

Abstract
Brain hemodynamics in cerebral malaria (CM) is poorly understood, with apparently conflicting data showing microcirculatory hypoperfusion and normal or even increased blood flow in large arteries. Using intravital microscopy to assess the pial microvasculature through a closed cranial window in the murine model of CM by Plasmodium berghei ANKA, we show that murine CM is associated with marked decreases (mean: 60%) of pial arteriolar blood flow attributable to vasoconstriction and decreased blood velocity. Leukocyte sequestration further decreased perfusion by narrowing luminal diameters in the affected vessels and blocking capillaries. Remarkably, vascular collapse at various degrees was observed in 44% of mice with CM, which also presented more severe vasoconstriction. Coadministration of artemether and nimodipine, a calcium channel blocker used to treat postsubarachnoid hemorrhage vasospasm, to mice presenting CM markedly increased survival compared with artemether plus vehicle only. Administration of nimodipine induced vasodilation and increased pial blood flow. We conclude that vasoconstriction and vascular collapse play a role in murine CM pathogenesis and nimodipine holds potential as adjunctive therapy for CM.
Cerebral malaria (CM) caused by Plasmodium falciparum claims the lives of nearly 1 million children every year.1 Despite antimalarial treatment, 10% to 20% of patients die, and one in every four survivors develops neurological sequelae,2,3 therefore adjunctive therapies are urgently needed. A number of clinical trials addressing potential adjunctive therapies for CM showed no proven benefits and some interventions were even deleterious,4 stressing the need for a better understanding of CM pathogenesis to develop effective therapies.
An unresolved issue of CM pathogenesis regards the role of brain hemodynamic perturbations and ischemia. Sequestration of parasitized red blood cells (pRBCs) containing mature forms of the parasite in the brain microvasculature is a characteristic postmortem finding in human CM cases5 and together with rosetting6 and reduced RBC deformability7 may result in the obstruction of blood flow potentially leading to ischemia and hypoxia. In vivo studies of the microcirculation in human CM support this mechanism, with direct observation of retinal microvasculature showing impaired perfusion, retinal whitening, vascular occlusion, and ischemia.8 Accordingly, microvascular obstruction observed in the rectal mucosa of CM patients was proportional to the severity of the disease.9 In addition, hypovolemia, shock and intracranial hypertension, commonly associated with poor outcomes in CM,4 reduce tissue perfusion, and tissue hypoxia is one of the likely explanations for the acidosis frequently observed in severe malaria.7,10 Ischemic damage has also been shown in children with CM and was associated with severe neurological sequelae.11 On the other hand, transcranial Doppler sonography studies showed normal or even increased cerebral blood flow (CBF) velocities12–15 in large arteries during CM, which associated with microcirculatory obstruction has been suggested to increase cerebral blood volume leading to intracranial hypertension.16 Alternatively, collateral flow has been proposed as a mechanism to reconcile the findings of normal or increased CBF velocities and impaired perfusion,17 an interpretation supported by findings of hyperdynamic flow in capillaries adjacent to obstructed vessels.9 Interventions that improve cerebral perfusion have been proposed to be beneficial in CM.8,18
The murine model of CM by Plasmodium berghei ANKA (PbA) shares many features with the human pathology,19 including the presence of multiple brain microhemorrhages and vascular obstruction, although the nature of the sequestered cell (leukocytes) differs. In murine CM, magnetic resonance imaging (MRI) and spectroscopy studies showed the presence of brain edema, decreased CBF, and ischemia.20,21 Lack of resolution in MRI, however, precludes detailed studies of the microcirculation, which is a major target and player in CM pathogenesis. A few studies have addressed the in vivo microcirculatory changes in murine models of severe malaria,22–24 however in sites other than the brain (cremaster muscle or skin). In the present work, we used for the first time brain intravital microscopy to follow the dynamic changes in the pial microcirculation during the course of PbA infection in mice and show that expression of CM is associated with microcirculatory dysfunctions characterized by vasoconstriction, profound decrease in blood flow, and eventually vascular collapse, events similar to postsubarachnoid hemorrhage (SAH) vasospasm.25 We also show that nimodipine, a calcium channel blocker used to treat post-SAH vasospasm,25,26 markedly increased survival when given off-label to mice with CM as adjunctive therapy to artemether.
Materials and Methods
Parasite, Infection, and Clinical Assessment
All protocols were approved by the La Jolla Bioengineering Institutional Animal Care and Use Committee. Eight- to 10-week-old C57Bl/6 (Jackson Laboratories, Bar Harbor, ME) were inoculated intraperitoneally (IP) with 1 × 106 PbA parasites expressing the green fluorescent protein (PbA-GFP, a donation from the Malaria Research and Reference Reagent Resource Center – MR4, Manassas, VA; deposited by C.J. Janse and A.P. Waters; MR4 number: MRA-865). Parasitemia, body weight, and rectal temperature were checked daily from day 4. A motor behavior assessment modified from the SHIRPA protocol was used to determine the clinical status of the animals.27 Five tests were performed: transfer arousal, locomotor activity, tail elevation, wire maneuver, and righting reflex. For each test, mice received an individual score, and the sum of scores was used to create a composite score (scale 0 to 23, where 0 represents complete impairment in all individual tests—usually comatose animals—and 23 represents maximum performance). CM was defined as the presentation of one or more of the following clinical signs of neurological involvement: ataxia, limb paralysis, poor righting reflex, seizures, roll-over, coma.
Cranial Window Surgery and Intravital Microscopy
We used the closed cranial window model as described.28 Briefly, mice were anesthetized with ketamine- xylazine and were administered dexamethasone (0.2 mg/kg), carprofen (5 mg/kg), and ampicillin (6 mg/kg) subcutaneously, to prevent swelling of the brain, inflammatory response, and infection. After shaving the head and cleansing with ethanol 70% and betadine, the mouse was placed on a stereotaxic frame and the head immobilized using ear bars. The scalp was removed with sterilized surgical instruments, lidocaine-epinephrine was applied on the periosteum, which was then retracted exposing the skull. A 3- to 4-mm-diameter skull opening was made in the left parietal bone using a surgical drill. Under a drop of saline, the craniotomy was lifted away from the skull with very thin tip forceps and gelfoam previously soaked in saline applied to the dura mater to stop any eventual small bleeding. The exposed area was covered with a 5-mm glass coverslip secured with cyanocrylate-based glue and dental acrylic. Carprofen and ampicillin were given daily for three to five days after recovery from surgery. Mice presenting signs of pain or discomfort were euthanized with 100 mg/kg of euthasol IP. Two to three weeks after surgery, mice were lightly anesthetized with isoflurane (4% for induction, 1% to 2% for maintenance) and held on a stereotaxic frame. A panoramic picture of the vessels under the window was taken, and then mice were transferred to an intravital microscope stage (customized Leica-McBain, San Diego, CA). Body temperature was maintained using a heating pad. Using water-immersion objectives (×20), blood vessel images were captured (COHU 4815, San Diego, CA) and recorded on video-tape. An image shear device (Image Shear, Vista Electronics, San Diego, CA) was used to measure baseline vessel diameters (D), and RBC velocities (V) were measured off line by cross correlation (Photo Diode/Velocity Tracker Model 102B, Vista Electronics, San Diego, CA). Measurements of 6 to 10 pial venules (diameter range: 22 to 80 μm, velocity range: 2 to 4 mm/s) and 2 to 6 pial arterioles (diameter range: 18 to 70 μm, velocity range: 3 to 6 mm/s) were performed in each animal, and blood flow (Q) in each individual vessel was calculated using the equation: Q = V × π(D/2)2. The next day mice were inoculated IP with 1 × 106 PbA-GFP pRBC. The intravital microscopy procedure was repeated daily from day 4 of infection until the mice died or were euthanized. Noninfected control mice were submitted to the same procedures. To enhance imaging of the vascular network, including poorly perfused vessels, in two experiments animals with clinical signs of CM (n = 8) and controls (n = 3) were infused i.v. with albumin-FITC (1 mg/kg; Molecular Probes, Irvine, CA). Adherent and rolling leukocytes were visualized by anti-CD45-TxR antibodies (CalTag, Carlsbad, CA), also infused i.v. Green fluorescence (518 nm) emitted by albumin-FITC and GFP (PbA-GFP pRBC) was captured using ALPHA Vivid: XF100-2 (Omega Optical, Brattleboro, VT), and anti-CD45-TxR fluorescence (615 nm) was exited and captured with a Vivid Standard: XF42. To evaluate the effect of nimodipine on pial blood flow, PbA-infected mice with clinical CM and noninfected controls were imaged, vessel diameter and RBC velocities were measured, and then they were injected with artemether plus nimodipine at 4 mg/kg (as described below) and measurements were repeated at 30, 60, and 120 minutes.
Treatment
PbA-infected mice presenting poor righting reflex, hypothermia, and/or other clinical signs of neurological involvement such as ataxia, limb paralysis, seizures, and/or roll-over were treated with artemether (Artesiane, Dafra Pharma, Belgium, a kind gift of Dr. Alberto Moreno, Emory University, Atlanta, GA) given IP at 50 mg/kg, in combination with nimodipine (Sigma, St Louis, MO) or vehicle. Nimodipine was dissolved in ethanol (EMD, NJ), dispersed with polyethyleneglycol 400 (PEG, Sigma), and then saline was added (1:1:8 v/v) and mixed thoroughly. This solution was administered IP in three different doses: 1.3 mg/kg, 4 mg/kg, and 12 mg/kg. Artemether was given daily for five days, and nimodipine or vehicle were given at 0, 12, 24, and 36 hours. Parasitemia, motor behavior, and rectal temperature were checked at each time point and daily afterward. After treatment, parasitemia was checked by microscopical examination of Giemsa-stained blood smears to differentiate viable from dead parasites.
Statistical Analysis
Statistical analyses were performed using the Student t test with Mann–Whitney correction when comparing two groups, analysis of variance with Kruskall–Wallis post hoc analysis when comparing more than two groups, and survival curves were compared with a nonparametric log-rank test, using the Graphpad Prism software (GraphPad Software Inc., La Jolla, CA). A P value <0.05 was considered significant. Reported data are the mean ± SEM unless otherwise indicated.
Results
Mice with CM Present Vasoconstriction and Marked Decreases in Pial Blood Flow
Mice with an implanted cranial window and infected with PbA presented an overall CM incidence of 83% (19/23 mice, four separate experiments), deaths occurring on days 5 to 8 (Figure 1A) with parasitemias between 10% and 30% (Figure 1B). Infected mice developed hypothermia, more intense in CM mice (Figure 1C). Pial vascular hemodynamics was sequentially studied in the 23 PbA-infected and 10 noninfected control mice. Three of the 19 PbA-infected mice that developed CM died before the measurements could be performed at the time of presentation of clinical signs of CM. Therefore, complete hemodynamics data are available for 16 mice with CM, with a total of 56 arterioles and 114 venules analyzed. In the four PbA-infected mice that did not develop CM, 8 arterioles and 38 venules were analyzed, and in the 10 control mice a total of 33 arterioles and 83 venules were analyzed. Marked decreases in arteriolar and venular blood flows were observed during infection, particularly in mice presenting clinical signs of CM (Figure 2, A and B; see also Supplemental Videos S1, S2, and S3 at http://ajp.amjpathol.org). In mice presenting clinical CM, the mean decrease in arteriolar blood flow was 60% of baseline, with one (6%) mouse showing preserved blood flow, 8/16 (50%) CM mice a decrease in blood flow between 40% and 60%, and 7/16 (44%) CM mice more than 75% decrease (Figure 2C). This last group includes three mice with vascular network collapse (see below), two of them with no visible patent vessels and whose blood flow was considered to be zero in the area under the cranial window. The observed decreases in blood flow in mice with CM were attributable to low RBC velocities and also to vasoconstriction in 11 (79%) of 14 mice (Figures 3, A and B). Three (21%) CM mice presented vasodilation instead. Interestingly, on the day before CM development, half the mice actually presented increased (6% to 25%) vessel diameters, and aneurism-like “balloon” vessel changes were observed in four mice. The outcome of the balloon lesions was not determined in three of the four affected mice because of death before next examination (two mice) and lack of record in one mouse; in the one mouse followed up, the balloon vessel evolved to vascular collapse. The four PbA-infected mice that did not develop CM presented maximal decreases of arteriolar blood flow between 14% and 37% (mean: 25%) during follow up (Figure 2, A–C). Noninfected control mice showed stable vessel diameters and a mean 10% decrease in RBC velocities on days 6 to 10 of follow-up, resulting in slight to moderate decreases in blood flow in this period (mean: 5% on day 6; 15% on day 8; 9% on day 10; Figures 2 and 3). Three of the 10 noninfected control mice were followed up to day 16, with measurements performed every other day, and showed stable blood flow during this period (data not shown).
Figure 1.
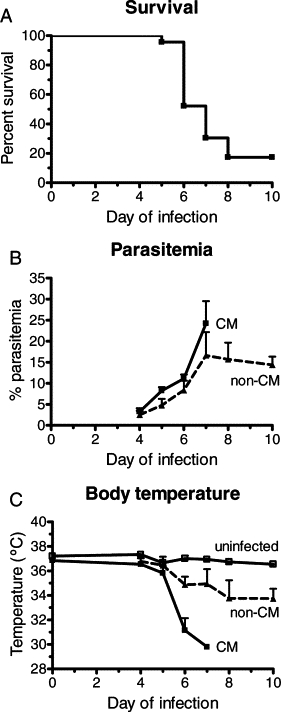
Cumulative survival (A), course of parasitemia (B), and rectal temperature (C) of PbA-infected mice that did (n = 19) or did not (n = 4) develop CM and in uninfected control mice (n = 10). Rectal temperature was lower in CM mice than in uninfected controls (P = 0.0004). Data in (B) and (C) are the mean ± SEM.
Figure 2.
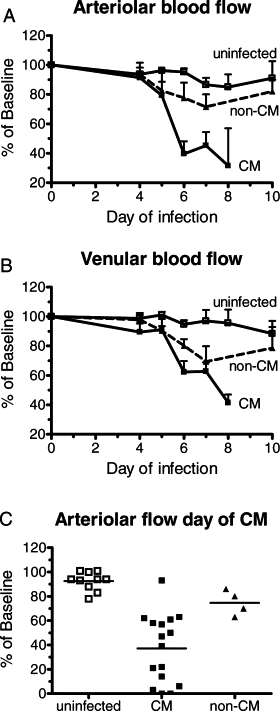
PbA infection leads to decreased blood flow in pial vessels. Arteriolar (A) and venular (B) blood flow in PbA-infected mice with or without CM and in uninfected control mice. Results are expressed as the percentage change in relation to baseline measurements performed before infection. Flow was significantly decreased on day 6 in mice that developed CM (arteriolar: P = 0.0003; venular: P = 0.0003). Data are the mean ± SEM. C: Arteriolar blood flow in mice with CM, at the time of presentation of clinical signs (irrespective of the day of infection). Flow was significantly decreased in mice with CM (P < 0.0001). Values shown for uninfected controls mice, and infected mice without CM are the lowest recorded for each mouse on days 5 to 8. Bars indicate the mean value.
Figure 3.
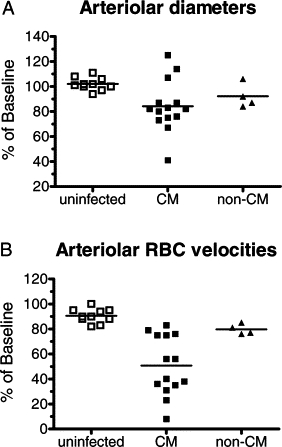
Changes in diameters and in RBC velocities in arterioles during PbA infection. A: Mice with CM showed a significant decrease in arteriolar diameters compared with uninfected controls (P = 0.0151). B: Mice with CM also showed significant decreases in RBC velocities in relation to uninfected controls (P < 0.0001). Bars indicate the mean value.
Adherent Leukocytes Cause Reduction of Luminal Diameters and Microvascular Blockade
In two experiments, after vessel diameter and RBC velocities were measured in mice presenting clinical CM, albumin-FITC and anti-CD45-TxR antibodies were injected in the tail vein to enhance imaging of the vascular network. In many venules of CM mice the large number of adherent leukocytes functioned as barriers to blood flow and caused marked reductions in luminal diameters (Figure 4, A–D; see also Supplemental Video S4 at http://ajp.amjpathol.org). RBC velocities were heterogeneous, with some larger vessels presenting sluggish RBC velocities and non-perfused feeding vessels (see Supplemental Videos S2 and S3 at http://ajp.amjpathol.org). Capillaries and smaller venules were frequently nonperfused, and in some cases adherent leukocytes were found to obstruct the lumen (Figure 4, E and F; see also Supplemental Videos S5 and S6 at http://ajp.amjpathol.org). We observed real-time obstruction of capillaries occurring during intravital microscopy (see Supplemental Video S6 at http://ajp.amjpathol.org). Nonperfused capillaries eventually collapsed (Figure 4, G and H). Most vessels with obstructed flow, showing no blood cell transit, still presented albumin-FITC–derived fluorescence and the adherent leukocytes, when present, showed anti-CD45 staining, suggesting that plasma flow was not completely impaired in such vessels. Sequestered pRBC were rarely observed and, in such cases, the trapped cells were usually attached to the surface of an endothelium-adherent leukocyte (Figures 5, A and B; see also Supplemental Video S7 at http://ajp.amjpathol.org), confirming previous observations by histology.29 It is conceivable that infusion of albumin and anti-CD45 antibodies might have beneficial effects in mice with CM and therefore altered outcome, albumin by expanding blood volume and improving perfusion,18,30 and anti-CD45 antibodies by potentially mediating destruction of leukocytes and decreasing inflammation.31 However, even if present, these effects might have been minor. The amount of albumin infused (1 mg/kg, or less than 20 μg/mouse) is almost insignificant compared with the albumin concentration in the plasma of mice (50 mg/ml), and the amount of anti-CD45 antibody infused (2 to 4 μg/mouse) was also small. In line with this interpretation, in the experiments in which albumin and anti-CD45 antibody infusion was performed, all CM mice died within 24 hours as expected, similar to the experiments in which infusion was not performed. In addition, no measurements of blood flow were performed after infusion.
Figure 4.
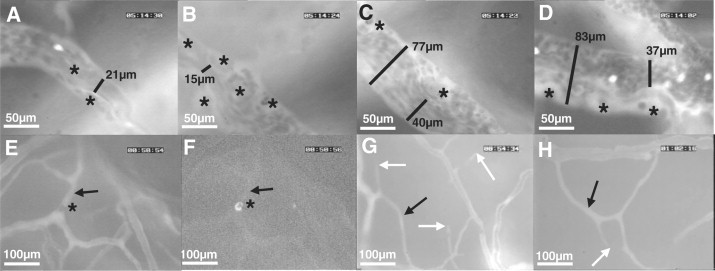
CM is associated with impaired perfusion in pial vessels. A–D: Four consecutive sections of the same venule showing how adherent leukocytes can cause marked reduction in luminal diameters and impair perfusion (a more detailed dynamic view is available at the supplemental video S3, at http://ajp.amjpathol.org). Asterisks show adherent leukocytes. The luminal diameter in specific sections is shown (black bars with the respective diameter value); note the large variations in diameter mostly attributable to adherent leukocytes, and a site of major constriction in B with apparent damage of the vessel structure. Bright spots in A and D are flowing fluorescent PbA-GFP pRBCs. Adherent leukocytes can completely block blood flow in small venules. E and F: Two consecutive frames showing vessels stained with albumin-FITC, one of them is nonperfused (arrow) blocked by an adherent leukocyte (asterisk; E) and the same area showing the leukocyte stained with anti-CD45-TxR antibodies (F; see dynamic view in supplemental video S5, at http://ajp.amjpathol.org). G and H: Collapse of nonperfused small vessels: disappearance of small vessels with no flow (white arrows), and associated nonflowing vessels (black arrows).
Figure 5.
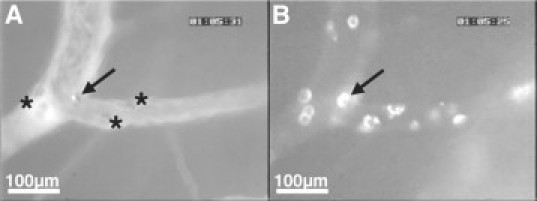
PbA pRBCs do not directly adhere to pial endothelial cells but may be found trapped by adherent leukocytes. A: A fluorescent PbA-GFP pRBC attached to a leukocyte (arrow). Other visible adherent leukocytes are indicated by asterisks. B: The same vessel section evidencing the adherent leukocytes, highlighted after changing the filter to detect TxR fluorescence. The arrow points to the leukocyte with the attached pRBC. See also supplemental video S7 at http://ajp.amjpathol.org.
CM can be Associated with Vascular Collapse
A striking feature observed in 7 of 16 (44%) mice with CM (three mice died before images could be taken), and in none of the control or non-CM mice, was the collapse of large pial vessels (Figures 6, A–C and D–F) or even of a microvascular network (Figures 6, G–I). Four mice presented one or few collapsed vessels, and three mice presented vascular network collapse. In two of the three cases of vascular network collapse, this was preceded by the occurrence of hemorrhage in a major vessel (Figures 6, J–L). Noteworthy, 71% (5/7) of the mice with vascular collapse presented decreases in blood flow over 75% in relation to baseline, against 22% (2/9) in the group of CM mice without vascular collapse. Mice with vascular collapse presented also more severe vasoconstriction, with a mean 32% decrease in overall arteriolar diameter of the remaining noncollapsed vessels in relation to baseline, against a mean 19% decrease in all mice with CM (P = 0.0162).
Figure 6.
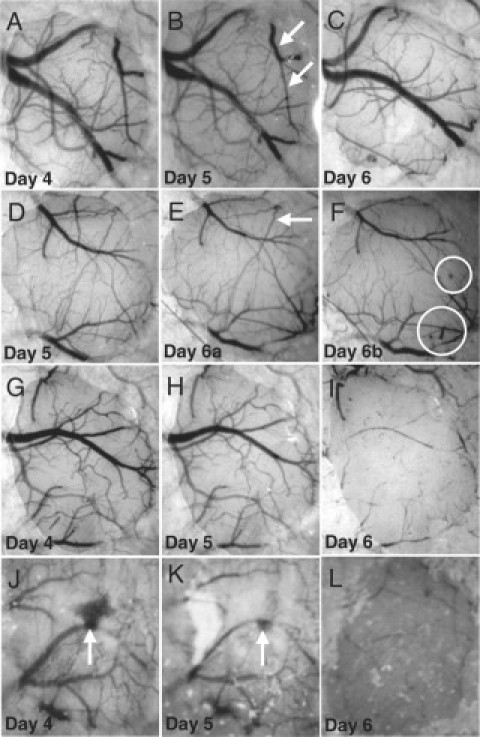
CM is associated with vascular collapse. A–C: Collapse of a major pial vessel and branches (arrows) on day 6 of infection. D–F: Collapse of two branches (arrows) of a vessel and microhemorrhages (encircled); note that vessel collapse occurred between two observations in the same day (6), in between which the mouse developed clinical CM. G–I: Collapse of virtually the entire pial vascular network under the cranial window on day 6 of infection. J–L: Collapse of the vascular network under the window on day 6 after a major hemorrhage (arrow) on days 4 and 5.
Nimodipine Increases Survival of Mice with CM on Rescue Treatment with Artemether
The observations of vasoconstriction and vascular collapse, which may occur in association with pial hemorrhage, are reminiscent of the vasospasm phenomenon that frequently occurs after SAH and is associated with neurological deterioration and poor prognosis.25,32 The standard drug for prevention and treatment of post-SAH vasospasm is nimodipine,26 a dihydropyridine that blocks specifically the L-type voltage-gated calcium channels, which in the vascular system mediate cytoplasmic calcium influx-dependent vasoconstriction.33 We asked whether nimodipine could be effective off-label if used as adjunctive therapy with artemether, a drug used to treat human CM, in mice with clinically well-defined CM, ie, with obligatory presentation of one or more of the neurological signs—ataxia, limb paralysis, seizures, roll-over, poor righting reflex—in addition to moderate or severe hypothermia (mean ± SD: 31.5 ± 1.1°C; uninfected controls: 37.1 ± 0.6°C) and low composite motor scores (mean ± SD: 5.8 ± 3.1; uninfected controls: 22.3 ± 0.7). The adjunctive administration of nimodipine at 4 and 12 mg/kg, but not 1.3 mg/kg, was able to rescue twice as many (60% to 66.6% survival) mice from CM and death than did artemether plus vehicle (32.2% survival; Figure 7A). There were no significant differences in motor scores, body temperature, and parasitemia levels between the different treatment groups. In addition, mice treated with nimodipine at 4 mg/kg but succumbing showed prolonged survival, with half of the deaths occurring over a period of 48 to 120 hours after the first dose, whereas in the vehicle-treated group 95.2% of deaths occurred in the first 36 hours. Finally, survivor mice treated with nimodipine showed faster recovery than did survivor mice treated with vehicle, with significantly higher motor scores at 48 and 72 hours (Figure 7B). There was no difference in the rate of parasite clearance in the nimodipine and vehicle groups (Figure 7C). Within one week of treatment, all but one (97.2%) of the survivor mice in all groups presented apparent full recovery from the neurological syndrome, only one mouse showing evidence of sequelae three weeks after treatment (head slightly leaned to the left after recovery from ataxia, rollover, hypothermia, hemiparesis, and a trend to walk in circles with poor coordination). Treatment interventions without artemether had no apparent beneficial effect, as all CM mice treated with either nimodipine 4 mg/kg (n = 8) or vehicle (n = 6) died within 12 hours of treatment (data not shown).
Figure 7.
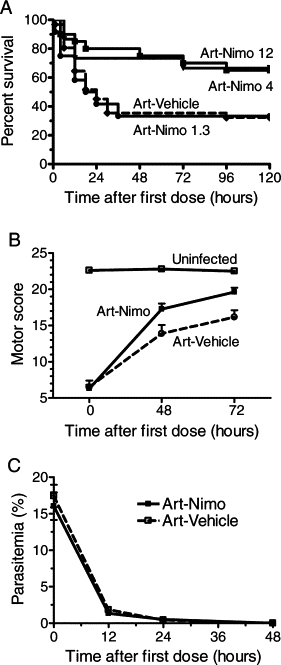
Nimodipine improves the death-rescuing capacity of artemether on mice with established CM. A: Cumulative survival of PbA-infected mice presenting clinical CM and treated with artemether-nimodipine (1.3 mg/kg, n = 12; 4 mg/kg, n = 20; and 12 mg/kg, n = 15) or artemether-vehicle (n = 31). A total of nine experiments were conducted. Survival was significantly increased in mice treated with 4 mg/kg (P = 0.0217) and 12 mg/kg (P = 0.0474), but not with 1.3 mg/kg. B: Survivor mice treated with artemether plus nimodipine (n = 20) presented faster clinical recovery than survivor mice treated with artemether-vehicle (n = 9) with significantly higher scores at 48 (P = 0.0402) and 72 (P = 0.0238) hours. C: Efficacy of artemether treatment: parasitemia decreased fast after artemether administration in mice treated with nimodipine or vehicle. Data are the mean ± SEM.
Nimodipine Induces Dilatation of Pial Arterioles
We asked whether the adjunctive effect of nimodipine was related to its vasorelaxation activity and ability to improve blood flow in the brain. When PbA-infected mice with an implanted cranial window and presenting clinical CM were given nimodipine 4 mg/kg IP, arteriolar blood flow increased by about 50% after 30 minutes of injection, then decreased but remained above baseline after 2 hours (Figure 8A). Vascular response in noninfected control mice was similar, but with a rebound at 60 minutes after the peak at 30 minutes. The increase in blood flow in mice with CM was attributable solely to a sustained increase in arteriolar diameters, because RBC velocities were even slightly decreased (Figure 8B).
Figure 8.
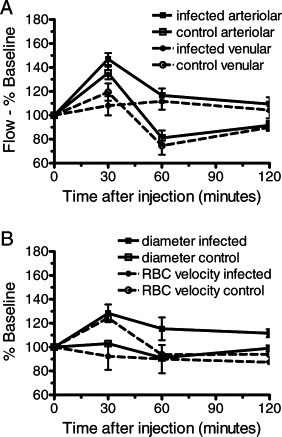
Nimodipine increases blood flow in CM mice through vasorelaxation. A: Administration of nimodipine at 4 mg/kg IP caused an increase in arteriolar blood flow in mice with clinical CM (n = 3) and in uninfected control mice (n = 3). B: Increase in arteriolar blood flow was attributable to a sustained increase in vascular diameter. Data are the mean ± SEM.
Discussion
The present study shows that brain microcirculatory hemodynamics and physiology are severely compromised during murine CM, with marked decreases in arteriolar blood flow at the time of CM manifestation, confirming that ischemia plays a significant role in murine CM.20,21 Remarkably, vasoconstriction and vascular collapse were characterized as novel mechanisms leading to ischemia in CM. The marked impact of the calcium channel blocker nimodipine on the survival rates of mice with CM as well as its effect in inducing vasodilation provide further support for the role of vasoconstriction and poor blood flow in murine CM pathogenesis, and generate a concept for a potential adjunctive therapy for CM.
Brain intravital microscopy has a number of unique advantages, mainly the direct in vivo dynamic observation of the brain vasculature and the possibility to conduct sequential studies in the same animal and follow the evolution of the pathogenic process. The most important limitation is that only a small area of the brain can be assessed, therefore we can only infer that the pathological events observed under the window occur in the remaining regions of the brain. However, the frequency at which key pathological manifestations such as the decreased blood flow, vasoconstriction, and the vascular collapse occurred in infected but not in control animals, as well as the beneficial effect of nimodipine, suggest that the events reported are indeed representative and relevant for the pathogenic process of CM.
Brain hemodynamic changes in mice with clinical CM have been described using MRI and showed brain hemodynamic perturbations with 30% to 40% decreases in CBF and voids in arterial blood signal.20 The presence of associated brain edema prompted the postulation of impaired CBF as caused by compression of arteries attributable to intracranial hypertension. Our results suggest a more complex picture, with a number of factors contributing to impaired perfusion. First, we showed that decreases in both vessel diameters and blood velocities contribute to decreased CBF in murine CM. Second, vasoconstriction itself does not seem to be solely a result of passive vascular compression but rather an active phenomenon, because blockade of calcium channels by nimodipine administration induced a vasodilation response in CM mice. This is in keeping with evidence in human and murine CM showing vascular dysfunction attributable to low nitric oxide (NO) bioavailability34 and potentially other vasoconstrictive mechanisms.35–37 Third, leukocyte adhesion in brain vessels acted as barriers for blood flow by decreasing luminal diameters and directly blocking capillaries. These dynamic observations provided by intravital microscopy support the long-disputed argument in favor of a role for microvascular sequestration in contributing to impaired CBF and ischemia in CM.38–40 Fourth, 44% of the CM mice presented some degree of vascular collapse, and these animals presented also more severe vasoconstriction, showing that in addition to a global decrease in blood flow, areas of more severe focal ischemia may develop in the brain of mice with CM. Indeed, 78% of CM mice presenting no vascular collapse showed blood flow levels compatible with a state of ischemic penumbra where damage is still potentially reversible,41 an interpretation supported by the beneficial effect of nimodipine. Conversely, 71% of mice with vascular collapse showed blood flow levels indicative of infarct and irreversible damage. The overall patterns of poor perfusion and vascular occlusion observed in mice pial vessels were similar to the findings by retinal angiography in human CM,8 and similarities can be drawn between the pial vascular collapse and the retinal whitening.
Overall, these data indicate that CBF is impaired in mice with CM attributable to increased cerebrovascular resistance42 resulting from vasoconstriction as well as capillary and venular nonperfusion, and also to decreased cerebral perfusion pressure resulting from low blood velocities likely attributable to hypotension, low cardiac output, and intracranial hypertension. Interestingly, although vasoconstriction was a hallmark in 79% of mice with clinical signs of CM, half of these animals presented vasodilation in the day before clinical CM manifested. The vasodilation may occur as an autoregulatory response to decreased blood velocity in an attempt to maintain blood supply. The reason this response was not sustained and instead turned into a vasoconstriction is unknown, but it may be related to low NO bioavailability.34
Vasoconstriction and vascular collapse have not been described in previous intravital studies of the skin and muscle microcirculation in rodent models of severe malaria,22–24 although in the P. yoelii model major decreases in blood flow were shown.22,23 However, these models and the sites of observations differ significantly from the present study and cannot be directly compared.
The findings of vasoconstriction and vascular collapse, in some cases preceded by aneurism-like vessel changes and vessel ruptures with major hemorrhages, led us to identify similarities between murine CM and SAH. In this case, blood released from ruptured vessels would play an active role in murine CM pathogenesis by causing vasospasm most likely through the potent NO-scavenging and pro-oxidant actions of hemoglobin, which also induces endothelin-1 synthesis.32 Although major hemorrhages were observed in only two of the seven mice with vascular collapse, it is remarkable that these two mice presented the most severe form of collapse. Subarachnoid hemorrhages have been described in murine43 and human44,45 CM, although the hallmark in both cases is the presence of disseminated cerebral ring microhemorrhages.43,46 We propose that systemic NO deficiency resulting from repeated cycles of hemolysis34 is a major cause of vasoconstriction in murine CM, which in association with low RBC velocities and vascular occlusion affects blood flow throughout the brain. Ischemia resulting from this global shortage in blood supply per se does not explain CM, as other plasmodial infections can induce similar or even more intense hemolysis and yet no CM. However, further cerebral vascular damage and rupture causes additional localized deprivation of blood supply, passively by the collapse of the ruptured vessel itself and actively by the vasospasm-inducing activity of the leaked blood on the adjacent vessels. The occurrence of multiple microhemorrhagic foci would lead to multifocal infarcts, and reversibility of this scenario would depend on the severity of ischemia, with widespread infarct areas indicating poor outcomes. Although this mechanism, as proposed, remains to be demonstrated, the marked efficacy of nimodipine, the only drug shown to improve outcome in patients with post-SAH vasospasm,25 in improving the CM-rescuing capacity of artemether, as well as its ability to induce vasodilation in pial vessels of mice with CM, provide support for this concept. The fact that exogenous NO prevents CM34 development also favors this interpretation, as do other interventions that induce vasodilation such as carbon monoxide therapy,47 or that have a beneficial effect on ischemic stroke, such as hyperbaric oxygen48 and erythropoietin.49 Most of these approaches have been used in the murine model to prevent, not reverse, CM, therefore other mechanisms such as the down-regulation of endothelial cell adhesion molecules and inhibition of inflammation preventing vascular damage are likely to take place. The primary beneficial effect of nimodipine can be attributed to its effect in reversing calcium-dependent vasoconstriction,33 hence decreasing cerebrovascular resistance and improving CBF, but nimodipine may also act through additional mechanisms. Ischemia, for instance, can cause deregulation of nerve cell membrane polarization, with large influx of calcium and ultimately neuronal cell death,50 and nimodipine could act by preventing or reversing this process.51 Calcium channel blockers have also been shown to decrease vascular inflammation and oxidative stress.52 A more thorough characterization of nimodipine effects in CM mice is therefore necessary.
Human CM is a complex pathophysiological entity with a wide range of presentations,4,53 therefore the murine model cannot mirror all its features,19,20 and translation of results obtained in the mouse model to the human situation is not straightforward. Cerebral hemodynamics is particularly difficult to compare because of few and apparently conflicting findings in human studies. Transcranial Doppler sonography studies showed mostly normal and sometimes increased CBF velocities in CM patients,12–15 although wide ranges and large differences between brain hemispheres were observed. In the larger study, although 30% of children initially presented increased CBF velocities, the children with fatal outcomes presented low CBF velocity recordings after admission.12 In another study, whereas Transcranial Doppler showed normal CBF velocities, computer tomography and near-infrared spectroscopy showed hypoperfusion in the same CM patient.15 Although not directly evidenced, cerebral vasodilation and increased blood volume have been proposed to occur during human CM, contributing to intracranial hypertension.4,11,17 On the other hand, the occurrence of hypoperfusion, vascular occlusion, and ischemia has been clearly demonstrated in vivo by retinal angiography in CM patients.8 Other findings suggestive of hypoperfusion such as increased cerebrovascular resistance,13 intracranial hypertension, hypovolemia, systemic hypotension, acidosis, and ischemic damage are associated with poor outcomes in human CM,4,11,18,53 and interventions that improve cerebral perfusion have been proposed to be beneficial.8,18 Definitive evidence of cerebral vasoconstriction in human CM is lacking, although histological findings suggestive of arteriolar spasm have been described in both human and murine CM,54 and raised serum levels of vasoconstrictive factors such as endothelin-1 have been associated with human and murine CM.35–37 It is also noteworthy that TNF-α, which is believed to play an important role in murine and human CM pathogenesis,19,40 has been shown to induce constriction of pial arterioles in piglets55 as well as vasoconstriction and reduction of cerebral blood volume in rats via an endothelin-dependent pathway.56 The apparent contradictory data regarding cerebral hemodynamics during human CM may arise from the heterogeneity of presentations of human CM. As we showed here, in murine CM vasoconstriction can be preceded by vasodilation, therefore the timing at which measurements are made is crucial. The nature of the methods used also generates different types of data and interpretations. Transcranial Doppler, for instance, estimates blood velocity in large cerebral arteries, and retinal angiography looks at the microcirculation. Collateral flow has been proposed to reconcile increased blood flow velocities in the presence of venular obstruction and impaired perfusion.16 Indeed, hyperdynamic flow has been shown to occur in vessels adjacent to nonflowing occluded vessels during severe malaria.9
Given the lack of consensus on the nature and the role of cerebral hemodynamic changes in human CM and the considerable heterogeneity of case presentations, potential adjunctive therapies targeting blood flow and vascular physiology must be taken with due caution. Caution is emphasized when considering that a number of adjunctive therapy trials with different interventions showed either no efficacy or even deleterious effects.4 In this regard, before any consideration for use in humans is taken, it is also necessary to perform a thorough evaluation of the potential adverse effects of nimodipine administration in mice with CM, for instance its potential hypotensive effect. In addition, given the observation of vasodilation in half of the mice the day before CM development, it is relevant to ask whether administration of nimodipine at this stage would be deleterious instead by causing effects such as facilitating the rupture of damaged vessels. Therefore, the timing of nimodipine administration is important. Noticeably, development of clinical CM prompting treatment in mice occurred mostly (80% of cases) when vasoconstriction rather than vasodilation was present. It is also worth mentioning that nimodipine has been safely used in humans for many years. The need for an effective adjunctive therapy for CM is imperative, and the marked improvement in both the rate and the pace of recovery afforded by nimodipine in murine CM indicates that it holds potential and deserves further investigation.
Acknowledgements
We thank Dr. Alberto Moreno (Emory University, Atlanta) for providing the artemether used in this study, Leah Clemmer (LJBI) for helping in one of the nimodipine treatment experiments, Dr. Antonio Teixeira and Norinne Queiroz (UFMG, Brazil) for providing initial training in the open cranial window surgery, and Dr. Claudio Tadeu Daniel Ribeiro and Yuri Chaves Martins (IOC-Fiocruz, Brazil) for providing training in the motor behavior assessment of mice.
Footnotes
Supported with funds from the United States National Heart, Lung, and Blood Institute grant to L.J.M.C. (R01-HL87290). G.M.Z. is recipient of a postdoctoral fellowship from the Brazilian Research Council (CNPq).
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
References
- 1.Rowe AK, Rowe SY, Snow RW, Korenromp EL, Schellenberg JR, Stein C, Nahlen BL, Bryce J, Black RE, Steketee RW. The burden of malaria mortality among African children in the year 2000. Int J Epidemiol. 2006;35:691–704. doi: 10.1093/ije/dyl027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McIntosh HM, Olliaro P. Artemisinin derivatives for treating severe malaria. Cochrane Database Syst Rev. 2000;2:CD000527. doi: 10.1002/14651858.CD000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.John CC, Bangirana P, Byarugaba J, Opoka RO, Idro R, Jurek AM, Wu B, Boivin MJ. Cerebral malaria in children is associated with long-term cognitive impairment. Pediatrics. 2008;122:e92–e99. doi: 10.1542/peds.2007-3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Idro R, Jenkins NE, Newton CR. Pathogenesis, clinical features, and neurological outcome of cerebral malaria. Lancet Neurol. 2005;4:827–840. doi: 10.1016/S1474-4422(05)70247-7. [DOI] [PubMed] [Google Scholar]
- 5.MacPherson GG, Warrell MJ, White NJ, Looareesuwan S, Warrell DA. Human cerebral malaria. A quantitative ultrastructural analysis of parasitized erythrocyte sequestration. Am J Pathol. 1985;119:385–401. [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandez V, Wahlgren M. Rosetting and autoagglutination in Plasmodium falciparum. Chem Immunol. 2002;80:163–187. [PubMed] [Google Scholar]
- 7.Dondorp AM, Pongponratn E, White NJ. Reduced microcirculatory flow in severe falciparum malaria: pathophysiology and electron-microscopic pathology. Acta Trop. 2004;89:309–317. doi: 10.1016/j.actatropica.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Beare NA, Harding SP, Taylor TE, Lewallen S, Molyneux ME. Perfusion abnormalities in children with cerebral malaria and malarial retinopathy. J Infect Dis. 2009;199:263–271. doi: 10.1086/595735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dondorp AM, Ince C, Charunwatthana P, Hanson J, van Kuijen A, Faiz MA, Rahman MR, Hasan M, Bin Yunus E, Ghose A, Ruangveerayut R, Limmathurotsakul D, Mathura K, White NJ, Day NP. Direct in vivo assessment of microcirculatory dysfunction in severe falciparum malaria. J Infect Dis. 2008;197:79–84. doi: 10.1086/523762. [DOI] [PubMed] [Google Scholar]
- 10.Maitland K, Newton CR. Acidosis of severe falciparum malaria: heading for a shock? Trends Parasitol. 2005;21:11–16. doi: 10.1016/j.pt.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Newton CR, Peshu N, Kendall B, Kirkham FJ, Sowunmi A, Waruiru C, Mwangi I, Murphy SA, Marsh K. Brain swelling and ischaemia in Kenyans with cerebral malaria. Arch Dis Child. 1994;70:281–287. doi: 10.1136/adc.70.4.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newton CR, Marsh K, Peshu N, Kirkham FJ. Perturbations of cerebral hemodynamics in Kenyans with cerebral malaria. Pediatr Neurol. 1996;1:41–49. doi: 10.1016/0887-8994(96)00115-4. [DOI] [PubMed] [Google Scholar]
- 13.Warrell DA, White NJ, Veall N, Looareesuwan S, Chanthavanich P, Phillips RE, Karbwang J, Pongpaew P, Krishna S. Cerebral anaerobic glycolysis and reduced cerebral oxygen transport in human cerebral malaria. Lancet. 1988;2:534–538. doi: 10.1016/s0140-6736(88)92658-x. [DOI] [PubMed] [Google Scholar]
- 14.Clavier N, Rahimy C, Falanga P, Ayivi B, Payen D. No evidence for cerebral hypoperfusion during cerebral malaria. Crit Care Med. 1999;27:628–632. doi: 10.1097/00003246-199903000-00047. [DOI] [PubMed] [Google Scholar]
- 15.Kampfl A, Pfausler B, Haring HP, Denchev D, Donnemiller E, Schmutzhard E. Impaired microcirculation and tissue oxygenation in human cerebral malaria: a single photon emission computed tomography and near-infrared spectroscopy study. Am J Trop Med Hyg. 1997;56:585–587. doi: 10.4269/ajtmh.1997.56.585. [DOI] [PubMed] [Google Scholar]
- 16.Newton CR, Kirkham FJ, Winstanley PA, Pasvol G, Peshu N, Warrell DA, Marsh K. Intracranial pressure in African children with cerebral malaria. Lancet. 1991;337:573–576. doi: 10.1016/0140-6736(91)91638-b. [DOI] [PubMed] [Google Scholar]
- 17.White NJ. Cerebral perfusion in cerebral malaria. Crit Care Med. 1999;27:478–479. doi: 10.1097/00003246-199903000-00020. [DOI] [PubMed] [Google Scholar]
- 18.Maitland K, Pamba A, English M, Peshu N, Marsh K, Newton C, Levin M. Randomized trial of volume expansion with albumin or saline in children with severe malaria: preliminary evidence of albumin benefit. Clin Infect Dis. 2005;40:538–545. doi: 10.1086/427505. [DOI] [PubMed] [Google Scholar]
- 19.Hunt NH, Grau GE. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003;9:491–499. doi: 10.1016/s1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- 20.Penet MF, Viola A, Confort-Gouny S, Le Fur Y, Duhamel G, Kober F, Ibarrola D, Izquierdo M, Coltel N, Gharib B, Grau GE, Cozzone PJ. Imaging experimental cerebral malaria in vivo: significant role of ischemic brain edema. J Neurosci. 2005;25:7352–7358. doi: 10.1523/JNEUROSCI.1002-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanni LA, Rae C, Maitland A, Stocker R, Hunt NH. Is ischemia involved in the pathogenesis of murine cerebral malaria? Am J Pathol. 2001;159:1105–1112. doi: 10.1016/S0002-9440(10)61786-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaul DK, Nagel RL, Llena JF, Shear HL. Cerebral malaria in mice: demonstration of cytoadherence of infected red blood cells and microrheologic correlates. Am J Trop Med Hyg. 1994;50:512–521. doi: 10.4269/ajtmh.1994.50.512. [DOI] [PubMed] [Google Scholar]
- 23.Kaul DK, Liu XD, Nagel RL, Shear HL. Microvascular hemodynamics and in vivo evidence for the role of intercellular adhesion molecule-1 in the sequestration of infected red blood cells in a mouse model of lethal malaria. Am J Trop Med Hyg. 1998;58:240–247. doi: 10.4269/ajtmh.1998.58.240. [DOI] [PubMed] [Google Scholar]
- 24.Martini J, Gramaglia I, Intaglietta M, van der Heyde HC. Impairment of functional capillary density but not oxygen delivery in the hamster window chamber during severe experimental malaria. Am J Pathol. 2007;170:505–517. doi: 10.2353/ajpath.2007.060433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keyrouz SG, Diringer MN. Prevention and therapy of vasospasm in subarachnoid hemorrhage. Crit Care. 2007;11:220–229. doi: 10.1186/cc5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dorhout Mees SM, Rinkel GJ, Feigin VL, Algra A, van den Bergh WM, Vermeulen M, van Gijn J. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev. 2007;18:CD000277. doi: 10.1002/14651858.CD000277.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lackner P, Beer R, Heussler V, Goebel G, Rudzki D, Helbok R, Tannich E, Schmutzhard E. Behavioural and histopathological alterations in mice with cerebral malaria. Neuropathol Appl Neurobiol. 2006;32:177–188. doi: 10.1111/j.1365-2990.2006.00706.x. [DOI] [PubMed] [Google Scholar]
- 28.Mostany R, Portera-Cailliau C. A craniotomy surgery procedure for chronic brain imaging. J Vis Exp. 2008;15:680. doi: 10.3791/680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martins YC, Smith MJ, Pelajo-Machado M, Werneck GL, Lenzi HL, Daniel-Ribeiro CT, Carvalho LJ. Characterization of cerebral malaria in the outbred Swiss Webster mouse infected by Plasmodium berghei ANKA. Int J Exp Pathol. 2009;90:119–130. doi: 10.1111/j.1365-2613.2008.00622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cabrales P, Tsai AG, Ananda K, Acharya SA, Intaglietta M. Volume resuscitation from hemorrhagic shock with albumin and hexaPEGylated human serum albumin. Resuscitation. 2008;79:139–146. doi: 10.1016/j.resuscitation.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wulf GG, Luo KL, Goodell MA, Brenner MK. Anti-CD45-mediated cytoreduction to facilitate allogeneic stem cell transplantation. Blood. 2003;101:2434–2439. doi: 10.1182/blood-2002-08-2379. [DOI] [PubMed] [Google Scholar]
- 32.Pluta RM, Hansen-Schwartz J, Dreier J, Vajkoczy P, Macdonald RL, Nishizawa S, Kasuya H, Wellman G, Keller E, Zauner A, Dorsch N, Clark J, Ono S, Kiris T, Leroux P, Zhang JH. Cerebral vasospasm following subarachnoid hemorrhage: time for a new world of thought. Neurol Res. 2009;31:151–158. doi: 10.1179/174313209X393564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ureña J, del Valle-Rodríguez A, López-Barneo J. Metabotropic Ca2+ channel-induced calcium release in vascular smooth muscle. Cell Calcium. 2007;42:513–520. doi: 10.1016/j.ceca.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 34.Gramaglia I, Sobolewski P, Meays D, Contreras R, Nolan JP, Frangos JA, Intaglietta M, van der Heyde HC. Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat Med. 2006;12:1417–1422. doi: 10.1038/nm1499. [DOI] [PubMed] [Google Scholar]
- 35.Wenisch C, Wenisch H, Wilairatana P, Looareesuwan S, Vannaphan S, Wagner O, Graninger W, Schönthal E, Rumpold H. Big endothelin in patients with complicated Plasmodium falciparum malaria. J Infect Dis. 1996;173:1281–1284. doi: 10.1093/infdis/173.5.1281. [DOI] [PubMed] [Google Scholar]
- 36.Dietmann A, Lackner P, Helbok R, Spora K, Issifou S, Lell B, Reindl M, Kremsner PG, Schmutzhard E. Opposed circulating plasma levels of endothelin-1 and C-type natriuretic peptide in children with Plasmodium falciparum malaria. Malar J. 2008;7:253–259. doi: 10.1186/1475-2875-7-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Machado FS, Desruisseaux MS, Nagajyothi, Kennan RP, Hetherington HP, Wittner M, Weiss LM, Lee SC, Scherer PE, Tsuji M, Tanowitz HB. Endothelin in a murine model of cerebral malaria. Exp Biol Med. 2006;1:1176–1181. [PubMed] [Google Scholar]
- 38.van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 2006;22:503–508. doi: 10.1016/j.pt.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 39.Berendt AR, Tumer GD, Newbold CI. Cerebral malaria: the sequestration hypothesis. Parasitol Today. 1994;10:412–414. doi: 10.1016/0169-4758(94)90238-0. [DOI] [PubMed] [Google Scholar]
- 40.Clark IA, Alleva LM. Is human malarial coma caused, or merely deepened, by sequestration? Trends Parasitol. 2009;25:314–318. doi: 10.1016/j.pt.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 41.Hossmann KA. Pathophysiology and therapy of experimental stroke. Cell Mol Neurobiol. 2006;26:1057–1083. doi: 10.1007/s10571-006-9008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kulik T, Kusano Y, Aronhime S, Sandler AL, Winn HR. Regulation of cerebral vasculature in normal and ischemic brain. Neuropharmacology. 2008;55:281–288. doi: 10.1016/j.neuropharm.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carvalho LJM, Lenzi HL, Pelajo-Machado M, Oliveira DN, Daniel-Ribeiro CT, Ferreira-da-Cruz MF. Plasmodium berghei: cerebral malaria in CBA mice is not clearly related to plasma TNF levels or intensity of histopathological changes. Exp Parasitol. 2000;95:1–7. doi: 10.1006/expr.2000.4508. [DOI] [PubMed] [Google Scholar]
- 44.Murugavel K, Saravanapavananthan S, Anpalahan A, James RF. Subarachnoid haemorrhage in Plasmodium falciparum malaria. Postgrad Med J. 1989;65:236–237. doi: 10.1136/pgmj.65.762.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gall C, Spuler A, Fraunberger P. Subarachnoid hemorrhage in a patient with cerebral malaria. N Engl J Med. 1999;341:611–613. doi: 10.1056/NEJM199908193410814. [DOI] [PubMed] [Google Scholar]
- 46.Marsden PD, Bruce-Chwatt LJ. Cerebral malaria. Contemp Neurol Ser. 1975;12:29–44. [PubMed] [Google Scholar]
- 47.Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, Chora A, Rodrigues CD, Gregoire IP, Cunha-Rodrigues M, Portugal S, Soares MP, Mota MM. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med. 2007;13:703–710. doi: 10.1038/nm1586. [DOI] [PubMed] [Google Scholar]
- 48.Blanco YC, Farias AS, Goelnitz U, Lopes SCP, Arrais-Silva WW, Carvalho BO, Amino R, Wunderlich G, Santos LMB, Giorgio S, Costa FTM. Hyperbaric oxygen prevents early death caused by experimental cerebral malaria. PLoS ONE. 2008;3:e3126. doi: 10.1371/journal.pone.0003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaiser K, Texier A, Ferrandiz J, Buguet A, Meiller A, Latour C, Peyron F, Cespuglio R, Picot S. Recombinant human erythropoietin prevents the death of mice during cerebral malaria. J Infect Dis. 2006;193:987–995. doi: 10.1086/500844. [DOI] [PubMed] [Google Scholar]
- 50.Krnjević K. Electrophysiology of cerebral ischemia. Neuropharmacology. 2008;55:319–333. doi: 10.1016/j.neuropharm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 51.Ouardouz M, Nikolaeva MA, Coderre E, Zamponi GW, McRory JE, Trapp BD, Yin X, Wang W, Woulfe J, Stys PK. Depolarization-induced Ca2+ release in ischemic spinal cord white matter involves L-type Ca2+ channel activation of ryanodine receptors. Neuron. 2003;40:53–63. doi: 10.1016/j.neuron.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 52.Yoshii T, Iwai M, Li Z, Chen R, Ide A, Fukunaga S, Oshita A, Mogi M, Higaki J, Horiuchi M. Regression of atherosclerosis by amlodipine via anti-inflammatory and anti-oxidative stress actions. Hypertens Res. 2006;29:457–466. doi: 10.1291/hypres.29.457. [DOI] [PubMed] [Google Scholar]
- 53.Newton CR, Hien TT, White N. Cerebral malaria. Neurol Neurosurg Psychiatry. 2000;69:433–441. doi: 10.1136/jnnp.69.4.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Polder TW, Jerusalem CR, Eling WM. Morphological characteristics of intracerebral arterioles in clinical (Plasmodium falciparum) and experimental (Plasmodium berghei) cerebral malaria. J Neurol Sci. 1991;101:35–46. doi: 10.1016/0022-510x(91)90016-z. [DOI] [PubMed] [Google Scholar]
- 55.Megyeri P, Abrahám CS, Temesvári P, Kovács J, Vas T, Speer CP. Recombinant human tumor necrosis factor alpha constricts pial arterioles and increases blood-brain barrier permeability in newborn piglets. Neurosci Lett. 1992;148:137–140. doi: 10.1016/0304-3940(92)90823-p. [DOI] [PubMed] [Google Scholar]
- 56.Sibson NR, Blamire AM, Perry VH, Gauldie J, Styles P, Anthony DC. TNF-alpha reduces cerebral blood volume and disrupts tissue homeostasis via an endothelin- and TNFR2-dependent pathway. Brain. 2002;125:2446–2459. doi: 10.1093/brain/awf256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.