

Abstract
Biliary obstruction results in a well-characterized cholestatic inflammatory and fibrogenic process; however, the mechanisms and potential for liver repair remain unclear. We previously demonstrated that Kupffer cell depletion reduces polymorphonuclear cell (neutrophil) (PMN) and matrix metalloproteinase (MMP)8 levels in repairing liver. We therefore hypothesized that PMN-dependent MMP activity is essential for successful repair. Male Sprague-Dawley rats received reversible biliary obstruction for 7 days, and the rat PMN-specific antibody RP3 was administered 2 days before biliary decompression (repair) and continued daily until necropsy, when liver underwent morphometric analysis, immunohistochemistry, quantitative RT-PCR, and in situ zymography. We found that RP3 treatment did not reduce Kupffer cell or monocyte number but significantly reduced PMN number at the time of decompression and 2 days after repair. RP3 treatment also blocked resorption of type I collagen. In addition, biliary obstruction resulted in increased expression of MMP3, MMP8, and tissue inhibitor of metalloproteinase 1. Two days after biliary decompression, both MMP3 and tissue inhibitor of metalloproteinase 1 expression declined toward sham levels, whereas MMP8 expression remained elevated and was identified in bile duct epithelial cells by immunohistochemistry. PMN depletion did not alter the hepatic expression of these genes. Conversely, collagen-based in situ zymography demonstrated markedly diminished collagenase activity following PMN depletion. We conclude that PMNs are essential for collagenase activity and collagen resorption during liver repair, and speculate that PMN-derived MMP8 or PMN-mediated activation of intrinsic hepatic MMPs are responsible for successful liver repair.
Extra- and/or intrahepatic bile duct obstruction induces a pattern of liver injury composed of bile duct epithelial cell hyperplasia, periportal fibrosis and an inflammatory cell infiltrate1,2 that includes monocytes3 and polymorphonuclear cells (neutrophils) (PMNs).4,5 A number of congenital and acquired illnesses result in obstruction of bile flow, which results in cholestatic liver injury and predisposes adults and children to the development of hepatic fibrosis and cirrhosis. Many studies have improved our understanding of the molecular regulation of hepatic fibrosis and until recently, hepatic fibrosis was considered to be the immobile extracellular matrix scar preceding cirrhosis. Clinical and experimental reports have since suggested the reversibility of liver fibrosis, but the mechanisms of fibrosis reversal are poorly understood. Consequently, therapeutic interventions to promote resolution of hepatic injury remain elusive.
Despite significant efforts to determine specific cellular and molecular mechanisms of cholestatic injury and intrinsic repair, our understanding is still far from complete.6,7 Current hypotheses have focused on the central role of resident tissue macrophages or Kupffer cells (KCs) as the key mediators of cholestatic liver injury.8–10 Activated KCs orchestrate the up-regulation of complex network of proinflammatory cytokines such as tumor necrosis factor α, transforming growth factor β, interleukin-1, and interleukin-6 and recruit systemic macrophages and PMNs to the site of injury. Subsequent to KC activation is the activation of hepatic stellate cells to α-smooth muscle actin (α-SMA)-positive myofibroblasts, which preferentially deposit collagen type I, leading to a marked increase in collagen type I relative to other matrix components in the fibrotic scar.11
Successful intrinsic repair of liver fibrosis involves remodeling and breakdown of multiple extracellular matrix components, with degradation of the predominant component, collagen type-I, being particularly important for recovery of normal liver architecture and function. Matrix metalloproteinases (MMPs) are zinc-dependent endoproteinases known to digest components of the extracellular matrix in a substrate-specific fashion. In the rat, MMP8 and MMP13 are the primary collagenases capable of digesting collagen type I. Hepatic stellate cells and KCs, located in the sinusoids, are the primary producers of MMP13. MMP8, also known as PMN collagenase, is known to be localized in PMNs, chondrocytes, endothelial cells, and rheumatoid synovial fibroblasts and can be induced in circulating mononuclear phagocytes.12
Using a rat model of biliary obstruction and decompression to simulate chronic cholestatic fibrotic injury and subsequent fibrinolytic repair we have used a strategy of KC and PMN depletion to study the cellular and molecular mechanisms involved in hepatic matrix metabolism. We have shown that KC depletion alone does not inhibit intrinsic repair mechanisms of fibrotic degradation. KC depletion in conjunction with a reduction in the number of PMNs in the liver, however, was sufficient to inhibit fibrotic degradation. Moreover, we have also shown that fibrosis resolution, following biliary decompression, is concurrent with a increase in matrix metalloproteinase (MMP)8 expression and activity13,14 without a contribution from the other main collagenase, MMP13 identified in rat liver. These findings may suggest a more meaningful effector role for PMNs and MMP8 in the resorption of matrix compared with KCs and MMP 13 during repair.
Cholestatic liver injury is further characterized by an inflammatory infiltrate that includes PMNs, which localize to portal regions.5,13,15 The peri-portal region is a prominent location of matrix deposition during injury and collagen resorption during repair. Our results reveal that unlike KC and recruited macrophage clearance from the liver after biliary decompression, PMNs persist after decompression and remain elevated during repair. The temporal association of PMNs colocalized in the portal region of collagen fibrosis and concurrent demonstration of elevated PMN collagenase (MMP8) gene expression and matrix degradation activity further supports an important biological role for PMN during resolution of hepatic fibrosis. We hypothesize that PMNs are crucial for successful hepatic repair following cholestatic injury. To test this hypothesis we used a unique rat model of reversible extrahepatic cholestatic injury consisting of bile duct obstruction that rapidly develops fibrosis during injury, followed by biliary decompression resulting in intrinsic resolution of fibrosis.14,16,17 Here we report that PMNs are necessary for liver repair in this model. We also show that PMN depletion results in decreased levels of direct collagenase activity, universally considered an important component involved in resolution of fibrosis. We conclude that PMNs are needed for successful repair either by directly degrading collagen fibrotic matrix via MMP8 release and activity or by activating and regulating intrinsic hepatic collagenase degradation.
Materials and Methods
Animals
Adult male rats (225 to 250 g; Harlan Sprague-Dawley, Indianapolis, IN) were housed in an artificial 12-hour light-dark cycle with access to rat chow and water ad libitum according to the National Institutes of Health publication Guide for the Care and Use of Laboratory Animals. Experiments were performed in compliance with guidelines prescribed by the Institutional Animal Care and Use Committee of Rhode Island Hospital and The Alpert Medical School of Brown University.
Biliary Obstruction (Liver Injury)
Animals (n = 64) were divided into saline (operated; n = 27 and sham; n = 16) or treated (operated; n = 15 or sham; n = 6) groups. Treated animals received mouse anti-rat PMN monoclonal IgM antibody ip (RP3, a gift from F. Sendo, Yamagata University School of Medicine, Yamagata, Japan)18 (5 ml of supernatant; n = 21) starting 2 days before decompression (see Figure 1A). Preliminary tests revealed that 5 ml of cultured supernatant had equivalent antibody potency to the 5 ml of ascites fluid used in previous work.18 This dose also demonstrated significant liver PMN depletion in preliminary experiments in our model (discussed later; preliminary data not shown).
Figure 1.
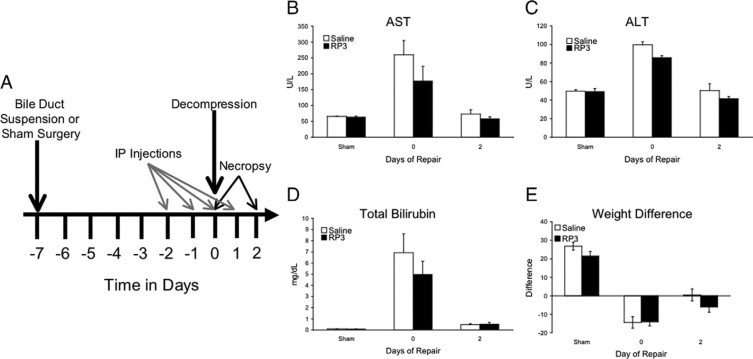
Experimental design and indicators of injury and repair. Biliary suspension surgery was performed, and cholestatic injury occurred over the following 7 days. Saline or RP3 (5 ml/rat) was administered i.p. 2 days before bile duct decompression and continued daily until necropsy. The day of decompression is defined here as day 0. Rats were sacrificed at day 0 and at 2 days of repair for each group (A). Measurements of the liver enzymes AST (B) and ALT (C) as well as bilirubin (D) were significantly (∗P ≤ 0.05) elevated following 7 days of injury and returned to sham levels following decompression. Although RP3 treatment resulted in an apparent attenuation in injury-induced increases in AST, ALT, and bilirubin, these differences were not statistically significant. Difference scores of presurgical weight and necropsy weight (E) show a significant (∗P ≤ 0.05) weight loss following injury with a significant weight recovery following decompression. No significant difference was noted with RP3 treatment.
The operated groups underwent bile duct loop suspension surgery for biliary obstruction as described previously.17,19 Briefly, animals were anesthetized with vaporized isofluorane, weighed, and prepared for surgery. A midline laparotomy was performed and the common bile duct was dissected sufficiently to allow passage of a 5-cm length of silicone vessel loop (Surg-I-Loop; Scanlan International, St. Paul, MN). A mid-segment of this loop was premarked to 1 cm, and the ends were brought through each side of the abdominal wall, 1 cm lateral to the midline at the costochondral margin. The vessel loop was stretched and sutured to the perichondrium bilaterally at the premarked points displacing the common bile duct ventrally. Bile duct obstruction was verified and the abdomen was closed. Sham-operated animals underwent an identical laparotomy, where the common bile duct was identified but not obstructed.
Biliary Decompression (Liver Repair)
Following 7 days of cholestasis, rats were anesthetized, weighed, and prepared for decompression/repair. The skin of the midline incision was opened exposing the vessel loop. The anchoring sutures were cut, and the vessel loop was removed allowing the bile duct to return to its decompressed natural resting position, thereby re-establishing bile flow. The abdominal skin was closed, and the animals were allowed to recover. This procedure is referred to as decompression and the time point as day 0 of repair (see Figure 1A).
Necropsy
Animals were necropsied following 7 days of cholestatic injury (day 0 of repair; n = 20) or 2 days following decompression (day 2 of repair; n = 21); all sham-operated groups (n = 22) were necropsied on day 0 of repair (see Figure 1A). Rats were anesthetized and weighed, the abdomen was opened with a U-shaped incision and the abdominal wall was reflected superiorly. Blood and serum (5 to 10 ml) was collected in K2 EDTA and clotting enhancement collection tubes, respectively, by venipuncture of the inferior vena cava. Samples were sent to Marshfield Laboratories (Marshfield, WI) for the independent determination of Complete Blood Count with differential, liver enzymes, and bilirubin. Hepatectomy was performed and livers were divided and processed as follows: frozen with dry ice in optimal cutting temperature (OCT) compound (Sakura, Torrance, CA); flash frozen in liquid nitrogen; fixed in 10% neutral phosphate buffered formalin (Fisher Scientific, Fair Lawn, NJ); or fixed in formalin free zinc fixative (BD Pharmingen, San Diego, CA). Weight, bilirubin, and liver enzymes were used as markers of the progression of cholestatic injury and subsequent repair. All animals exhibited obstruction as evidenced by bile duct dilatation, expected weight loss, and hyperbilirubinemia.13,14 In the 2-day repair group, 4 rats (of the 21 studied) did not demonstrate bile duct decompression grossly by clearing of jaundice and persistent ductal dilatation. They were excluded from the study.
Inflammatory Cell Identification and Quantification
Formalin- and zinc-fixed tissues were embedded in paraffin and sectioned at 7 μm. Inflammatory cells were identified for quantification using both histochemistry, naphthol AS-D chloroacetate esterase (esterase; Sigma-Aldrich, St. Louis, MO) specific for granulocytes,20 and immunohistochemistry, mouse anti-rat CD68 monoclonal antibody (ED1+; Serotec, Raleigh, NC) specific for the majority of macrophages21, and mouse anti-rat CD163 monoclonal antibody (ED2+; Serotec) specific for KCs.21,22 Liver sections, stained for ED1, ED2, or esterase, were digitally imaged at ×400 with a minimum of 10 fields/section, and every stained cell was counted within the field. Results are expressed as cells per square millimeters.
Matrix proteins, primarily collagen type I, were identified and quantified using Sirius red stained images. Sections were scanned (SprintScan 35 Plus; Polaroid, Cambridge, MA) into a PowerMac G4 (Apple Computer, Cupertino, CA) using a PathScan Enabler optical card (Meyer Instruments, Houston, TX). NIH ImageJ imaging software (National Institutes of Health, Bethesda, MD) was used to determine total tissue section area and collagen area. Results are expressed as a percentage of total section area.
MMP 8 Distribution and Localization
Liver sections were incubated at 4°C with polyclonal rabbit anti-human MMP 8 (AB8115; Chemicon International, Temecula, CA), followed by goat anti-rabbit IgG conjugated to Alexa Fluor 546 (Molecular Probes, Eugene, OR) to investigate the intrinsic hepatic protein expression of MMP8.
Real-Time RT-PCR
Total RNA was extracted with the RNeasy Mini Kit (Qiagen, Valencia, CA) from liver samples flash frozen in liquid nitrogen. RNA quality and quantity were determined using the RNA 6000 Nano LabChip kit on the 2600 BioAnalyzer (Agilent Technologies, Santa Clara, CA). Five micrograms of total RNA was converted into cDNA using First Strand cDNA Synthesis kit (GE Healthcare Life Sciences, Piscataway, NJ) with random primers. The Quantitect SYBR Green real-time PCR kit (Qiagen) was used for all quantitative PCR and run on the Stratagene MX4000 thermocycler. Primer sequences (Table 1) were chosen using appropriate Genbank sequences, National Center for Biotechnology Information (www.ncbi.nih.gov) and Primer 3 software (Whitehead Institute for Biomedical Research: source code available at http://fokker.wi.mit.edu/primer3/).23 Sequences were purchased from Integrated DNA Technologies (Coralville, IA). Melting curves validated the utility and specificity of each primer set. Data were evaluated using the Comparative Ct Method (2−ΔΔCt) of relative quantification standardized to 18S rRNA and are reported as fold increases over sham controls. Because of the relative abundance of 18S rRNA, standardizing samples were diluted 1/1000. The results were equivalent to those obtained using glyceraldehyde-3-phosphate dehydrogenase as an alternate reference gene (data not shown).
Table 1.
Primer List for Quantitative RT-PCR
| Primer name | Sequence |
|---|---|
| 18S-Up | 5′-CGAAAGCATTTGCCAAGAAT-3′ |
| 18S-Down | 5′-AGTCGGCATCGTTTATGGTC-3′ |
| MMP2-Up | 5′-TCCGCGTAAAGTATGGGAAC-3′ |
| MMP2-Down | 5′-CATCACTGCGACCAGTGTCT-3′ |
| MMP3-Up | 5′-TTGTCCTTCGATGCAGTCAG-3′ |
| MMP3-Down | 5′-AGACGGCCAAAATGAAGAGA-3′ |
| MMP8-Up | 5′-CCCACCTGAGATTTGATGCT-3′ |
| MMP8-Down | 5′-GGATGCCGTCTCCAGAAGTA-3′ |
| MMP9-Up | 5′-AGGGTCGGTTCTGACCTTTT-3′ |
| MMP9-Down | 5′-ATAAAAGGGCCGGTAAGGTG-3′ |
| MMP13-Up | 5′-TGACCTGGGATTTCCAAAAG-3′ |
| MMP13-Down | 5′-ACACGTGGTTCCCTGAGAAG-3′ |
| MMP14-Up | 5′-TGGGAACTTTGACACCGTGG-3′ |
| MMP14-Down | 5′-TTGGGTATCCGTCCATCACTTG-3′ |
| TIMP1-Up | 5′-TTCCCTGGCATAATCTGAGC-3′ |
| TIMP1-Down | 5′-ATGGCTGAACAGGGAAACAC-3′ |
| TIMP2-Up | 5′-AAGATCACACGCTGCCCTAT-3′ |
| TIMP2-Down | 5′-GTGCCCATTGATGCTCTTCT-3′ |
| αSMA-Up | 5′-GACACCAGGGAGTGATGGTT-3′ |
| αSMA-Down | 5′-CTTTTCCATGTCGTCCCAGT-3′ |
| Collagen type-I-Up | 5′-GAGAGCATGACCGATGGATT-3′ |
| Collagen type-I-Down | 5′-GCTACGCTGTTCTTGCAGTG-3′ |
In Situ Zymography
Liver tissue embedded in optimal cutting temperature compound (OCT) and frozen was sectioned at 7 μm for in situ zymography. Frozen sections were thawed and rinsed in Tris-buffered saline. DQ gelatin and DQ collagen-specific substrates (Invitrogen, Carlsbad, CA) were diluted as per manufacturer's instructions in agarose, overlain on the liver sections, and coverslipped. Sections were incubated at 4°C for 15 minutes, to solidify substrate, and then placed in a 37°C humidified incubator for 12 to 36 hours. Following incubation, fluorescent images were digitally acquired using QCapture software (Qimaging, Surrey, British Columbia, Canada), a Nikon Eclipse 80i fluorescent microscope, and a QImaging Retiga Exi Fast 1394 color digital camera. Regions of increased fluorescent activity were determined and delineated the location of active enzyme in situ.
Statistics
Data are expressed as means ± SEM and were analyzed by analysis of variance and Fisher's PSLD (protected least significant difference) (StatView 4.1+; SAS Institute, Cary, NC). A value of P < 0.05 was considered significant.
Results
Measuring Injury and Repair
Following 7 days of bile duct suspension, aspartate aminotransferase (AST), alanine aminotransferase (ALT), and bilirubin levels all significantly increased in both saline and RP3 treated animals, whereas animal weight declined. During obstruction, stools were light and urine was dark. After 2 days of repair, following biliary decompression, AST, ALT, and bilirubin levels declined when compared with injured and approached sham levels, and animal weight began to recover. These results indicate successful cholestatic injury and biliary decompression in this model of cholestatic injury and repair (Figure 1, B–E). Although RP3 appeared to reduce AST, ALT, and bilirubin in injured animals, there was no significant difference from saline-treated animals. There were no weight changes or other physical or autopsy findings to indicate other physiological changes relative to the relative neutropenia.
Assessment of PMN Depletion
Peripheral blood counts of lymphocytes, monocytes, and PMNs in saline-treated animals increased (2.1-, 3.1-, and 4-fold over sham, respectively) following 7 days of cholestatic liver injury (Figure 2A). Following 2 days of repair, lymphocyte and monocyte counts declined to sham levels while the PMNs continued to increase (5.9-fold over sham). RP3, a monoclonal antibody against PMNs, was designed to selectively deplete circulating blood PMNs to <100 cells/mm3 without effecting monocytes and lymphocytes.18 RP3 treatment did not completely eliminate circulating PMNs from peripheral blood but induced dramatic neutropenia early in this repair model. In RP3-treated animals, PMN counts were reduced in both injured (2.5-fold decrease versus sham) and in repairing (2.4-fold decrease versus sham) animals when compared with saline-treated controls (Figure 2B).
Figure 2.
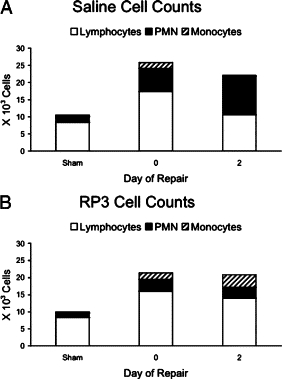
Complete blood cell counts (CBCs) with differential were performed on peripheral blood from all groups. Values from lymphocytes, PMNs, and monocytes are represented in stack bar graphs. The absolute number of lymphocytes, PMNs, and monocytes are significantly elevated after 7 days of injury (A); however, unlike lymphocytes and monocytes, PMN cells counts do not decline following decompression in saline treated animals. RP3 treatment (B) does not alter the injury-related increases in lymphocytes and monocytes; however, PMN cell counts are not different from sham controls. Repairing animals following RP3 treatment show sustained increases in lymphocytes and monocytes but not PMNs, which remain at sham levels.
We next looked at the inflammatory infiltrate within the liver to determine whether RP3 treatment was successful in selectively depleting liver PMNs. In saline control animals, infiltrating PMNs (Figure 3A), monocytes (Figure 3B), and resident KC (Figure 3C) counts revealed significant increases in inflammatory cell counts (4.9-, 3.6-, and 2.7-fold increase over sham, respectively) following 7 days of cholestatic injury. Infiltrating monocyte and KC populations then declined, approaching sham levels, following 2 days of repair. However, as we have previously demonstrated, PMN counts remained elevated (4.3-fold versus sham) during repair following biliary decompression. In the RP3 treatment group, however, RP3 monoclonal antibody significantly reduced PMN counts in sham, injured, and repairing animals by 4.3-, 11.3-, and 7.6-fold, respectively, compared with saline controls (Figure 3A). In contrast, infiltrating monocyte and KC populations remained elevated during injury and repair in RP3-treated animals (Figure 3, B and C). These data support the conclusion that RP3 treatment at the dose chosen specifically depleted circulating PMNs, making the RP3-treated animals markedly neutropenic and decreased infiltration of circulating PMNs into the liver. RP3 treatment did not impact monocyte or KC circulating numbers or infiltration in response to bile duct obstruction.
Figure 3.
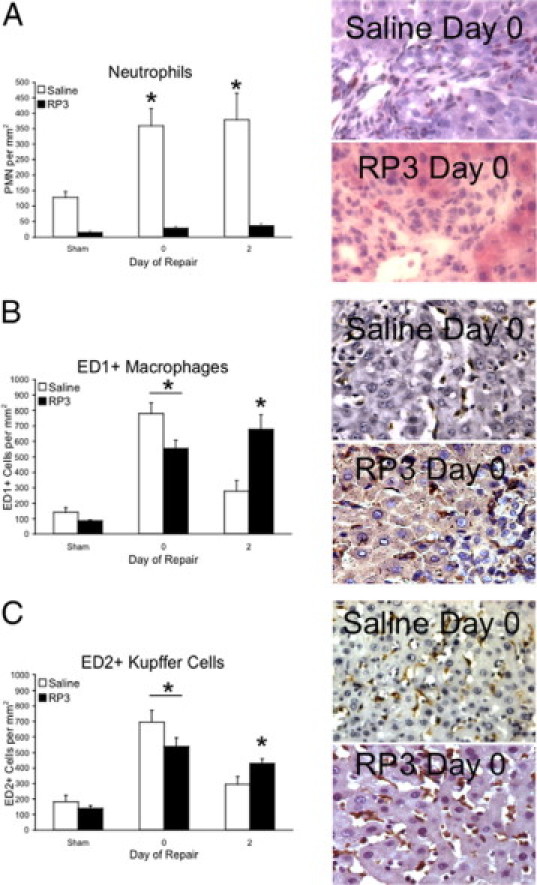
Inflammatory cells of the liver. Esterase (A)-, ED1+ (B)-, and ED2+ (C)-stained liver sections derived from sham, injured (day 0), and repairing (day 2) animals treated with saline or RP3 were digitally imaged and counted. Representative images of stained liver sections from injured animals accompany each graph and illustrate staining quality (×400). The number of esterase, ED1+, and ED2+ cells following cholestatic injury are significantly (∗P ≤ 0.05) elevated when compared with saline sham controls. Declines in ED1+ and ED2+ cells following decompression are in contrast with PMN cell counts that remain elevated. RP3 treatment significantly depletes PMNs from with liver without depleting ED1+ and ED2+ cells.
Fibrotic Injury and Repair
Having determined that RP3 successfully depleted PMNs from injured and repairing livers, we sought to investigate the impact of PMN depletion during cholestatic injury and following biliary decompression during repair by measuring the progression of extracellular matrix collagen fibrosis by Sirius red histology. Our findings demonstrate that PMN depletion with RP3 treatment did not alter the collagen deposition in response to biliary obstruction as evidenced by the development of fibrosis over 7 days compared with saline controls. (Figure 4A, top panels). PMN depletion with RP3 treatment, however, did specifically block early fibrotic degradation of the extracellular matrix following 2 days of biliary decompression (Figure 4A, bottom panels). Sirius red measurements of percent collagen fibrosis (Figure 4B) were similar and not significantly different for sham- and cholestatic-injured animals between the saline and RP3 treatment groups. However, following 2 days of biliary decompression-related intrinsic repair, the RP3-treated, PMN-depleted animals demonstrated percent collagen fibrosis by Sirius red histology that remained at injured levels despite successful surgical biliary decompression and the resolution of jaundice. In contrast, the intact, nonneutropenic saline control animals after biliary decompression had near complete resolution of collagen fibrosis (P ≤ 0.05) (Figure 4).
Figure 4.
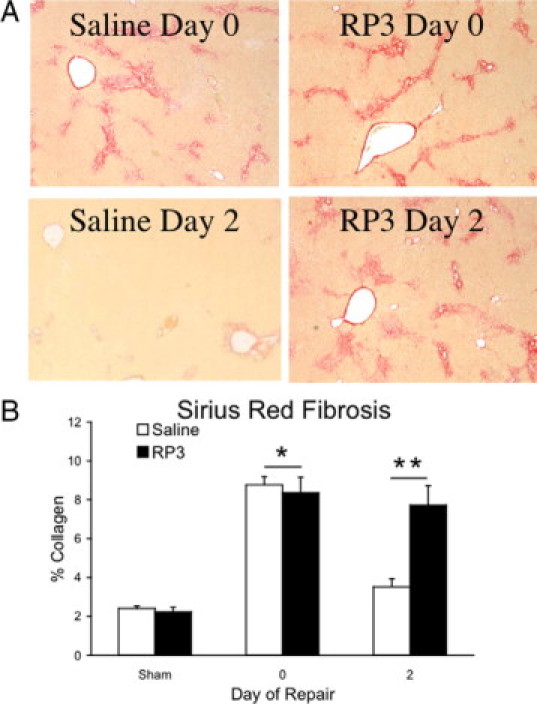
Fibrotic repair. Representative images of Sirius red-stained liver sections (A) derived from injured and repairing animals treated with either saline or RP3 (×40) illustrate the data shown in the bar graph. Collagen (B) is expressed as a percentage of total area section area. Biliary obstruction significantly (*P ≤ 0.05) increased the deposition of collagen both in saline- and RP3-treated groups following 7 days of injury. Collagen content, as measured by Sirius red, was significantly (*P ≤ 0.05) decreased in the saline-treated animals following decompression (day 2). RP3 treatment blocked the decompression related decline in collagen. Measures of collagen content were not different from injured animals and significantly (**P ≤ 0.05) elevated when compared with saline-treated matched controls.
Gene Expression
Finding that a significant decrease in hepatic PMNs was accompanied by inhibited early matrix degradation, we investigated MMPs related molecular changes that followed biliary decompression. To consider mechanisms whereby PMNs might influence the gene expression of proteins important for matrix degradation during repair, we measured the expression patterns of MMP and tissue inhibitors of metalloproteinases (TIMPs) during periods of cholestatic injury and repair by quantitative RT-PCR. Expression of MMP2, MMP9, MMP13, and MMP14 were not elevated during injury or repair nor was their expression altered by PMN depletion. TIMP2 expression was slightly increased during injury and repair in saline treated animals and PMN depletion resulted in only marginal reductions during repair (Figure 5, A, D, E, F, and H).
Figure 5.
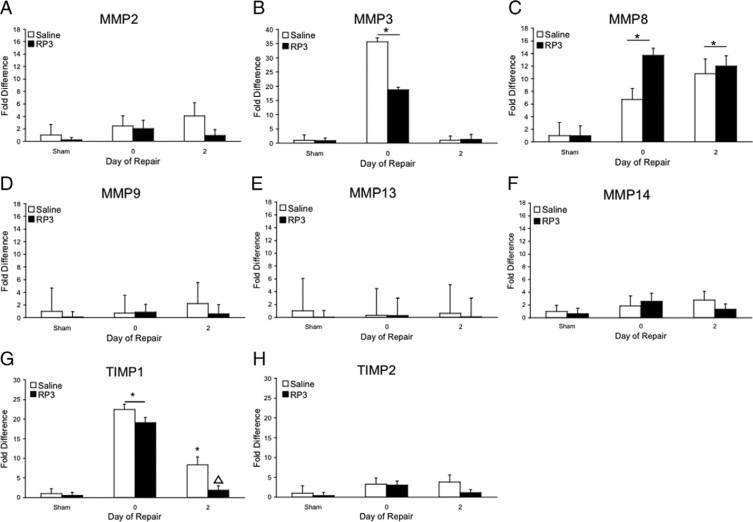
Quantitative real-time RT-PCR. MMP2 (A), MMP3 (B), MMP8 (C), MMP8 (D), MMP13 (E), MMP14 (F), TIMP1 (G), and TIMP2 (H) RNA expression was quantified in liver homogenates derived from sham, injured, and repairing animals following saline or RP3 treatment. Data are relative to 18S expression and are given as fold increases over saline sham. Significant increases over saline sham (∗P ≤ 0.05) are noted. ▵ represents significant difference in TIMP-1 expression (P < 0.05) from control repairing animals.
In contrast, MMP3, MMP8, and TIMP1 gene expression was significantly up-regulated (27.2-, 9.1-, and 30.5-fold, respectively, compared with sham controls) during injury (Figure 5, B, C, and G). MMP3 and TIMP1 gene expression retuned to baseline following biliary decompression-initiated repair in both saline- and RP3-treated, PMN-depleted animals, whereas MMP8 gene expression remained significantly elevated in both saline (9.4-fold)- and RP3 (15.7-fold)-treated animals (P ≤ 0.05) (Figure 5). A significant reduction in TIMP1 gene expression occurred in PMN-depleted animals compared with saline-treated animals following biliary decompression as well. Interestingly, there were no significant changes in the expression of the important gelatinase MMP9 to account for the potential changes in activation of intrinsic MMPs.
Attenuation of collagen fibrotic degradation following RP3-mediated PMN depletion may be reconciled either by a decreased production of degrading enzymes or by an increase in matrix production. Because RP3 treatment did not reduce the gene expression of the type I collagenase, MMP8, we investigated the expression of proteins associated with increased fibrosis. α-SMA and collagen type I expression were not elevated during repair relative to saline controls (Table 2).
Table 2.
α-SMA and Collagen Type I Expression Are Not Elevated Relative to Saline Controls following RP3-Mediated PMN Depletion
| Group | Treatment | Gene | Fold increase (± SD) |
|---|---|---|---|
| Sham | Saline | α-SMA | 1.00 ± 2.0 |
| Sham | RP3 | α-SMA | 0.81 ± 0.0 |
| Repair | Saline | α-SMA | 1.53 ± 2.0 |
| Repair | RP3 | α-SMA | 2.57 ± 3.5 |
| Sham | Saline | Coll-I | 1.00 ± 1.1 |
| Sham | RP3 | Coll-I | 0.87 ± 0.9 |
| Repair | Saline | Coll-I | 3.18 ± 1.3* |
| Repair | RP3 | Coll-I | 4.24 ± 3.2* |
Different from saline sham control by ANOVA; P ≤ 0.05.
MMP 8 Protein Localization
To confirm the MMP 8 gene expression results, immunohistochemistry was used to investigate the protein distribution of MMP8 during cholestatic injury. Polyclonal anti-MMP8 antibody raised against the hinge region of MMP 8 was used to immunolocalize MMP8 in liver sections from saline- and RP3-treated animals after 7 days of bile duct obstructive injury. Figure 6, A–D, demonstrates double immunofluorescent labeling of MMP8 in red and PMNs in green from bile duct obstructed animals treated with saline (Figure 6A) or RP3 (Figure 6B). Strong bile duct epithelial cell concentration of MMP8 protein is labeled red by immunofluoresence during injury whereas PMNs are immunolabeled green. The strongest immunofluoresence is noted in the portal tracts specifically the bile duct epithelial cells with infiltrating PMNs colocalizing to the same region. Faint sinusoidal immunostaining is also noted.
Figure 6.
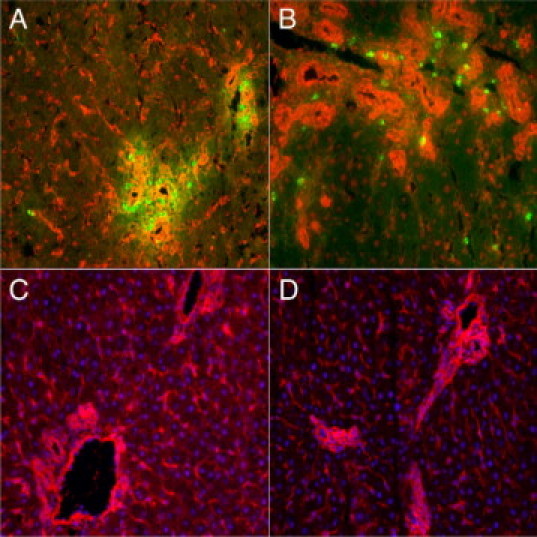
MMP 8 Immunohistochemistry. Representative images of liver sections from bile duct obstructed cholestatic-injured animals treated with saline (A and C) or RP3 (B and D). Double immunofluoresent labeling of MMP8 (red) and neutrophils (green) demonstrate MMP8 immunostained bile duct epithelial cells (red) and colocalized PMNs (green) in proximity (A and B). MMP8 immunostaining confirms the bile duct hepatic source of MMP8 protein during injury. Immunofluorescent labeling is contrasted with the nuclear counterstain 4′,6′-diamidino-2-phenylindole in C and D.
In Situ Zymography Gelatinase and Collagenase Activity
Our results suggested that hepatic PMN levels impact extracellular matrix metabolism after biliary decompression (repair). Furthermore, they revealed that gene expression of matrix-degrading MMP enzymes were not altered by PMN depletion conversely MMP8 expression actually increased and MMP8 protein was found localized to the bile duct epithelial cells. We next used gelatinase and collagenase-specific fluorescent substrates for in situ zymography to demonstrate the functional activity of matrix degradation in our model of cholestatic injury and subsequent repair. The essential biological activity of collagenase is necessary to localize and determine whether PMN depletion altered either the distribution or activity of either collagenases or gelatinases.
Gelatinase specific activity increased following 7 days of cholestatic liver injury and remained elevated through repair (Figure 7A). Gelatinase activity was localized primarily to the portal tracts surrounding bile ducts and extending into the sinusoids. Distribution of gelatinase activity followed portal tract morphology in both injured and repairing animals. RP3 treatment had no effect on gelatinase activity following 7 days of cholestatic liver injury (Figure 7B). After 2 days of repair gelatinase, activity in RP3-treated animals remained elevated compared with sham, and distribution was consistent with that in saline-treated animals despite the RP3-mediated attenuation of repair.
Figure 7.
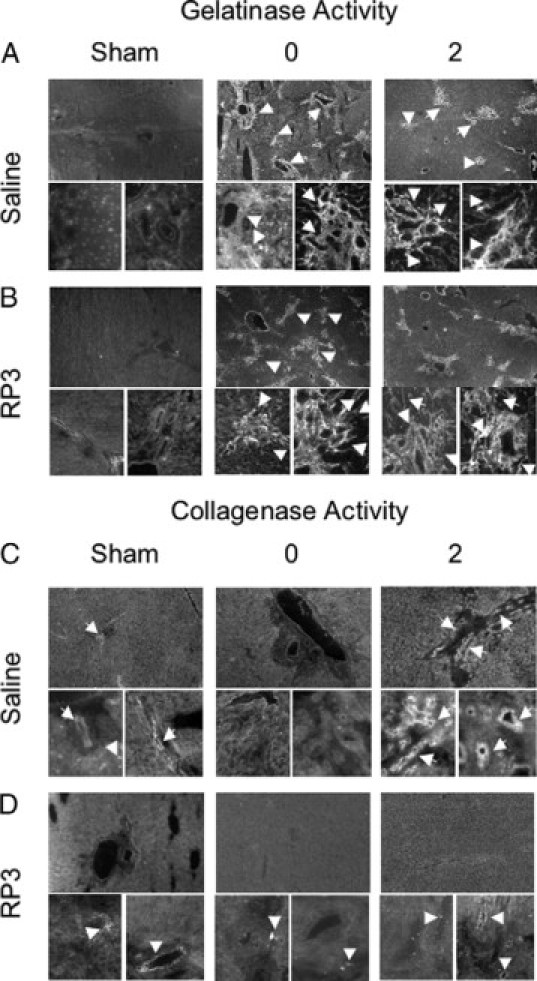
Gelatinase/collagenase in situ zymography. A: Representative images of gelatinase activity in sham, injured, and repairing animals are shown at both low (×40) and high magnification (×400). Arrows point to gelatinolytic activity in and around the injured and repairing portal tracts. B: RP3-mediated neutrophil depletion does not alter gelatinolytic activity or localization during injury or repair. C: Collagenolytic activity in saline-treated animals is evident (arrows) in highly restricted regions of the portal tract in sham animals, and this activity is absent during injury. Repairing animals show a substantial increase in collagenase activity (arrows) primarily in apparent bile duct epithelial cells but also in individual cells within the portal tracts. D: RP3 treatment reduces collagenase activity during repair to sham levels. Arrowheads indicate the regions of collagenase activity which is markedly reduced in the absence of neutrophils compared to saline controls in C. Note there is no difference collagenase activity in sham or repaired livers without neutrophils.
Collagenase activity essential for collagen resorption was constrained to the boundaries of biliary epithelial tracts of both saline (Figure 7C)- and RP3 (Figure 7D)-treated sham animals. This activity was not evident in saline- and RP3-treated animals during cholestatic injury; however, strong collagenase activity was visualized throughout the portal tracts adjacent to bile ducts only following biliary decompression in repairing saline, PMN-competent control animals. In contrast, significant and marked reduction in collagenase activity was noted in the decompressed repairing RP3-treated PMN-depleted animals. The minimal level of collagenase activity detected was at sham levels near baseline or background intensity. These results suggest that PMNs are important to collagenase, but not gelatinase activity, and that extracellular matrix degrading collagenase activity is only successful after biliary decompression in the presence of PMNs.
Discussion
Cholestatic liver injury from extra- or intrahepatic biliary obstruction and the majority of liver injuries share common inflammatory cascades and progressive hepatic fibrogenesis that leads to end stage liver disease. Biliary obstruction results in an increase of bile acids and toxins, normally excreted via bile, in the liver and results in the activation of KCs,3,6,7,9 the transformation of hepatic stellate cells to myofibroblasts,7,9,11,24,25 the proliferation of biliary epithelial cells and hepatocyte injury,6,7,9,11 the release of inflammatory cytokines,26–29 the infiltration of circulating leukocytes,9 and net collagen type I deposition by activated myofibroblasts and infiltrating fibroblasts, primarily in the sinusoids and portal triads.24,25,30–32 Unresolved, these profibrotic events lead to cirrhosis, end stage liver disease, and often the need for transplantation.24,29,33 Recent clinical and experimental evidence, however, suggest that there is a critical time period during which restoration of bile flow can halt or reverse fibrosis, indicating an intrinsic hepatic capacity for repair.24,29,33 Unlike wound healing, which replaces normal tissue with collagen scar, successful liver repair must be associated with removal of collagen matrix and restoration of normal hepatic architecture, hepatocyte gene expression, and metabolic function. Numerous studies have brought us closer to a better understanding of the nature of liver injury and the molecular regulation of hepatic fibrosis. Knowledge of the mechanisms that initiate and sustain liver repair is lacking and critical to developing reliable treatments for cholestatic injury.
Because of limited animal models that recapitulate the inflammatory and fibrogenic consequences of biliary obstruction and decompression, there have been few studies to examine the cellular and molecular mechanisms that control hepatic matrix metabolism and net resorption. Using a unique rat model of reversible extrahepatic cholestatic liver injury consisting of bile duct obstruction that rapidly develops collagen fibrosis during injury, followed by intrinsic resolution of fibrosis following release of obstruction,13,14 we have focused on an understanding of the cellular and molecular mechanisms involved in hepatic matrix metabolism. It is hypothesized that the KC plays a central role in the molecular regulation of hepatic fibrosis, and once activated, the KC elaborates a complex network of proinflammatory cytokines and cellular chemoattractants to initiate and sustain a profibrotic state. We further hypothesized that KCs might be the central effector cells in resolution of biliary fibrosis. In previous studies aimed at modifying KC function, we have shown that KCs apparently indirectly regulate successful resolution of collagen fibrosis repair. In prior studies, we demonstrated Gadolinium-mediated KC depletion inhibited matrix degradation in repairing livers.13,14 Remarkably, in addition to KC depletion, there was also a reduced number of PMNs in the liver sections of repairing decompressed livers. This reduction was significantly different from our previously published observations of increased PMNs in normal repairing livers and led us to consider the PMN as an effector of fibrotic repair. Moreover, we have previously shown that in situ MMP8 or PMN collagenase activity correlates with the collagen matrix resorption and resolution of fibrosis seen following decompression during repair.14 Taken together, the potential role of PMNs during intrinsic repair following biliary decompression is yet to be fully examined and determined. On the basis of this previous work implicating a contribution from PMNs during resolution of cholestatic liver injury, we aimed to deplete PMNs to test the question of PMN involvement in liver repair following obstructive cholestatic liver injury.
Treatment with the anti-rat PMN monoclonal antibody RP3 was specific and proficient to maintain a neutropenic environment in the liver during repair as evidenced by PMN reductions in peripheral blood and depletion in the liver without affecting monocyte or KC populations. PMN depletion in the liver was associated with inhibition of matrix degradation in repairing animals demonstrated by Sirius Red histology, collagenase zymography, and in situ zymography, supporting a role for the PMN in resolution of hepatic fibrosis in this model.
Mouse models of toxicity- and bile duct ligation-induced liver injury have previously suggested that PMNs contribute to liver injury; here our results propose that PMNs are necessary to initiate liver repair following relief of biliary obstruction. Previous work in our lab and in others have shown that biliary ligation in rats most closely replicates the proliferative and fibrogenic response to biliary obstruction found in humans.3,17,19,29,32 Mouse models of biliary obstruction, although advantageous secondary to the wide array of transgenic options available to investigate mechanisms of action, demonstrate rapid hepatic necrosis in response to biliary obstruction and unpredictable progression toward bridging fibrosis disparate to the human condition. The high fidelity of this rat model to simulate human hepatic fibrotic injury and spontaneous repair following decompression thus relies on alternative strategies for cellular depletion or chemokine blocking to duplicate genetic knockouts to attempt to evaluate cellular and molecular mechanisms during repair.
We next attempted to determine possible mechanisms by which PMNs impact resolution of hepatic fibrosis. The expression pattern of collagenase and gelatinase genes important for matrix metabolism during cholestatic injury and repair revealed that MMP3 and MMP8 expression was up-regulated in response to bile duct ligation injury; however, only MMP8 expression remained elevated during subsequent repair. Interestingly, PMN depletion with RP3 treatment did not reduce MMP8 expression at any time point. These results parallel the PMN infiltration pattern over the same timeframe. These data suggest two important points; MMP8 gene expression increases in response to bile duct obstruction and the liver may harbor a novel cellular source of PMN collagenase. This led us to investigate the protein distribution of MMP 8 from injured animals. The search for a novel MMP 8 source from the liver is supported by our PCR data (Figure 5C), and the fact that MMP 8 is one of the most potent and efficient interstitial collagenases. Our novel finding of bile duct epithelial cells as an intrinsic hepatic source of MMP8 suggests that the bile duct epithelial cell is more than a passive bystander during cholestatic injury and may harbor the potential to respond to injury, interact with inflammatory cells, PMNs in particular, and possess the capability to participate in matrix repair. PMNs recruited to the liver in response to injury and during repair are another possible source of MMP8 or MMP8 activators because they store pro-MMP8 protein in specific granules during their maturation in the bone marrow. Moreover, PMNs contain a number of proteases that can activate pro-MMP 8 to active MMP8 and proteases that inhibit TIMPs. Human PMNs were recently discovered to possess highly TIMP-resistant MMP 8 on the cell surface with pericellular collagenase activity likely to be an important form of MMP8-regulating inflammation and collagen turnover12. The ability to colocalize PMNs, which can temporally inhibit TIMP function and activate MMP8 in a region of the liver with a readily available cellular source of MMP8 from the bile duct epithelial cell, may shift the local stoichiometry to favor MMP balance over TIMPs and promote collagenase activity.
Our results further demonstrate that the expression of another well-known collagenase in the liver, MMP13, was unchanged in response to bile duct obstruction or reversal of cholestasis. Moreover, the expression of the two main gelatinases, MMP2 and MMP9, similarly were not elevated during injury nor repair further limits the potential MMP candidates available to degrade extracellular collagen matrix during repair.
Given this expression profile of the prominent gelatinases and collagenases in our model during injury, repair, and in response to PMN depletion, we questioned whether collagenolytic enzyme activity was altered during repair or by PMN depletion. We found, by in situ zymography, collagenolytic activity in the portal tracts only during native repair following biliary decompression but not during cholestatic injury despite high levels of MMP8 gene and protein expression. These data strengthen our hypothesis that PMN-derived MMP8 or intrinsic MMP8 is crucial for early degradation of extracellular collagen matrix. Our data demonstrate both a temporal and cellular proximity of collagen, PMNs, and MMP8 at the time of collagenase activity. Moreover, the alternative leading collagenase, MMP13, is neither up-regulated nor localized to the portal tracts by immunohistochemistry. PMN depletion diminished the expected vigorous collagenase activity following biliary decompression, which would imply that PMNs are essential for effective collagenase enzyme function early during liver repair, possibly as activators of intrinsic hepatic MMP8 collagenase or by direct PMN-derived collagenase MMP8 action. Gelatinase activity, which is known in some systems to activate collagenases, was found to be intrinsically active in response to injury and remained active throughout repair. This represents a marked increase in enzyme function compared with sham controls and points to alternative mechanisms of collagenase regulation.
These findings point to three possible mechanisms of matrix resorption in this model relative to PMNs. The first may be the obvious delivery of PMN derived, active MMP8 directly to the fibrotic matrix via granule release. This would follow if there were a direct correlation of PMN numbers and biological collagenase activity as demonstrated by these data. Furthermore, this would imply that pro-MMP8 stored in specific granules during bone marrow myeloblastic differentiation is activated to the active MMP8 form on degranulation. Knowledge of the mechanism of PMN degranulation and MMP8 activation under these circumstances is lacking and is the focus of ongoing experimental efforts on our laboratory. Recent compelling evidence points to coordinated PMN MMP8 and MMP9 activity in uterine cervical mucous during parturition as well as in other mouse models of liver injury and repair.34 If PMN gelatinase-collagenase synchronization were similarly responsible in our model, then we might expect that our results would demonstrate decreased gelatinase activity in PMN-depleted repairing livers via in situ zymography. Our results, however, clearly reveal in situ gelatinase activity during cholestatic injury and following decompression in the presence of PMNs or under neutropenic conditions. Moreover, other PMN proteases could be involved in the activation of PMN-derived pro-MMP8 or intrinsic hepatic MMP8. Alternatively, two other mechanisms that relate to other PMN functions might be speculated through PMN cell interactions via activation of membrane-bound MMP or through cytokine activation of the intrinsic MMP that we found to be up-regulated. Tumor necrosis factor α, for example, is a likely candidate. These pathways would further strengthen the association of PMN-dependent liver repair, and they will require further study. Ongoing studies in our laboratory are aimed at such a possibility.
In contrast to our findings, other studies using different models of liver injury have focused on the collagenase MMP13. MMP13 is the rodent analog of human MMP1 and, by virtue of its broad range of substrate activity, is generally considered to be the most potent of the MMP collagenases.35 MMP8 is found in both rodents and humans and also has a broad range of substrate activity. Although both MMP13 and MMP8 are capable of cleaving various forms of triple helical collagen, MMP8 is generally considered to more preferentially hydrolyze collagen type I, whereas MMP13 more preferentially hydrolyzes collagen type II.35
Recently, MMP13 producing infiltrating macrophage populations were found to localize to regions of hepatic scar formation and correlate with repair.36 However, this was observed in a mouse model of carbon tetrachloride toxicity with a different pattern of macrophage and PMN responses in a fundamentally hepatotoxic injury model disparate from the rat proliferative fibrogenic response to biliary obstruction. In our model, MMP13 expression is not elevated at any point or with any treatment. Conversely, we found that PMNs, and MMP8 specifically, colocalize to regions of collagen deposition13,14 in the portal tracts and are essential contributors for early degradation of extracellular collagen following biliary decompression. Using our model of cholestatic injury and repair, we have previously shown that macrophage depletion does not alter the ability of the liver to degrade matrix, unless PMN numbers are also reduced.13 Questions regarding the purpose of gelatinases activity during biliary obstruction injury in the presence of TIMPs remain. We speculate these same tissue inhibitors selectively inhibit or prevent collagenase activity until the onset of repair.
Taken together, the summary of our findings using cellular depletion strategies to impact the native inflammatory cellular participants as regards to collagen matrix metabolism during cholestatic injury and spontaneous repair after biliary decompression suggests mechanistic roles for an intact KC population and a coordinated series of cellular and molecular signals to achieve and sustain PMN migration to the liver and specifically to matrix embedded portal areas. Initiation of collagenase activation is essential to degrade the abundant extracellular collagen type I fibrosis yet only occurs following removal of the offending toxicity, ie, decompression of the obstructed biliary system in our model and requires an integral PMN presence for successful resolution of hepatic fibrosis.
Footnotes
Supported in part by National Institutes of Health grants R01 DK46831 (to T.F.T.), R01DK068097 (to S.H.G.), and RR-P20 RR17695 from the Institutional Development Award Program of the National Center for Research Resources.
References
- 1.Tracy TF, Jr, Goerke ME, Bailey PV, Sotelo-Avila C, Weber TR. Growth-related gene expression in early cholestatic liver injury. Surgery. 1993;114:532–537. [PubMed] [Google Scholar]
- 2.Slott PA, Liu MH, Tavoloni N. Origin, pattern, and mechanism of bile duct proliferation following biliary obstruction in the rat. Gastroenterology. 1990;99:466–477. doi: 10.1016/0016-5085(90)91030-a. [DOI] [PubMed] [Google Scholar]
- 3.Roggin KK, Kim JC, Kurkchubasche AG, Papa EF, Vezeridis AM, Tracy TF. Macrophage phenotype during cholestatic injury and repair: the persistent inflammatory response. J Pediatr Surg. 2001;36:220–228. doi: 10.1053/jpsu.2001.20059. [DOI] [PubMed] [Google Scholar]
- 4.Saito JM, Maher JJ. Bile duct ligation in rats induces biliary expression of cytokine-induced neutrophil chemoattractant. Gastroenterology. 2000;118:1157–1168. doi: 10.1016/s0016-5085(00)70369-6. [DOI] [PubMed] [Google Scholar]
- 5.Saito JM, Bostick MK, Campe CB, Xu J, Maher JJ. Infiltrating neutrophils in bile duct-ligated livers do not promote hepatic fibrosis. Hepatol Res. 2003;25:180–191. doi: 10.1016/s1386-6346(02)00247-4. [DOI] [PubMed] [Google Scholar]
- 6.Friedman SL. Seminars in medicine of the Beth Israel Hospital, Boston: the cellular basis of hepatic fibrosis Mechanisms and treatment strategies. N Engl J Med. 1993;328:1828–1835. doi: 10.1056/NEJM199306243282508. [DOI] [PubMed] [Google Scholar]
- 7.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aldana PR, Goerke ME, Carr SC, Tracy TF., Jr The expression of regenerative growth factors in chronic liver injury and repair. J Surg Res. 1994;57:711–717. doi: 10.1006/jsre.1994.1206. [DOI] [PubMed] [Google Scholar]
- 9.Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicol Sci. 2002;65:166–176. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- 10.Gehring S, Dickson EM, San Martin ME, van Rooijen N, Papa EF, Harty MW, Tracy TF, Jr, Gregory SH. Kupffer cells abrogate cholestatic liver injury in mice. Gastroenterology. 2006;130:810–822. doi: 10.1053/j.gastro.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 11.Tsukada S, Parsons CJ, Rippe RA. Mechanisms of liver fibrosis. Clin Chim Acta. 2006;364:33–60. doi: 10.1016/j.cca.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 12.Owen CA, Zhuma H, Lopez-Otin C, Shapiro SD. Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol. 2004;172:7791–7803. doi: 10.4049/jimmunol.172.12.7791. [DOI] [PubMed] [Google Scholar]
- 13.Harty MW, Papa EF, Huddleston HM, Young E, Nazareth S, Riley CA, Ramm GA, Gregory SH, Tracy TF., Jr Hepatic macrophages promote the neutrophil-dependent resolution of fibrosis in repairing cholestatic rat livers. Surgery. 2008;143:667–678. doi: 10.1016/j.surg.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 14.Harty MW, Huddleston HM, Papa EF, Puthawala T, Tracy AP, Ramm GA, Gehring S, Gregory SH, Tracy TF., Jr Repair after cholestatic liver injury correlates with neutrophil infiltration and matrix metalloproteinase 8 activity. Surgery. 2005;138:313–320. doi: 10.1016/j.surg.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Gujral JS, Farhood A, Bajt ML, Jaeschke H. Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology. 2003;38:355–363. doi: 10.1053/jhep.2003.50341. [DOI] [PubMed] [Google Scholar]
- 16.Svegliati-Baroni G, Ridolfi F, Caradonna Z, Alvaro D, Marzioni M, Saccomanno S, Candelaresi C, Trozzil L, Macarri G, Benedetti A, Folli F. Regulation of ERK/JNK/p70S6K in two rat models of liver injury and fibrosis. J Hepatol. 2003;39:528–537. doi: 10.1016/s0168-8278(03)00291-5. [DOI] [PubMed] [Google Scholar]
- 17.Roggin KK, Papa EF, Kurkchubasche AG, Tracy TF., Jr Kupffer cell inactivation delays repair in a rat model of reversible biliary obstruction. J Surg Res. 2000;90:166–173. doi: 10.1006/jsre.2000.5879. [DOI] [PubMed] [Google Scholar]
- 18.Sekiya S, Gotoh S, Yamashita T, Watanabe T, Saitoh S, Sendo F. Selective depletion of rat neutrophils by in vivo administration of a monoclonal antibody. J Leukoc Biol. 1989;46:96–102. doi: 10.1002/jlb.46.2.96. [DOI] [PubMed] [Google Scholar]
- 19.Posner MC, Burt ME, Stone MD, Han BL, Warren RS, Vydelingum NA, Brennan MF. A model of reversible obstructive jaundice in the rat. J Surg Res. 1990;48:204–210. doi: 10.1016/0022-4804(90)90214-m. [DOI] [PubMed] [Google Scholar]
- 20.Moloney WC, McPherson K, Fliegelman L. Esterase activity in leukocytes demonstrated by the use of naphthol AS-D chloroacetate substrate. J Histochem Cytochem. 1960;8:200–207. doi: 10.1177/8.3.200. [DOI] [PubMed] [Google Scholar]
- 21.Damoiseaux JG, Dopp EA, Calame W, Chao D, MacPherson GG, Dijkstra CD. Rat macrophage lysosomal membrane antigen recognized by monoclonal antibody ED1. Immunology. 1994;83:140–147. [PMC free article] [PubMed] [Google Scholar]
- 22.Dijkstra CD, Dopp EA, Joling P, Kraal G. The heterogeneity of mononuclear phagocytes in lymphoid organs: distinct macrophage subpopulations in the rat recognized by monoclonal antibodies ED1, ED2 and ED3. Immunology. 1985;54:589–599. [PMC free article] [PubMed] [Google Scholar]
- 23.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 24.Friedman SL. Liver fibrosis—from bench to bedside. J Hepatol. 2003;38(Suppl. 1):S38–S53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 25.Lewindon PJ, Pereira TN, Hoskins AC, Bridle KR, Williamson RM, Shepherd RW, Ramm GA. The role of hepatic stellate cells and transforming growth factor-β(1) in cystic fibrosis liver disease. Am J Pathol. 2002;160:1705–1715. doi: 10.1016/s0002-9440(10)61117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Decker K. Biologically active products of stimulated liver macrophages (Kupffer cells) Eur J Biochem. 1990;192:245–261. doi: 10.1111/j.1432-1033.1990.tb19222.x. [DOI] [PubMed] [Google Scholar]
- 27.Gaillard T, Mulsch A, Busse R, Klein H, Decker K. Regulation of nitric oxide production by stimulated rat Kupffer cells. Pathobiology. 1991;59:280–283. doi: 10.1159/000163663. [DOI] [PubMed] [Google Scholar]
- 28.Ishizaki-Koizumi S, Sonaka I, Takei Y, Ikejima K, Sato N. The glycine analogue, aminomethanesulfonic acid, inhibits LPS-induced production of TNF-α in isolated rat Kupffer cells and exerts hepatoprotective effects in mice. Biochem Biophys Res Commun. 2004;322:514–519. doi: 10.1016/j.bbrc.2004.07.147. [DOI] [PubMed] [Google Scholar]
- 29.Tracy TF, Jr, Dillon P, Fox ES, Minnick K, Vogler C. The inflammatory response in pediatric biliary disease: macrophage phenotype and distribution. J Pediatr Surg. 1996;31:121–125. doi: 10.1016/s0022-3468(96)90333-4. discussion 125–126. [DOI] [PubMed] [Google Scholar]
- 30.Schaefer B, Rivas-Estilla AM, Meraz-Cruz N, Reyes-Romero MA, Hernandez-Nazara ZH, Dominguez-Rosales JA, Schuppan D, Greenwel P, Rojkind M. Reciprocal modulation of matrix metalloproteinase-13 and type I collagen genes in rat hepatic stellate cells. Am J Pathol. 2003;162:1771–1780. doi: 10.1016/S0002-9440(10)64312-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benyon RC, Arthur MJ. Mechanisms of hepatic fibrosis. J Pediatr Gastroenterol Nutr. 1998;27:75–85. doi: 10.1097/00005176-199807000-00013. [DOI] [PubMed] [Google Scholar]
- 32.Ramm GA, Hoskins AC, Greco SA, Pereira TN, Lewindon PJ. Signals for hepatic figrogenesis in pediatric cholestatic liver disease: review and hypothesis. Comp Hepatol. 2004;3(Suppl. 1):S5. doi: 10.1186/1476-5926-2-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benyon RC, Iredale JP. Is liver fibrosis reversible? Gut. 2000;46:443–446. doi: 10.1136/gut.46.4.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Becher N, Hein M, Uldbjerg N, Christian Danielsen C. Balance between matrix metalloproteinases (MMP) and tissue inhibitors of metalloproteinases (TIMP) in the cervical mucus plug estimated by determination of free non-complexed TIMP. Reprod Biol Endocrinol. 2008;30:45–52. doi: 10.1186/1477-7827-6-45. 6: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. Ed. 2. Elsevier Academic Press; Amsterdam, The Netherlands: 2004. [Google Scholar]
- 36.Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, Iredale JP. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178:5288–5295. doi: 10.4049/jimmunol.178.8.5288. [DOI] [PubMed] [Google Scholar]