during phenotypic activation (Figure 6D). Collectively these data demonstrate that age-related resistance to TGF-β1-directed phenotypic activation is associated with age-related impaired ERK1/2 signaling.
Phenotype-Associated CD44 Relocation Is Impaired in Aged Cells
Having demonstrated the importance of HA in phenotypic activation by TGF-β1-dependent phosphorylation of ERK1/2, we sought to examine the role of the principal receptor for HA, CD44, in phenotypic activation, and its relationship to age-associated resistance to the effects of TGF-β1. Young fibroblasts were transfected with a specific siRNA for CD44 to determine its functional requirement for myofibroblast conversion. The efficacy of this approach was confirmed by Q-PCR assessment of CD44 mRNA suppression (Figure 7A). Suppression of CD44 expression was associated with abrogation of TGF-β1-dependent induction of α-SMA as assessed by Q-PCR (Figure 7B) and immunohistochemistry (Figure 7, D–G). In contrast, suppression of CD44 expression did not affect TGF-β1-mediated up-regulation of HAS2 (Figure 7C).
Figure 7.
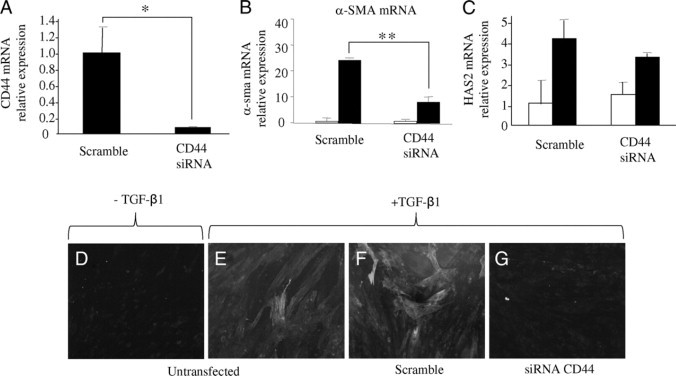
Effect of CD44 gene silencing on TGF-β1-dependent phenotypic activation. Confluent monolayers of young dermal fibroblasts were transfected with CD44 siRNA or scramble oligonucleotide control (scramble) for 48 hours. A: Total mRNA was extracted and CD44 mRNA expression was assessed by RT-Q-PCR. In parallel experiments after transfection the medium was replaced with serum-free medium for a further 24 hours before addition of serum-free medium alone (white bars) or serum-free medium containing 10 ng/ml TGF-β1 (black bars) for a further 72 hours, and α-SMA (B) or HAS2 (C) expression was assessed by RT-Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assessed relative to scramble (A) or control-scrambled (B and C) samples. The comparative CT method was used for relative quantification of gene expression, and the results represent the mean ± SE of six individual experiments using cells isolated from two different donors. Statistical analysis was performed by Student's t-test: *P < 0.05; **P < 0.01. D–G: Immunohistochemical analysis was also performed to assess dermal fibroblast phenotype in untransfected cells (D and E) or cells transfected with either scramble oligonucleotide control (scramble) (F) or CD44 siRNA (G) before addition of serum-free medium with (E–G) or without (D) 10 ng/ml TGF-β1 for a further 72 hours. The cells were fixed and stained for α-SMA, mounted in Vectashield fluorescent mountant and viewed under UV light. Original magnification: ×100.
Expression and localization of CD44 and its relationship to phenotypic activation as assessed by expression of α-SMA were examined by immunohistochemistry (Figure 8, A–F). In young cells, TGF-β1 increased expression of α-SMA (Figure 8B), compared with unstimulated conditions (Figure 8A), which was also associated with a relocalization of CD44. Under basal conditions CD44 was seen predominantly in a linear distribution around the cell periphery (Figure 8D). In contrast, after TGF-β1-dependent phenotypic activation, CD44 appeared in a diffuse pattern throughout the cell (Figure 8E). Aged cells are resistant to TGF-β1-dependent phenotypic activation (Figure 8C), and this resistance is associated with failure of CD44 relocalization, which remains predominantly in a linear distribution at the cell periphery (Figure 8F). Although these data demonstrate a defect in CD44 relocalization associated with age, there was no alteration in CD44 mRNA expression when young and in vitro aged cells were compared (Figure 8G), although in both cell types relocalization of CD44 was associated with a TGF-β1-dependent decrease in CD44 mRNA.
Figure 8.
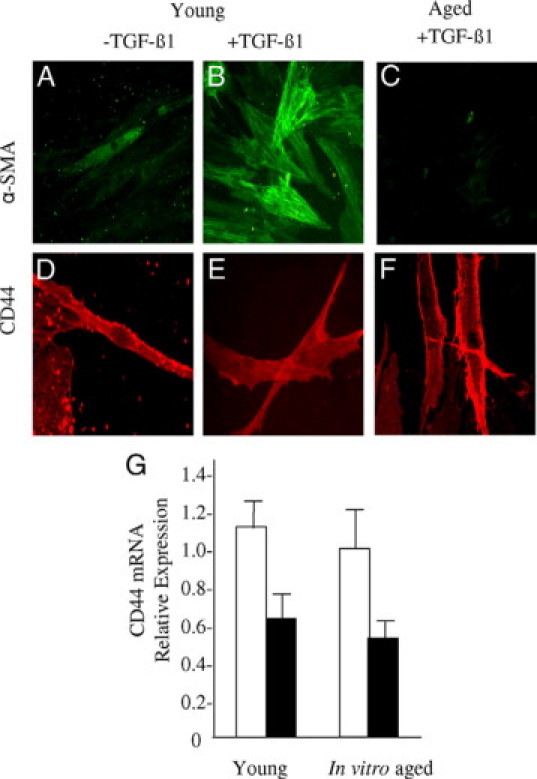
Relationship between fibroblast phenotype and age-related changed in CD44 expression. A–F: Confluent monolayers of patient-matched young (A, B, D, and E) and aged (C and F) dermal fibroblasts were growth-arrested in serum-free medium for 48 hours, before addition of either serum-free medium alone (A and D) or serum-free medium containing 10 ng/ml TGF-β1 (B, C, E, and F). The cells were fixed and stained for α-SMA (A–C) or CD44 (D–F). Cells were mounted in Vectashield fluorescent mountant and viewed under UV light. Representative images of three individual experiments using cells isolated from three different donors are shown. Original magnification: ×100. G: Monolayers of patient-matched young and in vitro aged dermal fibroblasts were growth-arrested in serum-free medium for 48 hours, before addition of either serum-free medium alone (white bars) or serum-free medium containing 10 ng/ml TGF-β (black bars). Total mRNA was extracted, and CD44 expression was assessed by RT-Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assessed relative to in vitro aged control cells. The comparative CT method was used for relative quantification of gene expression, and the results represent the mean ± SE of nine individual experiments using cells isolated from three different donors. Statistical analysis was performed by Student's t-test.
HA-Dependent CD44-EGF-R Colocalization Is Lost with in Vitro Aging
Given the dependence of CD44 and EGF-R in TGF-β1-induced phenotypic activation, we sought to examine their potential for interaction. Anti-CD44 immunoprecipitation followed by anti-EGF-R immunoblot analysis in young cells indicated that a low level of EGF-R is present in the anti-CD44 immunoprecipitated materials (Figure 9A). TGF-β1-dependent phenotypic activation in young cells was associated with recruitment of a significant amount of EGF-R to the CD44-EGF-R complex. Inhibition of HA synthesis using 4MU or digestion of HA using hyaluronidase resulted in a reduction of EGF-R accumulation in the CD44 immunoprecipitant. The effect of cellular aging on the CD44-EGF-R association was subsequently assessed by confocal microscopy (Figure 9, B and Q). The results clearly demonstrate colocalization of CD44 and EGF-R after TGF-β1-dependent phenotypic activation of young cells (Figure 9I) compared with nonstimulated cells (Figure 9E). In contrast, colocalization of CD44 and EGF-R was not apparent in aged fibroblasts (Figure 9, M and Q). Furthermore and consistent with earlier data, unstimulated (Figure 9L) and stimulated (Figure 9P) aged fibroblasts stained poorly for EGF-R. These results suggest that an age-dependent defect in TGF-β1-mediated phenotypic activation is associated with an age-dependent loss of EGF-R and subsequent CD44-EGF-R colocalization.
Figure 9.
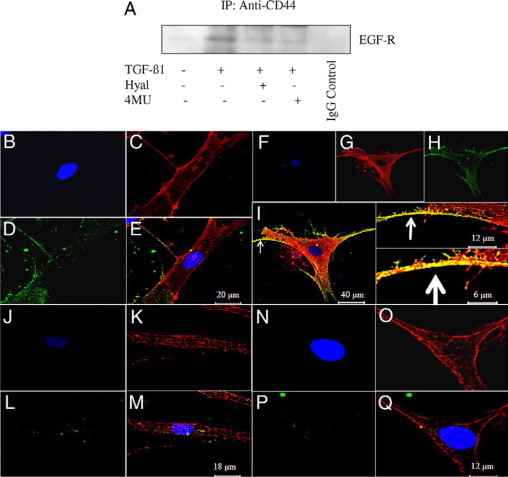
Colocalization of CD44 and EGFR and impact of HA disruption. A: Western blot analysis of the association of CD44 and EFGR receptors. Cell protein was extracted from confluent monolayers of serum-deprived young dermal fibroblasts exposed to either serum-free medium alone (lane 1) or serum-free medium containing 10 ng/ml TGF-β1 in isolation (lane 2) or in combination with hyaluronidase (Hyal) (200 μg/ml) (lane 3) or 4MU (0.5 mmol/L) (lane 4) for 30 minutes. Subsequently samples were immunoprecipitated (IP) with anti-CD44 antibody, followed by immunoblotting with anti-EGF-R antibody. Specificity of immunoprecipitation was confirmed by negative control reactions performed with an IgG control (lane 5). B–Q: Immunohistochemical analysis of CD44 and EGF-R colocalization. Confluent monolayers of patient-matched young (B–I) and aged (J–Q) dermal fibroblasts were growth-arrested in serum-free medium for 48 hours, before addition of either serum-free medium alone (B–E and J–M) or serum-free medium containing 10 ng/ml TGF-β1 (F–I and N–Q). Cells were fixed and the expression of CD44 (C, G, K, and O) and EGF-R (D, H, L, and P) was examined by confocal section series using the Leica TCS SP2 AOBS confocal microscope, and their association was examined by merging of individual images (E, I, M, and Q). CD44 label is red, EGF-R is green, nuclei are counterstained blue with 4,6-diamidino-2-phenylindole (B, F, J, and N), and colocalization is represented by yellow. Arrows indicate colocalization at different magnification.
The importance of HAS2-dependent HA in loss of phenotypic activation and CD44 relocalization was examined by TGF-β1 stimulation of cells transiently transfected with HAS2 (Figure 10, D–F) or EGF-R (Figure 10, G–I) overexpression vectors either alone or in combination (Figure 10, J–L). Transfection with GFP was used as a control (Figure 10, A–C). In these experiments the combination of EGF-R and HAS2 overexpression (but not independently) restored colocalization of CD44 and EGF-R in aged cells. To identify whether this CD44-EGF-R interaction plays a role in signaling to ERK young cells were treated with an anti-CD44 blocking antibody, and ERK1/2 phosphorylation was assessed (Figure 10M). Both basal and TGF-β1-augmented ERK1/2 activation was inhibited by the anti-CD44 blocking antibody; this result together with earlier data implies that ERK1/2 activation is downstream of HA-dependent CD44 EGF-R association.
Figure 10.
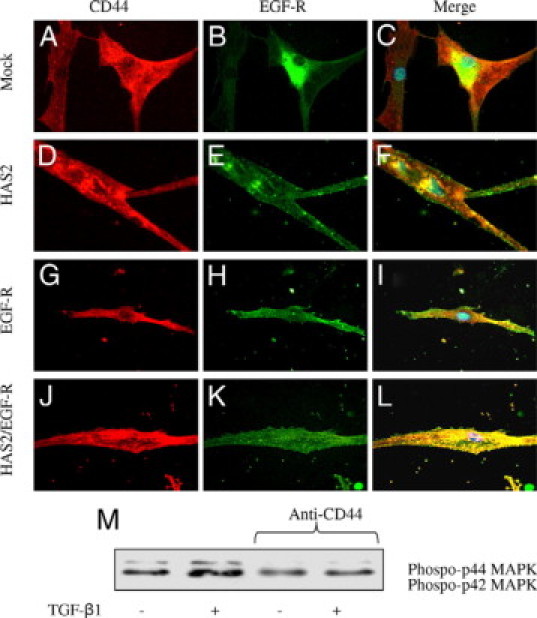
Effect of HAS2 and EGF-R overexpression in aged cells on colocalization of CD44 and EGF-R. A–L: Aged dermal fibroblasts were transfected either with GFP (mock transfected) (A–C), HAS2-pCR3.1 (HAS2 transfected) (D–F), EGF-R-pCR3.1 (EGF-R transfected) (G–I), or the combination of HAS2-pCR3.1 and EGF-R-pCR3.1 (J–L). Cells were growth-arrested in serum-free medium for 48 hours before addition of serum-free medium containing 10 ng/ml TGF-β1 for a further 72 hours. The cells were fixed, and the expression of CD44 (red) and EGF-R (green) was examined by immunocytochemical analysis and their association was examined by merging of individual images (yellow). In merged images 4,6-diamidino-2-phenylindole was used to visualize the nuclei (blue). Original magnification: ×100. M: confluent monolayers of young dermal fibroblasts were growth-arrested in serum-free medium for 48 hours, before addition of either serum-free medium alone or serum-free medium containing 10 ng/ml TGF-β1 as indicated for 10 minutes in the presence or absence of a blocking antibody to CD44 (final concentration 5 μg/ml). Cell lysates were prepared and subjected to Western blot analysis for the phosphorylated form of ERK1/2.
Discussion
Impaired wound healing in elderly individuals is characterized by delayed re-epithelialization and an excessive inflammatory response, the latter leading to matrix degradation by inflammatory cell-derived proteases.41 It has been long established that there is marked alteration in fibroblast function with increasing age. For example in young patients, the fibroblast is an active secretory cell, with a well developed endoplasmic reticulum. In contrast, in old patients (physiological or pathological aging) it becomes a quiescent cell, with a poorly developed endoplasmic reticulum.42,43 The mitogenic and stimulatory effects of growth factors, hormones, and other agents are reduced significantly during cellular aging, and the sensitivity of aging cells to toxic agents including antibiotics, phorbol esters, radiation, and heat shock increases.44 In normal wound healing, fibroblasts are recruited from the surrounding intact tissue into the granulation tissue to proliferate, and mitogenesis is critical for wound repair. Fibroblast proliferation is regulated by activation of the EGF-R, the levels of which in dermal fibroblasts have been seen to decline in aging related to alterations in receptor processing and reduced EGF-R mRNA transcription with this decline correlating with decreased mitogenic responsiveness to EGF.4,45
In addition to the regulation of cell proliferation, the regulation of fibroblast phenotype and differentiation during tissue injury is an important determinant of wound healing outcomes. It is well established that activated fibroblasts, called myofibroblasts, are ultimately responsible for closure of wounds and for the formation of the collagen-rich scar. These cells represent a subpopulation of specialized fibroblasts that have developed a contractile phenotype expressed in a number of pathological settings associated with wound healing and fibrosis.3 We have previously demonstrated an age-dependent resistance to TGF-β1-mediated phenotypic activation of dermal fibroblasts, a functional change that may contribute to age-related impaired wound healing.6 We have demonstrated that phenotypic conversion of fibroblasts to myofibroblasts is associated with major changes in the production and metabolism of HA.46 Acquisition of the myofibroblast phenotype is associated with induction of the HAS2 isoform and accumulation of large HA pericellular matrices. Furthermore, inhibition of HA synthesis results in abrogation of TGF-β1-mediated myofibroblastic phenotypic conversion.7 In addition, we have demonstrated that maintenance of fibroblast phenotype also requires continued HA synthesis.47 Age-associated resistance to phenotypic activation is associated with decreased HAS2 expression and failure of its induction by TGF-β1. Although the role of HAS2 is integral to phenotypic activation, restoration of HAS2 expression in aged cells failed to rescue the myofibroblastic phenotype.6
This article demonstrates that, in addition to HAS2, functional EGF-R is required for TGF-β1-stimulated phenotypic activation of fibroblasts, and both are lost with in vitro aging. Furthermore, the results demonstrate the need for a synergistic activation of TGF-β receptor-associated signaling and indirect activation of the EGF-R by TGF-β1. This synergy is supported by suppression of TGF-β1-driven phenotypic activation by either inhibition of Smad signaling or inhibition of the activity of the EGF-R using AG1478. These data therefore further emphasize the importance of the age-dependent decrease in the expression of EGF-R in age-dependent alterations in fibroblast functions such as proliferation as well as regulation of phenotypic activation, two aspects of dermal fibroblast behavior that are fundamentally important to wound healing. The data also suggest that activation of the EGF-R is related to TGF-β1-dependent stimulation of EGF. Age-associated decreased activation of the EGF-R is related to both decreased expression of EGF-R, as demonstrated previously,4 and also abrogation of TGF-β1-stimulated EGF synthesis. In support of this result we were able to partially restore age-dependent TGF-β1 phenotypic activation by overexpression of the EGF-R and costimulation with TGF-β1 and EGF.
With regard to the role of HAS2-dependent HA regulation in the regulation of fibroblast phenotypic activation, the data presented build on our previously published results, demonstrating loss of HAS2 induction by TGF in aged cells.6 We now report that HAS2-dependent HA synthesis is regulated by EGF-R signaling, as pharmacological blockade by AG1478 attenuates its TGF-dependent up-regulation. Furthermore, the data suggest that EGF represents an intermediate signaling molecule for TGF-β1-dependent induction of HAS2. The involvement of the EGF-R pathway was implicated previously in mediation of HA synthesis in epidermal keratinocytes,37 and here we demonstrate that age-associated EGF-R loss may directly disrupt HAS2-dependent HA accumulation.
The data presented also suggest that HA itself facilitates subsequent EGF-R signaling, which may occur in a ligand/EGF-independent way. The potential involvement of HA and its principle receptor CD44 in modulation of EGF-dependent responses is well recognized in tumor cells. For example, in the absence of exogenous EGF, the EGF-R can be directly activated by HA through EGF-R interactions with CD44.40 In the context of wound healing, EGF-dependent motogenic responses of dermal fibroblasts require functionality of the HA receptor CD44.48 The relationship between EGF signaling and HAS2-dependent HA synthesis, however, has not been previously explored in the context of age-associated defects in wound healing and fibroblast function. The demonstration that the need for exogenous EGF could be overcome by overexpression of HAS2 in this study also suggests that the EGF-R could be activated by the extracellular matrix polysaccharide HA, although whether this activation is the result of direct receptor activation or occurs through HA-dependent activation of its own receptor remains to be determined. CD44 is the main hyaluronan-binding receptor and with HA it participates in many vital functions such as migration, adhesion proliferation, and differentiation. Using a CD44-specific siRNA strategy, we showed that down-regulation of CD44 inhibits HA-mediated phenotypic activation of young cells. With age, there is a failure of CD44 relocalization, which in the young cells, is associated with TGF-β1 stimulation and phenotypic activation. In the young cells, this relocalization is associated with colocalization of CD44 and EGF-R as demonstrated by our immunoprecipitation and immunohistochemistry. Inhibition of HA synthesis, however, prevents colocalization of CD44 and EGF-R, suggesting that HA binding to CD44 directly regulates this functional coupling. Taken together these data demonstrate that HA may drive an increase in the association of CD44 and the EGF receptor, which subsequently facilitates EGF-R signaling. Disruption of this mechanism due to a deficit in HAS2 and EGF-R contributes to age-related impairment of fibroblast to myofibroblast activation because overexpression of both components in aged cells restores phenotypic activation with associated restoration of CD44-EGF-R colocalization as illustrated by our immunofluorescence data.
Previous studies on the pathogenesis of head and neck squamous cell carcinomas have demonstrated direct activation of EGF-R tyrosine kinase activity by the addition of exogenous HA, which in turn stimulates downstream MAPK (in particular ERK1 and ERK2) pathways and carcinoma cell growth.40 As with our data this was coupled with an increase in the association of CD44 and the EGF-R. Studies in rat hepatocytes have demonstrated an age-related decline in EGF-stimulated MAPK activation and in particular ERK2 is associated with an age-related decline in proliferative capacity.49 Consistent with these studies, we have demonstrated phosphorylation of ERK1 and ERK2 downstream of EGF-mediated EGF-R activation because AG1478 and anti-EGF antibody suppressed ERK1/2 phosphorylation. The activation pattern of ERK1/2 was rapid and short-lived in response to TGF-β1, but, despite this, using the ERK inhibitor PD98059 we clearly demonstrated the significance of this pathway in phenotypic conversion. TGF-β1-dependent activation of ERK1/2 is dependent on HA and CD44 because HA disruption or CD44 inhibition using an anti-CD44 blocking antibody suppressed ERK1/2 phosphorylation. Furthermore, consistent with the age-dependent decrease in EGF-R expression and its association with CD44, there is an age-dependent decrease in TGF-β1-dependent activation of ERK1 and 2. This signaling pathway is, however, distinct from EGF/ligand activation of EGF-R, because CD44 blockade, although it inhibits ERK1/2 phosphorylation, does not influence HAS2 expression, which is also EGF-R-dependent.
When phosphorylated, ERK may translocate to the nucleus and activate transcription factors such as signal transducer and activator of transcription 3, which has been reported to facilitate expression of HAS2, hyaluronan synthesis, and CD44 expression in epidermal keratinocytes.50 In a recent study HAS2 activation was reported to be downstream of ERK signaling,51 but although this mechanism may provide a more rational explanation for the rapid activation of ERK demonstrated in this article, use of the ERK inhibitor PD98059 had no impact on HAS2 induction (data not shown). Collectively the results of the current study suggest that HA-dependent CD44-EGFR signaling is a prerequisite for ERK signaling and subsequent phenotypic activation in fibroblasts. Further investigation is needed, therefore, to elucidate the signaling events downstream of ERK1/2 activation and the kinetics involved in regulation of fibroblast phenotype by TGF-β1 together with the interactions of the various signaling intermediates (EGF-HAS2-HA-CD44-EGF-R) implicated in this study.
The EGF-R is a transmembrane glycoprotein that constitutes one of four closely related members of the erbB family of tyrosine kinase receptors: EGF-R (ErbB-1), HER2/c-neu (ErbB-2), Her 3 (ErbB-3), and Her 4 (ErbB-4).52 Although this study has focused on EGF-R (ErbB-1), its potential for interaction with other members of the erbB superfamily has been demonstrated.53 There is growing evidence that in glioma cell lines, for example, there is dual interaction with CD44 by both EGF-R and ErbB-2 that is comparable with the mechanism proposed in this communication.27 Furthermore, ErbB-2-ERK signaling and HAS regulation are functionally coupled in human ovarian tumor cells.51 It would therefore be interesting to extend the present data to include the regulation of fibroblast phenotype by HER2.
In the current study and consistent with our previous observations,36 Smad2 but not Smad3 activation was demonstrated to be essential for TGF-β1-mediated cellular activation. TGF-β1 mobilizes both Smad-dependent and Smad-independent signaling.24 In the current study we have demonstrated that activation of Smad2 and EGF-R/ERK1/2 pathways work in a complementary way to coordinate TGF-β1-mediated α-SMA induction. Phosphorylation of Smad2 was not affected by the anti-EGF antibody AG1478, HA disruption by hyaluronidase, or 4MU, signifying that Smad signaling is independent of EGF-R signaling and its modulation by HA. Consistent with this finding is the divergent effect of aging on these pathways as the TGF-β1-dependent activation of Smad is unaffected by cellular aging. We and others have demonstrated that cross-talk between ERK MAPK and Smad signaling pathways may enhance TGF-β1-dependent responses in other cell systems.54,55 To our knowledge this is the first time that the synergistic involvement of these signaling pathways has been implicated in the impaired regulation of dermal fibroblast-myofibroblast activation in the context of cellular aging.
In summary, the data presented provide a mechanism to explain age-dependent resistance of dermal fibroblasts to phenotypic activation. Figure 11 illustrates our current proposed model.
Figure 11.
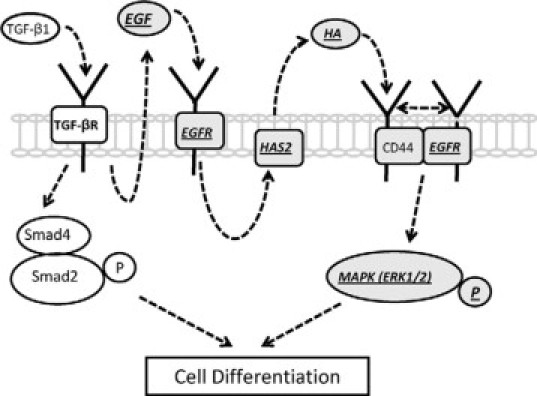
Proposed model for the signal transduction pathways involved in TGFβ1-mediated fibroblast-myofibroblast differentiation. In young dermal fibroblasts TGF-β1-dependent phenotypic activation uses two distinct but cooperating pathways that involve TGF-β receptor (TGF-βR)/Smad2 activation and EGF-mediated EGF-R/ERK1/2 signaling, and the latter is compromised with in vitro aging. We propose that induction of HAS2-dependent HA is mediated through EGF-R via EGF and subsequent binding of HA to CD44 facilitates its association with the EGF-R. This association triggers phosphorylation of the p42/p44 MAPKs, ERK1 and ERK2, necessary for promoting cellular differentiation. Various aspects of this signaling cascade (underlined and highlighted in italic text) are impaired with in vitro aging. This impairment includes decreased expression of EGF-R, suppression of TGF-β1-dependent EGF synthesis, and a resultant failure of induction of HAS2-dependent HA. We propose that deficient HA-CD44 interaction leads to loss of CD44-EGFR colocalization and subsequent downstream ERK signaling. Collectively, age-related defects in this pathway confer resistance to cellular differentiation, and this may play an important role in age-related impaired wound healing.
TGF-β1-dependent fibroblast-myofibroblast differentiation involves two distinct but cooperating pathways that involve TGF-β1-Smad2 activation and HA-dependent EGF-R-ERK1/2 signaling and the latter is compromised during in vitro aging. In a young cell TGF-β1-mediated activation of EGF-R via EGF is associated with accumulation of HAS2-induced HA. We propose that binding of HA to its receptor CD44 promotes its association with EGF-R, leading to ERK1/2 phosphorylation and subsequent cellular differentiation. With age, numerous aspects of this process are impaired, including decreased expression of EGF-R, suppression of TGF-β1-dependent EGF synthesis, and the resultant failure of induction of HAS2. The result of this is selective impairment of the ERK1/2 signaling pathway and resistance to phenotypic maturation. These findings may have a direct implication in impaired wound healing associated with aging. Our data add further support to the hypothesis that the matrix polysaccharide HA is a key component of the regulation of fibroblast phenotype and that its dysregulation may be casually related to age-dependent failure of a fibroblast to acquire a myofibroblastic phenotype.
Acknowledgements
We acknowledge the help of Mr. Marc Isaacs, Dr. Anthony Hayes, and Dr. Jim Ralphs (Cardiff University School of Biosciences) for generation of the confocal images.
Footnotes
Supported in part by a Ph.D. studentship from Research into Ageing (to R.S.).
R.S. and A.P. contributed equally to this work.
References
- 1.Ashcroft GS, Mills SJ, Ashworth JJ. Ageing and wound healing. Biogerontology. 2002;3:337–345. doi: 10.1023/a:1021399228395. [DOI] [PubMed] [Google Scholar]
- 2.Ashcroft GS, Roberts AB. Loss of Smad3 modulates wound healing. Cytokine Growth Factor Rev. 2000;11:125–131. doi: 10.1016/s1359-6101(99)00036-2. [DOI] [PubMed] [Google Scholar]
- 3.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- 4.Shiraha H, Gupta K, Drabik K, Wells A. Aging fibroblasts present reduced epidermal growth factor (EGF) responsiveness due to preferential loss of EGF receptors. J Biol Chem. 2000;275:19343–19351. doi: 10.1074/jbc.M000008200. [DOI] [PubMed] [Google Scholar]
- 5.Ashcroft GS, Horan MA, Ferguson MW. The effects of ageing on cutaneous wound healing in mammals. J Anat. 1995;187:1–26. [PMC free article] [PubMed] [Google Scholar]
- 6.Simpson R, Meran S, Thomas D, Stephens P, Bowen T, Steadman R, Phillips A. Age related changes in peri-cellular hyaluronan organisation leads to impaired dermal fibroblast to myofibroblast differentiation. Am J Pathol. 2009;175:1915–1928. doi: 10.2353/ajpath.2009.090045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meran S, Thomas D, Stephens P, Martin J, Bowen T, Phillips A, Steadman R. Involvement of hyaluronan in regulation of fibroblast phenotype. J Biol Chem. 2007;282:25687–25697. doi: 10.1074/jbc.M700773200. [DOI] [PubMed] [Google Scholar]
- 8.Meran S, Thomas DW, Stephens P, Enoch S, Martin J, Steadman R, Phillips AO. Hyaluronan facilitates transforming growth factor-β1-mediated fibroblast proliferation. J Biol Chem. 2008;283:6530–6545. doi: 10.1074/jbc.M704819200. [DOI] [PubMed] [Google Scholar]
- 9.Webber J, Jenkins RH, Meran S, Phillips A, Steadman R. Modulation of TGFβ1-dependent myofibroblast differentiation by hyaluronan. Am J Pathol. 2009;175:148–160. doi: 10.2353/ajpath.2009.080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spicer AP, Kaback LA, Smith TJ, Seldin MF. Molecular cloning and characterisation of the human and mouse UDP-glucose dehydrogenase genes. J Biol Chem. 1998;271:25117–25124. doi: 10.1074/jbc.273.39.25117. [DOI] [PubMed] [Google Scholar]
- 11.Spicer AP, McDonald JA. Characterisation and molecular evolution of a vertebrate hyaluronan synthase gene family. J Biol Chem. 1998;272:1923–1932. doi: 10.1074/jbc.273.4.1923. [DOI] [PubMed] [Google Scholar]
- 12.Kosaki R, Watanabe K, Yamaguchi Y. Overproduction of hyaluronan by expression of the hyaluronan synthase Has2 enhances anchorage independent growth. Cancer Res. 1999;59:1141–1145. [PubMed] [Google Scholar]
- 13.Itano N, Atsumi F, Sawai T, Yamada Y, Miyaishi O, Senga T, Hamaguchi M, Kimata K. Abnormal accumulation of hyaluronan matrix diminishes contact inhibition of cell growth and promotes cell migration. Proc Natl Acad Sci USA. 2002;99:3609–3614. doi: 10.1073/pnas.052026799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ito T, Williams JD, Al-Assaf S, Phillips GO, Phillips AO. Hyaluronan and proximal tubular epithelial cell migration. Kidney Int. 2004;65:823–833. doi: 10.1111/j.1523-1755.2004.00457.x. [DOI] [PubMed] [Google Scholar]
- 15.Legg JW, Lewis CA, Parsons M, Ng T, Isacke CM. A novel PKC-regulated mechanism controls CD44 ezrin association and directional cell motility. Nat Cell Biol. 2002;4:399–407. doi: 10.1038/ncb797. [DOI] [PubMed] [Google Scholar]
- 16.Zoltan-Jones A, Huang L, Ghatak S, Toole BP. Elevated hyaluronan production induces mesenchymal and transformed properties in epithelial cells. J Biol Chem. 2003;278:45801–45810. doi: 10.1074/jbc.M308168200. [DOI] [PubMed] [Google Scholar]
- 17.Brecht M, Mayer U, Schlosser E, Prehm P. Increased hyaluronate synthesis is required for fibroblast detachment and mitosis. Biochem J. 1986;239:445–450. doi: 10.1042/bj2390445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evanko SP, Angello JC, Wight TN. Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:1004–1013. doi: 10.1161/01.atv.19.4.1004. [DOI] [PubMed] [Google Scholar]
- 19.Ito T, Williams JD, Fraser DJ, Phillips AO. Hyaluronan attenuates TGF-β1 mediated signalling in renal proximal tubular epithelial cells. Am J Pathol. 2004;164:1979–1988. doi: 10.1016/s0002-9440(10)63758-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito T, Williams JD, Fraser DJ, Phillips AO. Hyaluronan regulates TGF-β1 receptor compartmentalisation. J Biol Chem. 2004;279:25326–25332. doi: 10.1074/jbc.M403135200. [DOI] [PubMed] [Google Scholar]
- 21.Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 22.Miyazawa K, Shinozaki M, Hara T, Furuya T, Miyazono K. Two major Smad pathways in TGF-β superfamily signalling. Genes Cells. 2002;7:1191–1204. doi: 10.1046/j.1365-2443.2002.00599.x. [DOI] [PubMed] [Google Scholar]
- 23.Bourguignon LY, Singleton PA, Zhu H, Zhou B. Hyaluronan promotes signaling interaction between CD44 and the transforming growth factor β receptor I in metastatic breast tumor cells. J Biol Chem. 2002;277:39703–39712. doi: 10.1074/jbc.M204320200. [DOI] [PubMed] [Google Scholar]
- 24.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 25.Grände M, Franzen A, Karlsson JO, Ericson LE, Heldin NE, Nilsson M. Transforming growth factor-β and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J Cell Sci. 2002;115:4227–4236. doi: 10.1242/jcs.00091. [DOI] [PubMed] [Google Scholar]
- 26.Song K, Krebs TL, Danielpour D. Novel permissive role of EGF in TGF-β signaling and growth suppression: Mediation by stabilization of TGF-β receptor type II. J Biol Chem. 2006;281:7765–7774. doi: 10.1074/jbc.M511781200. [DOI] [PubMed] [Google Scholar]
- 27.Docherty NG, O'Sullivan OE, Healy DA, Murphy M, O'Neill AJ, Fitzpatrick JM, Watson RWG. TGF-β1-induced EMT can occur independently of its proapoptotic effects and is aided by EGF receptor activation. Am J Physiol Renal Physiol. 2006;290:F1202–F1212. doi: 10.1152/ajprenal.00406.2005. [DOI] [PubMed] [Google Scholar]
- 28.Tian Y-C, Chen Y-C, Chang C-T, Hung C-C, Wu M-S, Phillips A, Yang C-W. Epidermal growth factor and transforming growth factor-β1 enhance HK-2 cell migration through a synergistic increase of matrix metalloproteinase and sustained activation of ERK signaling pathway. Exp Cell Res. 2007;313:2367–2377. doi: 10.1016/j.yexcr.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 29.Uttamsingh S, Bao X, Nguyen KT, Bhanot M, Gong J, Chan JL-K, Liu FY, Chu TT. Synergistic effect between EGF and TFG-β1 in inducing oncogenic properties of intestinal epithelial cells. Oncogene. 2008;27:2626–2634. doi: 10.1038/sj.onc.1210915. [DOI] [PubMed] [Google Scholar]
- 30.He J, Bazan HEP. Epidermal growth factor synergism with TGF-β1 via PI-3 kinase activity in corneal keratocyte differentiation. Invest Ophthalmol Vis Sci. 2008;49:2936–2945. doi: 10.1167/iovs.07-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsatas D, Kanagasundaram V, Kaye A, Novak U. EGF receptor modifies cellular responses to hyaluronan in glioblastoma cell lines. J Clin Neurosci. 2002;9:282–288. doi: 10.1054/jocn.2001.1063. [DOI] [PubMed] [Google Scholar]
- 32.Vigetti D, Viola M, Karousou E, Rizzi M, Moretto P, Genasetti A, Clerici M, Hascall VC, De Luca G, Passi A. Hyaluronan-CD44-ERK1/2 regulate human aortic smooth muscle cell motility during aging. J Biol Chem. 2008;283:4448–4458. doi: 10.1074/jbc.M709051200. [DOI] [PubMed] [Google Scholar]
- 33.Vigetti D, Moretto P, Viola M, Genasetti A, Rizzi M, Karousou E, Pallotti F, De Luca G, Passi A. Matrix metalloproteinase 2 and tissue inhibitors of metalloproteinases regulate human aortic smooth muscle cell migration during in vitro aging. FASEB J. 2006;20:1118–1130. doi: 10.1096/fj.05-4504com. [DOI] [PubMed] [Google Scholar]
- 34.Stephens P, Davies KJ, Occleston N, Pleass RD, Kon C, Daniels J, Khaw PT, Thomas DW. Skin and oral fibroblasts exhibit phenotypic differences in extracellular matrix reorganization and matrix metalloproteinase activity. Br J Dermatol. 2001;144:229–237. doi: 10.1046/j.1365-2133.2001.04006.x. [DOI] [PubMed] [Google Scholar]
- 35.Cristofalo VJ, Allen RG, Pignolo RJ, Martin BG, Beck JC. Relationship between donor age and the replicative lifespan of human cells in culture: a reevaluation. Proc Natl Acad Sci USA. 1998;95:10614–10619. doi: 10.1073/pnas.95.18.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans RA, Tian YC, Steadman R, Phillips AO. TGF-β1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp Cell Res. 2003;282:90–100. doi: 10.1016/s0014-4827(02)00015-0. [DOI] [PubMed] [Google Scholar]
- 37.Saavalainen K, Pasonen-Seppanen S, Dunlop TW, Tammi R, Tammi MI, Carlberg C. The human hyaluronan synthase 2 gene is a primary retinoic acid and epidermal growth factor responding gene. J Biol Chem. 2005;280:14636–14644. doi: 10.1074/jbc.M500206200. [DOI] [PubMed] [Google Scholar]
- 38.Wang SJ, Bourguignon LYW. Hyaluronan and the interaction between CD44 and epidermal growth factor receptor in oncogenic signaling and chemotherapy resistance in head and neck cancer. Arch Otolaryngol Head Neck Surg. 2006;132:771–778. doi: 10.1001/archotol.132.7.771. [DOI] [PubMed] [Google Scholar]
- 39.Humes HD, Beals TF, Cieslinski DA, Sanchez IO, Page TP. Effects of transforming growth factor-β, and transforming growth factor-α, and other growth factors on renal proximal tubule cells. Lab Invest. 1991;64:538–545. [PubMed] [Google Scholar]
- 40.Bourguignon LYW, Gilad E, Brightman A, Diedrich F, Singleton P. Hyaluronan-CD44 interaction with leukemia-associated RhoGEF and epidermal growth factor receptor promotes Rho/Ras co-activation. Phospholipase Cɛ-Ca2+ signaling, and cytoskeleton modification in head and neck squamous cell carcinoma cells. J Biol Chem. 2006;281:14026–14040. doi: 10.1074/jbc.M507734200. [DOI] [PubMed] [Google Scholar]
- 41.Ashcroft GS, Horan MA, Ferguson MW. Aging alters the inflammatory and endothelial cell adhesion molecule profiles during human cutaneous wound healing. Lab Invest. 1998;78:47–58. [PubMed] [Google Scholar]
- 42.Bruce SA, Deamond SF. Longitudinal study of in vivo wound repair and in vitro cellular senescence of dermal fibroblasts. Exp Gerontol. 1991;26:17–27. doi: 10.1016/0531-5565(91)90058-t. [DOI] [PubMed] [Google Scholar]
- 43.Pieraggi MJ, Bouissou H, Angelier C, Uhart D, Magnol J, Kokolo J. Le Fibroblaste. Ann Pathol. 1985;5:65–76. [PubMed] [Google Scholar]
- 44.Rattan SI, Derventzi A. Altered cellular responsiveness during ageing. BioEssays. 1991;13:601. doi: 10.1002/bies.950131111. 601. [DOI] [PubMed] [Google Scholar]
- 45.Reenstra WR, Yaar M, Gilchrest BA. Aging affects epidermal growth factor receptor phosphorylation and traffic kinetics. Exp Cell Res. 1996;227:252–255. doi: 10.1006/excr.1996.0274. [DOI] [PubMed] [Google Scholar]
- 46.Jenkins RH, Thomas GJ, Williams JD, Steadman R. Myofibroblastic differentiation leads to hyaluronan accumulation through reduced hyaluronan turnover. J Biol Chem. 2004;279:41453–41460. doi: 10.1074/jbc.M401678200. [DOI] [PubMed] [Google Scholar]
- 47.Webber J, Meran S, Steadman R, Phillips AO. Hyaluronan orchestrates TGF-β1 dependent maintenance of myofibroblast phenotype. J Biol Chem. 2009;284:9083–9092. doi: 10.1074/jbc.M806989200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ellis IR, Schor AM, Schor SL. EGF AND TGF-α motogenic activities are mediated by the EGF receptor via distinct matrix-dependent mechanisms. Exp Cell Res. 2007;313:732–741. doi: 10.1016/j.yexcr.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 49.Liu Y, Guyton KZ, Gorospe M, Xu Q, Kokkonen GC, Mock YD, Roth GS, Holbrook NJ. Age-related decline in mitogen-activated protein kinase activity in epidermal growth factor-stimulated rat hepatocytes. J Biol Chem. 1996;271:3604–3607. [PubMed] [Google Scholar]
- 50.Pasonen-Seppänen SM, Maytin EV, Torronen KJ, Hyttinen JM, Hascall VC, MacCallum DR, Kultti AH, Jokela TA, Tammi MI, Tammi RH. All-trans retinoic acid-induced hyaluronan production and hyperplasia are partly mediated by EGFR signaling in epidermal keratinocytes. J Invest Dermatol. 2008;128:797–807. doi: 10.1038/sj.jid.5701098. [DOI] [PubMed] [Google Scholar]
- 51.Bourguignon LYW, Gilad E, Peyrollier K. Heregulin-mediated ErbB2-ERK signaling activates hyaluronan synthases leading to CD44-dependent ovarian tumor cell growth and migration. J Biol Chem. 2007;282:19426–19441. doi: 10.1074/jbc.M610054200. [DOI] [PubMed] [Google Scholar]
- 52.Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59:S21–S26. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 53.Wells A. EGF receptor. Int J Biochem Cell Biol. 1999;31:637–643. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- 54.Hayashida T, Decaestecker M, Schnaper HW. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-β-dependent responses in human mesangial cells. FASEB J. 2003;17:1576–1578. doi: 10.1096/fj.03-0037fje. [DOI] [PubMed] [Google Scholar]
- 55.Zhang M, Fraser D, Phillips A. ERK, p38, and Smad signaling pathways differentially regulate transforming growth factor-β1 autoinduction in proximal tubular epithelial cells. Am J Pathol. 2006;169:1282–1293. doi: 10.2353/ajpath.2006.050921. [DOI] [PMC free article] [PubMed] [Google Scholar]