

Abstract
Cystic fibrosis (CF), which is caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR), is characterized by multiorgan pathology that begins early in life. To better understand the initial stages of disease, we studied the gastrointestinal pathology of CFTR−/− pigs. By studying newborns, we avoided secondary changes attributable to environmental interactions, infection, or disease progression. Lesions resembling those in humans with CF were detected in intestine, pancreas, liver, gallbladder, and cystic duct. These organs had four common features. First, disease was accelerated compared with that in humans, which could provide a strategy to discover modifying factors. Second, affected organs showed variable hyperplastic, metaplastic, and connective tissue changes, indicating that remodeling was a dynamic component of fetal life. Third, cellular inflammation was often mild to moderate and not always present, which raises new questions as to the role of cellular inflammation in early disease pathogenesis. Fourth, epithelial mucus-producing cells were often increased, producing a striking accumulation of mucus with a layered appearance and resilient structure. Thus, mucus cell hyperplasia and mucus accumulation play prominent roles in early disease. Our findings also have implications for CF lung disease, and they lay the foundation for a better understanding of CF pathogenesis.
Early in the twentieth century, pioneering researchers established a framework for understanding the pathology of cystic fibrosis (CF). In 1905, Landsteiner described meconium ileus associated with pancreatic disease in a newborn.1 By 1938, Andersen documented the common pancreatic pathology in a cohort of CF patients and applied the term “cystic fibrosis of the pancreas” in her description of 49 pediatric cases.2 Later, Farber recognized significant involvement of multiple organs by a tenacious, thick mucus, which led to his descriptive terminology of “mucoviscidosis.”3,4 Almost a decade later, Bodian published a seminal record of young pediatric CF disease with detailed medical history and images that arguably have not been equaled since.5 At that time, CF was a pediatric disease with a high mortality, and the study of pathological changes in CF tissues from young infants and children5–8 generated hypotheses about pathogenesis and therapies. Fortunately, that era has passed with advances in medical management that have improved median lifespan to more than 37 years.9,10 Hence, early lesions are now rarely accessible for pathological examination and study.
It has been twenty years since the gene responsible for CF was identified and more than fifteen years since the first cystic fibrosis transmembrane conductance regulator (CFTR−/−) mouse models were developed.11,12 Although much progress has been made,9,13,14 there remains a lack of understanding regarding the pathogenic events that orchestrate disease in affected organs. To provide a model in which to investigate CF, we recently developed CFTR−/− pigs and found that they display gastrointestinal lesions consistent with CF disease.15,16 These pigs provide several advantages for pathology studies and avoid limitations of human studies. We are able to study newborn CFTR−/− pigs before they develop secondary changes attributable to interactions with their environment and infection. We can also avoid changes attributable to severe terminal disease and postmortem alterations. In addition, pathological data from humans can be skewed by many factors, including toward the most severe cases that come to autopsy, whereas with newborn pigs we have the opportunity to assess the spectrum of disease at one point in time. Thus, the goal of this study was to assess the gastrointestinal pathology in newborn CFTR−/− pigs to provide insight into the early changes of the disease.
Materials and Methods
Animals
All studies were approved by the University of Iowa Animal Care and Use Committee. Neonatal pigs (eight to twenty-four hours old) were used for the study to minimize environmental influences on tissues. Littermates from CFTR+/− matings were obtained from Exemplar Genetics (Sioux Center, IA). Piglets included CFTR+/+ (n = 12), CFTR+/− (n = 12), and CFTR−/− (n = 26) genotypes. For determining the frequency of fetal perforation/peritonitis, we examined the necropsy records of the 26 CFTR−/− pigs along with the necropsy records of six additional CFTR−/− pigs whose tissues were not available for subsequent histopathology.
Tissues and Pathology Examination
After euthanasia (Euthasol, Virbac, Fort Worth, TX), all tissues were collected and immersed in fixative for 48 to 96 hours depending on tissue size, routinely processed, embedded, sectioned (4 micrometers), and stained with hematoxylin and eosin. Some sections were further stained with periodic acid-Schiff (PAS) stain, which detects neutral mucins, Alcian Blue/Pyronine Y (ABPY) stain, which detects acidic mucins, or Masson trichrome stain for identification of collagen deposition.16–18 All tissues were examined by a pathologist (D.K.M.) familiar with the species and CF. Human CF tissues samples were identified from autopsy archival material in the Department of Pathology, University of Iowa.
Morphometry
After histopathological examination, high-resolution digital images were collected (DP71 digital camera and BX41 microscope, Olympus, Center Valley, PA) and these images were morphometrically analyzed with MicroSuite Pathology Edition software (Olympus, Center Valley, PA). Thickness of the intestinal smooth muscle layer (ie, tunica muscularis) was consistently defined as the minimal diameter of the layer (to avoid sectioning artifacts) in tissue cross-sections and reported in micrometers. Pancreas parenchyma was assessed by low magnification examination, and the lobular area of pancreatic parenchyma (containing a mixture of exocrine with endocrine tissue) was defined, area quantified, and then reflected as a percentage of the total pancreatic area. For gallbladder diameter assessment, all sections were consistently sectioned in the same plane at the widest portion of the intact gallbladder. To account for “collapsed” gallbladders postsectioning, and to provide consistent/reproducible measurements, the histopathological circumference of the gallbladder lumen was enumerated by software analysis and the diameter calculated (diameter = circumference/π).
Statistics
Analysis for genotypic (CFTR+/+, CFTR+/−, and CFTR−/−) differences was performed using one-way analysis of variance, and intergroup post tests were made with a Bonferroni multiple comparison test. Statistical analysis was performed using Prism software (Graphpad, La Jolla, CA).
Results
Intestinal Disease
Meconium Ileus Obstruction
Meconium ileus involves an obstruction of the intestine that occurs in about 15% of CF neonates and may be detected as early as 17 weeks gestation.19–22 In all neonatal CFTR−/− pigs, meconium ileus was present (Figure 1, A and B). The site of obstruction ranged from the distal jejunum to the proximal spiral colon (Figure 1, A–C), an elongated and coiled equivalent to the human ascending colon. This distribution is similar to that reported in humans with meconium ileus.23 In CFTR−/− pigs, the site of obstruction consisted of a dilated meconium-filled proximal bowel segment, a transition zone (abrupt to gradual), and a distal bowel that was diffusely small in caliber (Figure 1A) and variably filled with white to gray-green firm cords of mucus. The small caliber distal bowel was firm and lacked pliability compared with normal bowel and may have contributed to the intestinal obstruction. In addition, CFTR−/− meconium was sticky and adherent to the intestinal wall, and it may have contributed to the formation of the obstruction.
Figure 1.
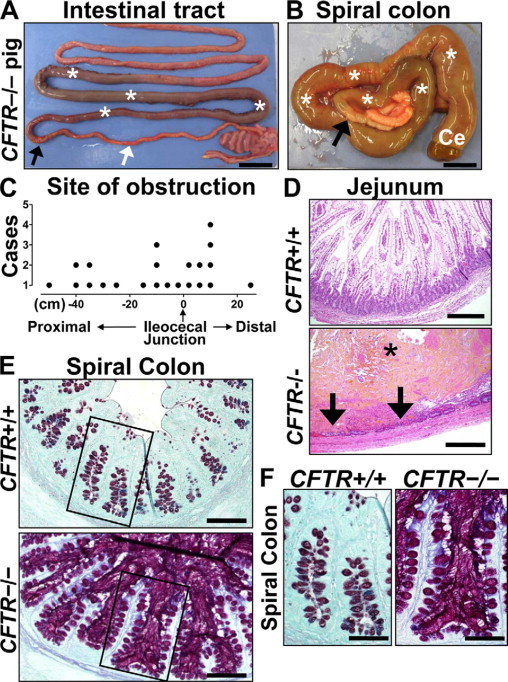
Meconium ileus in CFTR−/− intestine. A: Meconium-filled bowel (asterisks) was proximal to the obstruction interface (black arrow); distal to the interface, the bowel was hypoplastic (white arrow). Scale bar = 2.45 cm. B: Meconium (white asterisks) dilated the spiral colon (ventral view) and cecum (Ce) proximal to the obstruction interface (black arrow). Microcolon appeared distally. Scale bar = 1.34 cm. C: Sites of obstruction in CFTR−/− pigs were distributed on either side of the ileocecal junction extending from the small intestine (proximal) into the spiral colon (distal). D:CFTR−/− jejunum showing luminal meconium (asterisk) causing severe distension and mucosal thinning with focal hemorrhage, necrosis (arrows), and neutrophilic inflammation, HE stain. Scale bars = 0.29 mm. E:CFTR−/− spiral colon had luminal mucus accumulation with hyperplastic mucus-producing cells especially noticeable compared with CFTR+/+ along the surface epithelium (inset boxes, see F). ABPY stain. Scale bars = 117 μm. F:CFTR−/− spiral colon glands were distended by stringy mucus that extends from stout mucus-producing cells in the epithelium, ABPY stain. Scale bars = 69 μm.
Inflammation was not observed in CFTR−/− intestine compared with controls, except as a secondary feature near sites of perforation and necrosis (Figure 1D); there, neutrophils were the predominant leukocytes in acute lesions. Studies of human CF have reported intestinal perforation, peritonitis, and/or volvulus as in utero and postnatal complications of meconium ileus.24,25 Likewise in CFTR−/− pigs, we observed intestinal perforation and peritonitis that occurred in the fetal and early postnatal period usually in severely dilated small intestine. Fetal perforation was uncommon (2 of 32, 6%) and when present was characterized by fibrin, adhesions, exudative fluid filling the abdomen, and granulomatous peritonitis centered on meconium material.
The extent of mucus production depended on the intestinal location. In the spiral colon distal to the obstruction, the lumen contained mucocellular accumulations (Figure 1E), and the adjacent colonic glands were often distended by stringy mucus (Figure 1, E and F). The CFTR−/− colonic epithelium showed mild to moderate mucinous hyperplasia, especially toward the surface epithelium, and the cells were relatively stout and distended by mucus (Figure 1F). The presence of luminal mucus and epithelial mucinous change in the colon did not appear to depend on the proximity to the site of intestinal obstruction. The histochemical staining quality of the CFTR−/− mucus often showed enhanced production of sulfated mucus (Figure 1, E and F; red coloration) similar to that reported in human CF.26 The CFTR−/− colon with mucinous hyperplasia lacked detectable inflammation.
In the proximal small intestine, focal thin strands of eosinophilic mucus were uncommonly detected within the crypts and between villi with some minor focal luminal accumulation. Mucinous hyperplasia in the small intestine proximal to the obstruction was lacking to focal and mild. Brünner glands of the duodenum were focally ectatic, filled by wispy mucus, and had a thin epithelial border (Figure 2A). In the small intestine distal to the obstruction, luminal mucus was more readily detected and combined with sloughed cells in a layered fashion to form mucocellular cords (pellets; Figure 2B). The presence of these cords did not appear to be dependent on the proximity to the obstruction. Patches of meconium were occasionally intermixed with these mucocellular cords, which sometimes dislodged from the intestinal wall to accumulate distally, especially in the hours after CFTR−/− pigs consumed their first meal. The morphological appearance of these dislodged cords (Figure 2B, right panel) was similar to their appearance before dislodgement (Figure 2B, left panel), suggesting resilience in physical structure. The proximal spiral colon was a common site of accumulation for dislodged cords, which in some cases formed large luminal masses (Figure 2, C and D). These large aggregates occasionally caused severe distension, which led to necrosis and perforation in CFTR−/− pigs. Although the small intestine is the most common site of perforation in CF infants (and CFTR−/− pigs), colonic perforations have been reported to occur in children due to numerous cords filling the colon and causing direct irritation and injury.27
Figure 2.
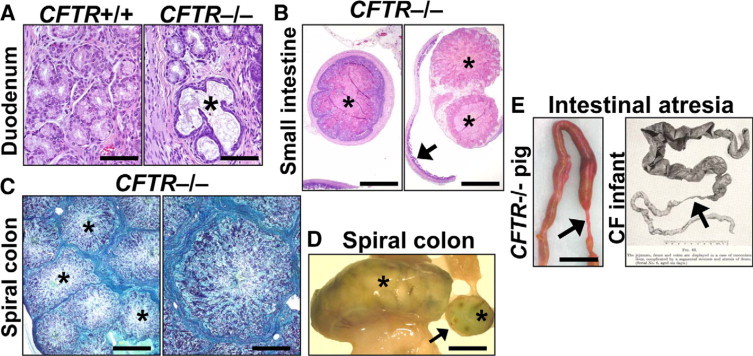
Mucus accumulation and atresia in CFTR−/− intestine. A: Duodenal Brünner glands were focally distended by stringy mucus (asterisk) with flattening of adjacent epithelium. HE stain. Scale bars = 80 μm. B: Distal to the obstruction interface, mucocellular cords (asterisks) filled the CFTR−/− intestinal lumen (left panel) and sometimes became dislodged from the intestinal wall to aggregate (right panel) distally in the bowel (arrow). HE stain. Scale bars = 1.2 mm. C: Cross section of a distended CFTR−/− spiral colon filled with large aggregates of mucocellular cords (asterisks) that retained their morphology from the small intestine (see also B, left panel). ABPY stain. Scale bars = 1.7 mm (left panel) and 0.7 mm (right panel). D: Aggregates of mucocellular cords (asterisks) distended a loop of CFTR−/− spiral colon and were detectable in cross section (arrow). Scale bar = 11.1 mm. E: Segmental atresia (arrows) was present distal to the obstruction interface in the CFTR−/− pig (Scale bar = 11.5 mm) and is similar to what Bodian described in CF infants.5 Reprinted from Fibrocystic Disease of the Pancreas: a Congenital Disorder of Mucus Production-Mucosis, Martin Bodian, Page 84, Copyright 1953.
We observed no difference between CFTR+/+ and CFTR+/− animals at any site along the intestinal tract or in any of the other organs we describe below. We also observed no differences between lesions in male and female piglets.
Additional Intestinal Lesions
CF infants are prone to intestinal atresia with a >200-fold increased risk compared with the general Caucasian population.23,28 Atresias were also detected in CFTR−/− pigs (Figure 2E, left panel). Stenotic and atretic segments of bowel were commonly detected distal to the site of meconium obstruction. Most CFTR−/− pig atresias were small gauged (less than 2 mm outer diameter), continuous with adjacent bowel and had rare serosal twisting. The atresia formation was similar to that described for CF infants (Figure 2E, right panel).5,29
The mechanisms that produce atresia remain uncertain. Atresias might result from in utero vascular compromise caused by lesions such as volvulus.21,29,30 Although we did not observe overt volvulus in the CFTR−/− pigs, it is possible that obstruction predisposed to secondary vascular compromise, manifested as serosal twisting at birth. It is also possible that intestine distal to the site of obstruction failed to develop normally because of a lack of mechanical distention by luminal contents or because obstruction prevented distal delivery of luminal contents that were required for intestinal growth.31
Diverticulosis has been reported in the appendix and colon of CF patients,32,33 and George33 found that CF patients had a 7- to 14-fold increased incidence of diverticulosis of the vermiform appendix (median age ≈13 years, including one infant). Histologically, the lesions were classified as acquired pulsion or pressure type diverticula. The pathogenesis was proposed to result from increased pressure/distension by luminal material and concurrent smooth muscle hypertrophy. Increased intraluminal pressure may then lead to herniation of the mucosa through the weakest part of the muscular wall at sites of vascular penetration along the mesenteric border.33,34 Diverticulosis was a common intestinal lesion in CFTR−/− pigs. It appeared as saccular bulges along the mesenteric border of dilated bowel (Figure 3A). Diverticulosis usually occurred in the meconium-filled jejunum when there was severe obstruction. Histologically, intestinal diverticula were composed of tunica mucosa and submucosa that were herniated through a hypertrophic tunica muscularis (Figure 3A, right panel). In fact, the tunica muscularis was hypertrophied throughout the intestinal tract including the duodenum and the spiral colon (Figure 3A, right panel, and Figure 3B). In pathology studies of CF infants, the tunica muscularis has also been described as “well-formed” to “hypertrophic.”5,8
Figure 3.
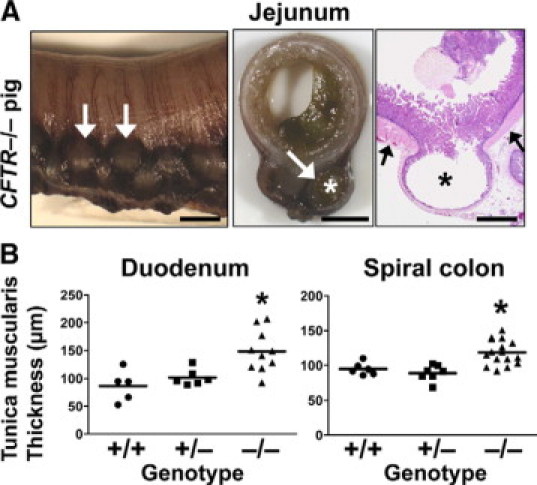
Diverticuli and smooth muscle thickening in CFTR−/− intestine. A: Diverticula (asterisks and white arrows) formed along the mesenteric border and were lined by a thin mucosa and submucosa, which herniated through a thickened tunica muscularis (black arrows). Scale bars = 3.8 mm (left and middle panel) and 1.4 mm (right panel, HE stain). B: Tunica muscularis of the CFTR−/− pig intestine was generally hypertrophied, including in the duodenum (left, P < 0.01 versus CFTR+/+, P < 0.05, versus CFTR+/−) and the spiral colon (right, P < 0.01 versus CFTR+/+, P < 0.001 versus CFTR+/−, one-way analysis of variance with Bonferroni post test).
A review of the literature did not identify reports of jejunoileal diverticula (excluding Meckel's diverticula) in CF. Although this may reflect the true clinical situation, some have suggested the overall incidence of diverticula may be under-represented because incidental identification often requires high vigilance during examination.33,34 In people without CF, jejunoileal diverticula are typically observed in elderly patients.34 Pigs have been reported to have smooth muscle hypertrophy and diverticulosis of the small intestine as incidental findings at necropsy and rarely as a cause of perforation. A genetic predisposition has also been speculated for some pig breeds.35 Consistent with the proposed pathogenesis in humans, diverticula in CFTR−/− pigs may have occurred in the dilated meconium filled bowel because of the combined increased intraluminal pressure and hypertrophic smooth muscle during fetal life.
Pancreatic Disease
The pancreas is affected in ≈85% to 90% of CF patients,36,37 and pancreatic lesions have been reported in neonates and fetuses as young as seventeen weeks gestation. These lesions consist of luminal dilation of acini/ducts by eosinophilic zymogen material. With increased luminal material there is progressive thinning of the lining epithelium, and mucus metaplasia may be detected in the larger ducts.20,38,39 In fact, the classic lesion of the CF pancreas consists of dilated (“cystic”) ducts filled by stringy zymogen material and mucus.2,4,5,40 In neonatal CFTR+/+ pig pancreas, the exocrine tissue contained zymogen-rich acini (Figure 4A), and on occasion, we observed ducts filled with normal appearing zymogen secretions. Mucus cells were typically restricted to large ducts (Figure 4A), and small submucosal glands were noted in the terminal duct near the duodenum.
Figure 4.
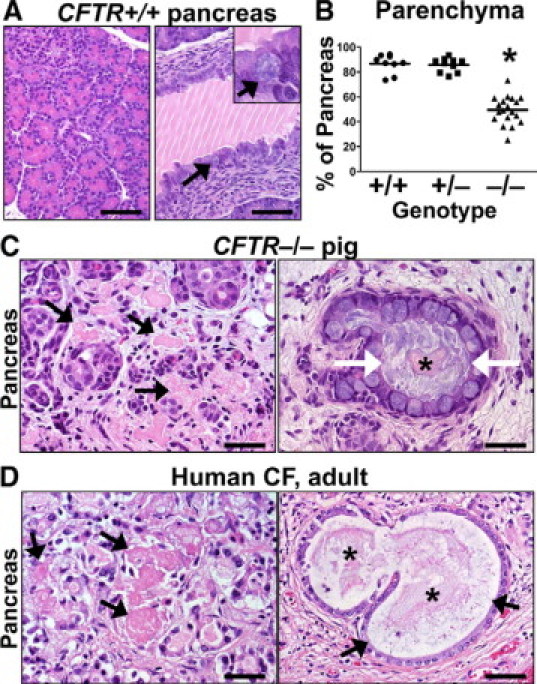
Exocrine pancreatic destruction in CFTR−/− pigs. A:CFTR+/+ pig pancreas was rich in exocrine tissue (left panel) with large ducts (right panel) that were partially filled with normal zymogen secretions (white lines are sectioning artifact). Mucus cells were uncommon in the epithelium (arrows and inset). HE stain. Scale bars = 72 μm. B:CFTR−/− pancreata had reduced lobular parenchyma (P < 0.001 vs. CFTR+/+ and CFTR+/−, one-way analysis of variance with Bonferroni post test). Scale bar = mean. C: Degenerative pools of eosinophilic zymogen secretions were detected “free” within the interstitium (black arrows). Within dilated ducts, centrally oriented zymogen secretions (asterisk) were often surrounded by stringy mucus (white arrows). HE stain. Scale bars = 37 μm. D: Adult human CF pancreas (with autolysis making some features less appreciable) showed similar degenerative pools of interstitial zymogen secretions (arrows, left panel) and similar secretions (asterisk) were detected in dilated ducts surrounded by mucus (arrows, right panel). HE stain. Scale bars = 35 μm and 53 μm (left and right panel, respectively).
In CFTR−/− pigs, pancreatic lesions were ubiquitous and there was a range in severity of exocrine tissue destruction (Figure 4B). Endocrine tissue was spared from the destruction and appeared morphologically intact.16 Acinar cells were reduced in number and had decreased amounts of cytoplasmic zymogen granules. Zymogen secretions often filled the acinar and ductular lumens of CFTR−/− pancreata (Figure 4C). In some ducts, stringy zymogen material was surrounded by mild to moderate amounts of mucus (Figure 4C). With increased filling of acinar and ductal lumens by altered zymogen material, the adjacent acinar and ductular epithelium became thinner. Adjacent to the residual exocrine tissue were pools of eosinophilic zymogen material that appeared to be free within the interstitium (Figure 4C). The interstitial zymogen material was morphologically degenerate (eg, hyalinized, fragmented) compared with zymogen material still confined by an epithelial border. Because the pancreas is not easily accessible to biopsy and tissues from young children with CF are no longer common, we examined autopsy tissue from an adult with clinically mild CF disease. We found remnant exocrine tissue that had zymogen material in a similar interstitial distribution with degenerative characteristics as that in CFTR−/− pigs (Figure 4D). In both pigs and humans, the interstitial pools of zymogen material elicited minimal direct cellular inflammatory response even with the appearance of morphological degeneration. The loss of exocrine tissue was replaced by increased connective tissue in the pancreata of CF infants41,42 and CFTR−/− pigs.
In addition, foci of duct proliferation were detected in exocrine tissue (Figure 5A), especially noted in pancreata with more severe exocrine destruction. In her detailed description of CF pancreas pathology, Sturgess38 noted that proliferation of ducts was common in CF infant pancreas. Recent evidence suggests that ductular proliferation may be a manifestation of acinar to ductular metaplasia, a reaction to pancreatic injury.43,44 An additional mechanism of degenerative change seen in a few rare ducts included purple granular material consistent with dystrophic calcification (Figure 5A). In pancreata with mild disease, mucinous metaplasia of ducts was only observed in uncommon large ducts. This is in contrast to pancreata with severe destruction where mucinous metaplasia was especially prominent and seen in large, medium, and small ducts (Figure 5B). The proliferative mucus cells lining ectatic ducts were often plump and full of mucus.
Figure 5.
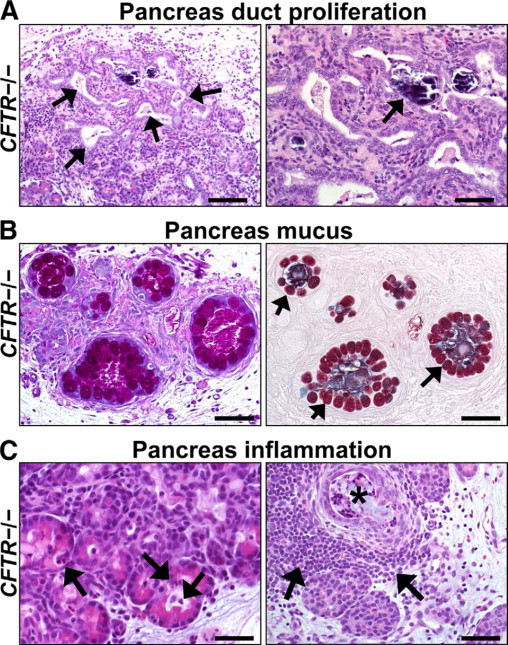
Duct proliferation, mucus accumulation, and inflammation in CFTR−/− pancreas. A: Foci of proliferative (left panel, arrows) and dilated ducts were common in CFTR−/− pancreata. Dystrophic calcification was rarely detected (right panel, arrow). HE stain. Scale bars = 110 μm and 55 μm (left and right panel, respectively). B: In severely disease pancreata, increased mucinous metaplasia of ducts was detected. Note the large mucus cells (arrows) circumferentially lining variably sized ducts that are moderately distended by stringy mucus. PAS (left panel) and ABPY (right panel) stains. Scale bars = 55 μm. C: Acini and ducts dilated by zymogen material had scattered infiltrates of neutrophils (arrows, left panel) and to a lesser extent, macrophages. Severe exocrine destruction was often associated with interstitial lymphoid aggregates (arrows, right panel) adjacent to dilated, cyst-like ducts (black asterisk). HE stain. Scale bars = 36 μm (left panel) and 55 μm (right panel).
In CF infants, pancreas inflammation was described by Andersen2 as mild with a small number of neutrophils and macrophages in the acinar tissue and scattered mononuclear aggregates in the interstitium. This was also the case in porcine CFTR−/− pancreas. Although all CFTR−/− pancreata had detectable cellular inflammation, most of this was patchy in distribution with increased detection noted with progressive disease severity. Scattered neutrophils and macrophages were detected principally within dilated acini (Figure 5C, left panel) and ectatic ducts. There, they infrequently formed loose cellular aggregates, which morphologically could be described as a “pancreatitis.” Interstitial inflammation usually consisted of scattered lymphocytic aggregates that became more prominent around ectatic obstructed ducts in areas of severe pancreatic destruction (Figure 5C, right panel).
Liver Disease
Liver disease is now the second leading cause of CF mortality.45–47 Focal biliary cirrhosis (FBC), the hallmark lesion in patients with CF, is characterized by biliary proliferation, fibrosis, and inflammation.2,5,41,48,49 FBC may progress to bridging and multilobular cirrhosis in up to 17% of patients, with most being diagnosed before age fourteen.50 In CFTR−/− pigs, biliary proliferation was the most readily detected parameter of FBC (Table 1). Fibrosis (fibroplasia) was typically mild in severity and usually adjacent to proliferative ducts. Lymphocytic inflammation, when present, was detected adjacent to proliferative ducts and ranged from scattered cells to moderate cellular aggregates expanding the triad. Bridging of triads by these lesions was rare, but when present was accentuated by biliary proliferation (Figure 6A, top left panel) with fibroplasia and inflammation—all suggestive of early multilobular change (Figure 6B, Table 1). In the pig, peribiliary glands appear principally in larger ducts, thus should mucus obstruction occur here, it could result in proliferation of the smaller bile ducts. Uncommonly, alterations were found in biliary ducts including mucinous change (increased epithelial mucus staining), obstruction by mucocellular plugs (inflammatory cells, cell debris, and variable mucus depending on the size of duct), and concretions of bile (choleliths; Figure 6A).
Table 1.
Liver and Gallbladder Lesion Parameters according to Pig Genotype
| Genotype | Biliary proliferation | Fibrosis | Inflammation | Triad bridging | Extra-medullary hematopoiesis | Steatosis | GB mucinous change |
|---|---|---|---|---|---|---|---|
| CFTR+/+ | 0% (1/12) | 0% (0/12) | 0% (0/12) | 0% (0/12) | 100% (12/12) | 17% (2/12) mild | 8% (1/12) mild |
| CFTR+/− | 0% (0/12) | 0% (0/12) | 0% (0/12) | 0% (0/12) | 92% (11/12) | 0% (0/12) | 0% (0/12) |
| CFTR−/− | 58% (15/26) | 42% (11/26) | 54% (14/26) | 8% (2/26) | 100% (24/24) | 8% (2/26) mild | 94% (17/18) |
GB indicates gallbladder.
Figure 6.
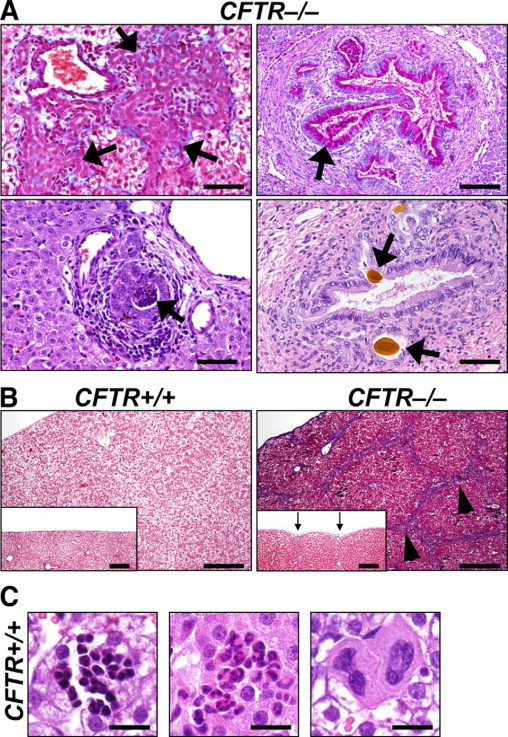
Rare lesions in CFTR−/− liver and incidental findings in all genotypes. A: Infrequent to rare findings in CFTR−/− liver. Biliary tracts had expansive and florid proliferation (arrow, top left panel, MT stain) with adjacent fibrosis (blue) and inflammation. Intrahepatic ducts had mucinous change (arrow, top right panel, PAS stain), mucocellular plugs (arrow, left bottom panel, HE stain), or choleliths (arrows, right bottom panel, HE stain). Scale bars = 55 μm. B: Widespread bridging (arrowheads) of triads by biliary hyperplasia, inflammation, and fibrosis (blue staining) was rarely detected in CFTR−/− pigs. Scale bars = 220 μm. In these cases, the serosal surface was often retracted causing a slightly irregular surface (arrows, inset, Scale bars = 197 μm). MT stain. C: Foci of extramedullary hematopoiesis were composed of erythroid, granulocytic, and megakaryocytic lineage aggregates (respective panels) in liver of all genotypes. HE stain. Scale bars = 17.5 μm.
There were no detectable differences between CFTR+/+ and CFTR−/− livers in steatosis, hemosiderosis, or cholestasis. Likewise, in CF infants, steatosis is generally considered to be secondary to the various chronic metabolic changes in postnatal life.51 Independent of genotype, neonatal pigs had multifocal cellular aggregates of hematopoietic cells (ie, extramedullary hematopoiesis) in the liver (Figure 6C). Cellular aggregates were often grouped according to lineage, they were localized in the parenchyma and less commonly adjacent to hepatic triads, and they required differentiation from inflammation.
Gallbladder Disease
Gallbladder lesions are reported in ≈20% to 30% of CF infants and typically consist of a small gallbladder (microgallbladder) with proliferative epithelium that sometimes form mucosal folds or “cysts.”2,5,51–53 The CF infant gallbladder can also manifest features such as luminal mucus, thickened bile, choleliths, and/or cholecystitis.52 Incidentally, most CF patients lack clinical consequences of microgallbladder.51 All CFTR−/− pigs had a microgallbladder, which was readily apparent at necropsy and histopathology (Figure 7, A and B). It resembled that seen in humans with CF (Figure 7A, right panel).54
Figure 7.
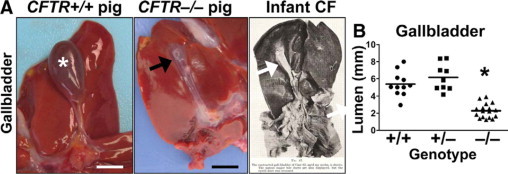
Microgallbladder in CFTR−/− pigs. A:CFTR+/+ pig gallbladders (asterisk, Scale bar = 10.5 mm) were typically distended with bile, whereas microgallbladder (arrows, Scale bar = 7.4 mm) was seen in the CFTR−/− pig. Right panel shows microgallbladder from infant with CF as reported by Bodian.5 Reprinted from Fibrocystic Disease of the Pancreas: a Congenital Disorder of Mucus Production-Mucosis, Martin Bodian, Page 112, Copyright 1953. B: Histological measurements showed that porcine CFTR−/− gallbladder was smaller than CFTR+/+ or CFTR+/− (P < 0.001 respectively, one-way analysis of variance with Bonferroni post test, Scale bar = mean).
We stained the gallbladder with PAS, which detects neutral mucins and ABPY histochemical stains, which detect sialylated (blue), sulfated (red), or mixed (purple) acidic mucins.17,18 The CFTR+/+ gallbladders had minor staining (Figure 8A, top panels), whereas the gallbladder epithelium of CFTR−/− pigs showed diffuse epithelial mucinous change in most cases (Table 1), although the extent of luminal mucus accumulation varied. In the CFTR−/− gallbladder, the PAS stain accentuated concentrically lamellar striations in the mucus that were parallel to the epithelial surface and that were reminiscent of the growth rings in a tree (Figure 8A, bottom left). CFTR−/− gallbladders stained with ABPY showed a heterogeneous cellular production of sialylated, sulfated, and some mixed acidic mucus with a detectable preference toward sulfated mucus. This histochemical stain also highlighted distinct ribbons of mucus extending out perpendicularly from the epithelium. These ribbons often retained their form and structure with minimal amalgamation of adjacent mucus ribbons (Figure 8A, bottom right). It was difficult to obtain serial sections because of frequent tissue folds associated with sectioning gallbladders filled with mucus plugs. When available, serial sections using both stains demonstrated similar morphological features (Figure 8B).
Figure 8.
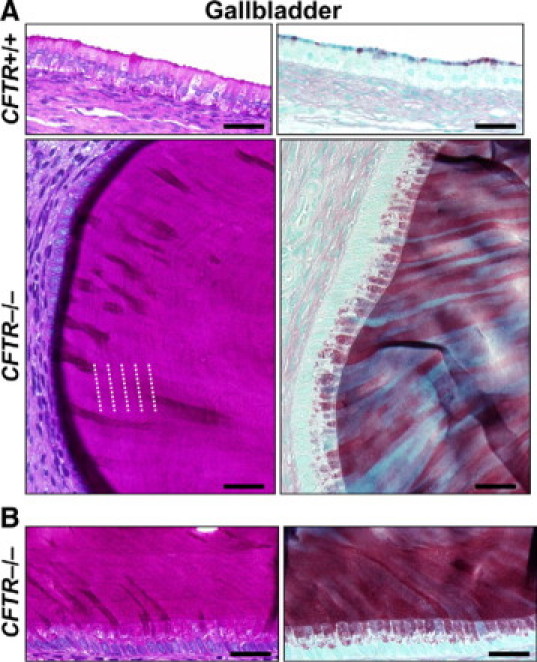
Mucus cell proliferation and mucus accumulation in CFTR−/− gallbladder. A: Nominal apical mucus staining in CFTR+/+ gallbladder epithelium (top panels, Scale bars = 38 μm). CFTR−/− gallbladder typically had diffuse mucinous change (bottom panels, Scale bars = 26 μm) with contrasting morphological characteristics of mucus including lamellar striations (see white dotted lines) parallel to the epithelium (left panel, PAS stain) and ribbons of resilient mucus perpendicular to the epithelium (right panel, ABPY stain). B: Serial sections of CFTR−/− gallbladder contained similar morphological changes. Scale bars = 32 μm.
Additional microscopic changes in the CFTR−/− gallbladder included luminal obstruction by a mixture of mucus and altered bile with proliferative mucosal folds (Figure 9A). Occasionally, small focal aggregates of neutrophils were detected within the luminal mucus near the epithelium. Mild mononuclear inflammation was sometimes detected in the lamina propria and connective tissue of the gallbladder wall, which also had mild to moderate thickening by increased collagen and smooth muscle. The apparent discrepancy between the severity of the gallbladder disease and the milder liver disease in neonatal CFTR−/− pigs could suggest that their pathogenesis is not sequential or directly related. Alternatively, given the focal nature of CF liver lesions (eg, FBC), we cannot rule out that every CFTR−/− liver may have some degree of FBC that is not readily detectable in routine tissue sampling.
Figure 9.
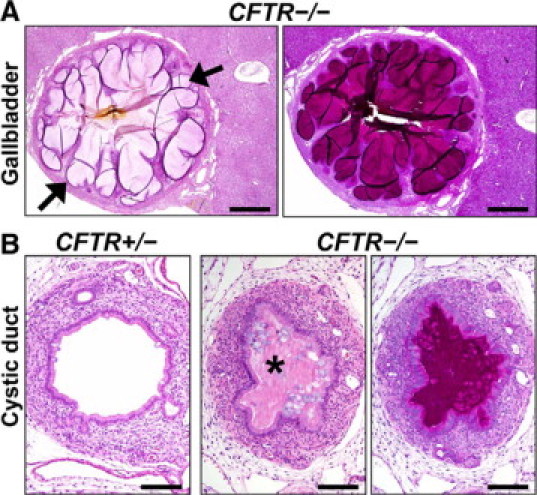
Gallbladder and cystic duct lesions. A: The CFTR−/− gallbladder mucosa sometimes formed folds of proliferative epithelium to form cyst-like structures (arrows) along the mucosa. HE (left) and PAS (right) stain. Scale bars = 550 μm. B:CFTR−/− cystic ducts were variably obstructed (asterisk) and stenotic compared with controls. HE (left and middle) and PAS (right) stains. Scale bars = 116 μm.
Cystic Duct Disease
The cystic duct connects the gallbladder to the common bile duct. The occurrence of cystic duct lesions paralleled gallbladder lesions and included mucinous change with obstruction and stenosis.5,52,53 Bodian5 noted that in fourteen cases of CF gallbladder disease, the cystic duct was consistently obstructed, yet the extrahepatic ducts were patent. Similarly, CFTR−/− pig cystic ducts were consistently obstructed by mucocellular material (Figure 9B), bile, and at times proliferative mucosa folds that formed cystic-like structures containing luminal material. Inflammatory changes were similar to that seen in the gallbladder. We examined the common bile duct at necropsy in a few cases, and no obstruction was detected.
Discussion
Disease Severity in Humans with CF and CFTR−/− Pigs
The most prominent difference between disease in newborn babies with CF and newborn CFTR−/− pigs was that the disease was more severe in the pigs. For example, meconium ileus occurred in 100% of the pigs, but ≈15% of humans19–22; gallbladder abnormalities were present in 100% of CFTR−/− pigs, but ≈20% to 30% of humans2,51–53; up to 42% of newborn CFTR−/− pigs had lesion parameters consistent with FBC, whereas ≈14% to 43% of CF infants show similar morphological changes2,5,48,52; and all CFTR−/− pig pancreata (100%) were readily distinguished from controls by gross inspection at necropsy, whereas in CF infants, morphological detection of pancreatic disease is up to 93% successful only when using extensive histological examination and quantitative morphometric analysis.55
What accounts for the increased disease severity in the CFTR−/− pigs? There are several considerations. First, the pigs have a null mutation, whereas the majority of humans have at least one ΔF508 or one missense mutation, either of which might generate a very small amount of CFTR function. Perhaps only a small amount of CFTR function is sufficient to slow the progression of disease in humans relative to CFTR null pigs. Consistent with this possibility, many aspects of the disease, including meconium ileus, have a significant genotype/phenotype correlation.56 Second, genetic modifiers might be distinct in humans and these pigs. Genetic modifiers are known to influence the course of CF in humans.56–59 Although not inbred to the extent defined for mice, the CFTR−/− pigs are currently much more inbred than humans, and it is possible that by out-breeding the pigs we will be able to modify their phenotype. Consistent with this speculation, the severity of pathological changes varied between individual animals. Furthermore, study of human CFTR genotype to phenotype correlation in twins suggests modifier genes play an important role for both causative and preventive effects.56 Third, it is possible that humans express another anion channel that compensates, at least in small measure, for the loss of CFTR and thereby partly mitigates the pathological developments. In this regard, it has been hypothesized that an alternative Cl− channel partly replaces CFTR function in CFTR−/− mice.60 Fourth, it has been hypothesized that a misfolded CFTR protein, especially CFTR-ΔF508, might elicit an unfolded protein response that contributes to CF disease in humans.61 Because CF disease tends to be milder in humans than in pigs, our data suggest that if an unfolded protein response contributes to differences in severity between the species, then it may attenuate disease severity. Fifth, it seems unlikely that anatomical differences between the species (eg, the spiral colon in pigs versus the ascending colon in humans) are entirely responsible because differences in severity between the species occurred in multiple organs. In fact, the greater severity of disease in pigs versus humans suggest that whatever the cause, it likely involves a global difference in phenotype of CFTR-deficient epithelia that is manifest in many organs before birth. The same is likely to be true in mice. Although CFTR−/− mice do not develop classical meconium ileus like humans with CF, at an early age, most CFTR−/− mice develop intestinal mucus obstruction. Histopathological changes in the pancreas, liver, and gallbladder are nominal and may show some disease progression in advanced age.60,62 We propose that comparing the pathophysiological features of these three species may yield novel insight into the varying lesion severity (pig > human ≫ mice) and hence, the pathogenesis of CF disease.
Tissue Remodeling and Mucus Accumulation
In general, affected organs in newborn pigs shared four features: a) prominent epithelial mucus producing cells, b) mucus accumulation, c) tissue remodeling, and d) inflammation.
Epithelial Mucus Cells
Epithelia in the affected porcine organs often showed a prominent increase in mucus cells. Mucinous epithelial changes also occur in human tissues, which led Bodian5 to use the term “mucosis” to describe the changes in various CF organs.
Mucus Accumulation
Accumulation of mucus in pancreatic ducts, gallbladder/cystic duct, and intestine was a consistent finding in CFTR−/− pigs. Likewise, early observations in CF infants reported mucus accumulation as a common pathological feature.3,4 The prominent mucus accumulation even caused some early investigators to suggest the cause of CF was a “mucinase deficiency.”4 In CFTR−/− pigs, the morphological character of the mucus was striking. The concentrically lamellar striations of mucus (gallbladder, pancreas ducts, and intestine) suggest that luminal accumulation formed by serial secretions. Consistent with this speculation, the cytoplasm of mucus-producing cells was usually distended with mucus suggesting that the secretion process occurred at a modest rate that did not deplete mucus from the cells.63 The mucus secretions were also congealed and resilient, retaining a relatively fixed macroscopic structure even when it migrated to a new location. Whether the structure of the mucus was a result of stasis attributable to a long-lasting obstruction or a consequence of altered transepithelial ion transport attributable to the lack of CFTR is unknown.
Tissue Remodeling
Tissue remodeling (eg, hyperplastic, metaplastic, connective tissue changes) occurs as a response to chronic disease, and many of these parameters were evident in the neonatal CFTR−/− organs. This implies that affected tissues undergo significant remodeling even in the absence of environmental stimuli and that remodeling was a dynamic component of fetal life.
Inflammation
Inflammation was detected in affected organs and was characterized by mild to moderate patchy cellular infiltrates adjacent to tissue lesions. The heterogeneous character and localization of cellular infiltrates suggests that multiple mechanisms/roles for inflammation may be at work.
The observations of increased epithelial mucus cells, luminal mucus accumulation, tissue remodeling, and inflammation in multiple organs raise questions about the pathogenic sequence of events. One idea might be that inflammation triggered the conversion of a normal epithelium to one with increased numbers of mucus cells, and that they then secreted mucus that obstructed the lumen. In that case, the stimulus for inflammation would presumably be an inherent inflammatory state caused by lack of CFTR; such an event has been postulated to exist in CF.64 Although this study cannot fully address the role of inflammation, our finding that profound intestinal mucinous change occurred in the absence of overt cellular inflammation suggests that the presence of leukocytes may not be a requirement for mucus production in all CF tissues. Another possibility is that CF luminal mucus had an intrinsic abnormal structure thereby causing tissue injury and inflammation, which then further drove mucus cell production. However, mucus cells are not routinely present in epithelia at some anatomical sites (eg, small ducts of the pancreas), and thus mucus production would be absent or limited in the initial stages of the disease pathogenesis. Nevertheless, the quantity of mucus required for obstruction might be very small. A third, related scenario is that defective electrolyte transport attributable to loss of CFTR might change the consistency of luminal secretions, thereby causing luminal mucoid plugs. Obstruction by altered secretions could cause direct physical irritation of lining cells leading to a cycle of mucinous metaplasia and further mucus production. Consistent with this scenario, Tucker et al40 suggested that altered zymogen secretions, rather than mucus, caused the initial detectable obstruction of pancreatic ducts; then, mucus metaplasia occurred and contributed to the obstruction and exocrine atrophy. In addition, mucinous metaplasia of the gallbladder epithelium is chronologically correlated with gallstone formation, and the physical presence of luminal gallstones may synergistically perpetuate metaplastic change in the gallbladder epithelium.65 Studies of fetal CFTR−/− pigs may shed further light on some of these issues.
We also found that smooth muscle layers (eg, intestinal tunica muscularis and gallbladder wall) were hypertrophic in CFTR−/− pigs. Recently, CFTR has been detected in smooth muscle, and functional studies suggested that CFTR may play a role in their relaxation.66–68 Although hypertrophy could be a secondary response of tissue remodeling, it could also represent an intrinsic consequence due to loss of CFTR (ie, a persistent contracted state). Thus, the absence of CFTR might cause altered smooth muscle physiology that could contribute to CF disease pathogenesis; however, the mechanism and extent of this effect remain elusive.
Relationship to Lung Disease
Even though CFTR is absent, the lungs of newborn CFTR−/− pigs appeared similar to those of their CFTR+/+ littermates.16 The discrepancy between the marked pathology in the gastrointestinal organs described here and the lack of pulmonary disease in newborn pigs may provide some hints about CF pathogenesis and allows for some speculation.
There are several feature(s) of the fetal lung that might potentially prevent it from developing CF disease before birth. The fetal lung is filled with liquid that originates from respiratory epithelia and amniotic fluid.69 The composition of fetal lung liquid may be less prone to congealing and causing obstruction than that in the ducts of gastrointestinal organs. Furthermore, impaired mucociliary clearance is speculated to initiate CF airway disease,70 but in the relatively quiescent fetal lung, there is no apparent requirement for mucociliary clearance. This situation contrasts with the functional activity of fetal gastrointestinal organs, as hepatobiliary and pancreatic secretions are constituents of meconium that is moved through the intestines via peristalsis. It is also possible that compared with affected gastrointestinal organs, the composition or concentration of material in fetal lung liquid is insufficient to incite disease. Perhaps only after breathing air, when the volume of airway surface liquid is very small, is abnormal transepithelial electrolyte transport able to modify its composition in a way that initiates CF airway disease.
Comparing gastrointestinal organ pathology with the lack of abnormalities in lungs also has implications for two other aspects of CF that are well known to contribute to the disease, submucosal glands and inflammation. Submucosal glands have been hypothesized to generate mucus with abnormal properties that contribute to lung disease.71 Mucus metaplasia or hyperplasia with dramatic mucus accumulation was detected in affected gastrointestinal organs. In some cases (eg, pancreatic ducts and intestinal epithelia), the luminal mucus derived from metaplastic surface epithelia, and glands were not apparently required for disease. In contrast, the CFTR-rich Brünner glands of the duodenal submucosa showed only minimal abnormalities compared with other portions of the intestine. These observations lead us to wonder about the relative contribution of mucus derived from submucosal glands versus surface epithelia in the development of CF lung disease.
Inflammation was a feature in some affected gastrointestinal organs of newborn pigs, but not in the airways, despite the fact that epithelia in all these organs express CFTR. While it is certainly possible that lack of CFTR may initiate an overly exuberant inflammatory response,72 our results lead us to speculate that inflammation may not be an inherent property of a CFTR−/− cell or tissue in the absence of a stimulus, but with a stimulus it plays an important role in progression of the disease.73
This work sets the stage for studies that look forward as animals mature and look backward to the fetus to assess early pathogenic changes. Similarities and differences between pathological changes in the pig, humans, and mice should provide an opportunity to better understand the pathogenesis of this devastating disease.
Acknowledgements
We thank Tiffany Fagan, Michelle Griffin, Chris Hochstedler, Paul Naumann, Janice Rodgers, Ed Solin, and Tanner Wallen for technical assistance and Dr. Marcus Nashelsky for assistance with autopsy material.
Footnotes
Supported by the National Heart Lung and Blood Institute (grant HL51670 and HL091842), the National Institute of Diabetes and Digestive and Kidney Diseases (grant DK54759), the Cystic Fibrosis Foundation, and HHMI. D.A.S. is a Parker B. Francis Fellow and was supported by the National Institute of Allergy and Infectious Diseases (grant AI076671). M.J.W. is an Investigator of the HHMI.
M.J.W. was a co-founder of Exemplar Genetics, a company that is licensing materials and technology related to this work.
References
- 1.Landsteiner K. Darmverschluss durch eingedictes Meconium Pankreatitis. Zentralbl Allg Pathol. 1905;16:903–907. [Google Scholar]
- 2.Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. Am J Dis Child. 1938;56:344–399. [Google Scholar]
- 3.Farber S. Pancreatic function and disease early in life V. Pathologic changes associated with pancreatic insufficiency in early life. Arch Path. 1944;37:238–250. [Google Scholar]
- 4.Farber S. Some organic digestive disturbances early in life. J Mich Med Soc. 1946;44:587–594. [Google Scholar]
- 5.Bodian M. Fibrocystic Disease of the Pancreas: a Congenital Disorder of Mucus Production-Mucosis. Grune & Stratton; New York, NY: 1953. pp. 67–146. [Google Scholar]
- 6.Levy E. A case of fibrocystic disease of the pancreas with intestinal obstruction. Arch Dis Child. 1951;26:335–339. doi: 10.1136/adc.26.128.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menten ML, Middleton TO. Cystic fibrosis of the pancreas: report of eighteen proved cases. Am J Dis Child. 1944;67:355–359. [Google Scholar]
- 8.Zuelzer WW, Newton WA. The pathogenesis of fibrocystic disease of the pancreas: a study of 36 cases with special reference to the pulmonary lesions. Pediatrics. 1949;4:53–69. [PubMed] [Google Scholar]
- 9.Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med. 2006;173:475–482. doi: 10.1164/rccm.200505-840OE. [DOI] [PubMed] [Google Scholar]
- 10.Welsh MJ, Ramsey BN, Accurso F, Cutting GR. In: Cystic fibrosis. The Metabolic and Molecular Bases of Inherited Disease. ed 8. Scriver CR, Beaudet AL, Sly WS, Valle D, editors. McGraw-Hill; New York: 2001. pp. 5121–5188. [Google Scholar]
- 11.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzi MC, Collins F, Tsui L. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 12.Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH. An animal model for cystic fibrosis made by gene targeting. Science. 1992;57:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- 13.Quinton PM. Physiological basis of cystic fibrosis: a historical perspective. Physiol Rev. 1999;79:S3–S22. doi: 10.1152/physrev.1999.79.1.S3. [DOI] [PubMed] [Google Scholar]
- 14.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 15.Rogers CS, Hao Y, Rokhlina T, Samuel M, Stoltz DA, Li Y, Petroff E, Vermeer DW, Kabel AC, Yan Z, Spate L, Wax D, Murphy CN, Rieke A, Whitworth K, Linville ML, Korte SW, Engelhardt JF, Welsh MJ, Prather RS. Production of CFTR-null and CFTR- DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J Clin Invest. 2008;118:1571–1577. doi: 10.1172/JCI34773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, Rogan MP, Pezzulo AA, Karp PH, Itani OA, Kabel AC, Wohlford-Lenane CL, Davis GJ, Hanfland RA, Smith TL, Samuel M, Wax D, Murphy CN, Rieke A, Whitworth K, Uc A, Starner TD, Brogden KA, Shilyansky J, McCray PB, Jr, Zabner J, Prather RS, Welsh MJ. 2008. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321:1837–1841. doi: 10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyerholz DK, Rodgers J, Castilow EM, Varga SM. Alcian Blue and Pyronine Y histochemical stains permit assessment of multiple parameters in pulmonary disease models. Vet Pathol. 2009;46:325–328. doi: 10.1354/vp.46-2-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inglis SK, Corboz MR, Taylor AE, Ballard ST. Effect of anion transport inhibition on mucus secretion by airway submucosal glands. Am J Physiol. 1997;272:L372–L377. doi: 10.1152/ajplung.1997.272.2.L372. [DOI] [PubMed] [Google Scholar]
- 19.Oppenheimer EH, Esterly JR. Observations in cystic fibrosis of the pancreas. II. Neonatal intestinal obstruction. Bull Johns Hopkins Hosp. 1962;111:1–13. [PubMed] [Google Scholar]
- 20.Szeifert GT, Szabo M, Papp Z. Morphology of cystic fibrosis at 17 weeks of gestation. Clin Genet. 1985;28:556–561. doi: 10.1111/j.1399-0004.1985.tb00427.x. [DOI] [PubMed] [Google Scholar]
- 21.Gaillard D, Bouvier R, Scheiner C, Nessman C, Delezoide AL, Dechelotte P, Leheup B, Cordier MP, Carles D, Lallemand A. Meconium ileus and intestinal atresia in fetuses and neonates. Pediatr Pathol Lab Med. 1996;16:25–40. [PubMed] [Google Scholar]
- 22.Wilschanski M, Durie PR. Pathology of pancreatic and intestinal disorders in cystic fibrosis. J R Soc Med. 1998;91 Suppl 34:40–49. doi: 10.1177/014107689809134s07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mushtaq I, Wright VM, Drake DP, Mearns MB, Wood CBS. Meconium ileus secondary to cystic fibrosis: the East London experience. Pediatr Surg Int. 1998;13:365–369. doi: 10.1007/s003830050341. [DOI] [PubMed] [Google Scholar]
- 24.Murshed R, Spitz L, Kiely E, Drake D. Meconium ileus: a ten-year review of thirty-six patients. Eur J Pediatr Surg. 1997;7:275–277. doi: 10.1055/s-2008-1071170. [DOI] [PubMed] [Google Scholar]
- 25.Rescorla FJ, Grosfeld JL, West KJ, Vane DW. Changing patterns of treatment and survival in neonates with meconium ileus. Arch Surg. 1989;124:837–840. doi: 10.1001/archsurg.1989.01410070095019. [DOI] [PubMed] [Google Scholar]
- 26.Mendicino J, Sangadala S. Synthesis of sulfated oligosaccharides by cystic fibrosis trachea epithelial cells. Mol Cell Biochem. 1999;201:141–149. doi: 10.1023/a:1007014613768. [DOI] [PubMed] [Google Scholar]
- 27.Siddiqui MM, Burge DM. Neonatal spontaneous colonic perforation due to cystic fibrosis. Pediatr Surg Int. 2008;24:863–864. doi: 10.1007/s00383-008-2164-2. [DOI] [PubMed] [Google Scholar]
- 28.Sweeney B, Surana R, Puri P. Jejunoileal atresia and associated malformations: correlation with timing of in utero insult. J Pediatr Surg. 2001;36:774–776. doi: 10.1053/jpsu.2001.22958. [DOI] [PubMed] [Google Scholar]
- 29.Bernstein J, Vawter G, Harris GBC, Young V, Hillman LS. The occurrence of intestinal atresia in newborns with meconium ileus. AMA J Dis Child. 1960;99:804–818. doi: 10.1001/archpedi.1960.02070030806016. [DOI] [PubMed] [Google Scholar]
- 30.Roberts HE, Cragan JD, Cono J, Khoury MJ, Weatherly MR, Moore CA. Increased frequency of cystic fibrosis among infants with jejunoileal atresia. Am J Med Genet. 1998;78:446–449. doi: 10.1002/(sici)1096-8628(19980806)78:5<446::aid-ajmg9>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 31.Amodio J, Berdon W, Abramson S, Stolar C. Microcolon of prematurity: a form of functional obstruction. AJR Am J Roentgenol. 1986;146:239–244. doi: 10.2214/ajr.146.2.239. [DOI] [PubMed] [Google Scholar]
- 32.Benya EC, Nussbaum-Blask AR, Selby DM. Colonic diverticulitis causing partial bowel obstruction in a child with cystic fibrosis. Pediatr Radiol. 1997;27:918–919. doi: 10.1007/s002470050271. [DOI] [PubMed] [Google Scholar]
- 33.George DH. Diverticulosis of the vermiform appendix patients with cystic fibrosis. Hum Pathol. 1987;18:75–79. doi: 10.1016/s0046-8177(87)80197-1. [DOI] [PubMed] [Google Scholar]
- 34.Makris K, Tsiotos GG, Stafyla V, Sakorafas GH. Small intestinal nonmeckelian diverticulosis. J Clin Gastroenterol. 2009;43:201–207. doi: 10.1097/MCG.0b013e3181919261. [DOI] [PubMed] [Google Scholar]
- 35.Cordes DO, Dewes HF. Diverticulosis and muscular hypertrophy of the small intestine of horses, pigs and sheep. N Z Vet J. 1971;19:108–111. doi: 10.1080/00480169.1971.33943. [DOI] [PubMed] [Google Scholar]
- 36.Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration. 2000;67:117–133. doi: 10.1159/000029497. [DOI] [PubMed] [Google Scholar]
- 37.Kulczycki LL, Kostuch M, Bellanti JA. A clinical perspective of cystic fibrosis and new genetic findings: relationship of CFTR mutations to genotype-phenotype manifestations. Am J Med Genet. 2003;116A:262–267. doi: 10.1002/ajmg.a.10886. [DOI] [PubMed] [Google Scholar]
- 38.Sturgess JM. Structural and developmental abnormalities of the exocrine pancreas in cystic fibrosis. J Pediatr Gastroenterol Nutr. 1984;3:S55–S66. doi: 10.1097/00005176-198400031-00011. [DOI] [PubMed] [Google Scholar]
- 39.Thomaidis TS, Arey JB. The intestinal lesions in cystic fibrosis of the pancreas. J Pediatr. 1963;63:444–453. doi: 10.1016/s0022-3476(63)80434-5. [DOI] [PubMed] [Google Scholar]
- 40.Tucker JA, Spock A, Spicer SS, Shelburne JD, Bradford W. Inspissation of pancreatic zymogen material in cystic fibrosis. Ultrastruct Pathol. 2003;27:323–335. [PubMed] [Google Scholar]
- 41.Farber S. The relation of pancreatic achylia to meconium ileus. J Pediatr. 1944;24:387–392. [Google Scholar]
- 42.Durie PR, Forstner GG. Pathophysiology of the exocrine pancreas in cystic fibrosis. J R Soc Med. 1989;82 Suppl 16:2–10. [PMC free article] [PubMed] [Google Scholar]
- 43.Bockman DE. Morphology of the exocrine pancreas related to pancreatitis. Microsc Res Tech. 1997;37(5–6):509–519. doi: 10.1002/(SICI)1097-0029(19970601)37:5/6<509::AID-JEMT13>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 44.Strobel O, Dor Y, Alsina J, Stirman A, Lauwers G, Trainor A, Castillo CF, Warshaw AL, Thayer SP. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology. 2007;133:1999–2009. doi: 10.1053/j.gastro.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oppenheimer EH, Esterly JR. Hepatic changes in young infants with cystic fibrosis: possible relation to focal biliary cirrhosis. J Pediatr. 1975;86:683–689. doi: 10.1016/s0022-3476(75)80351-9. [DOI] [PubMed] [Google Scholar]
- 46.Efrati O, Barak A, Modan-Moses D, Augarten A, Vilozni D, Katznelson D, Szeinberg A, Yahav J, Bujanover Y. Liver cirrhosis and portal hypertension in cystic fibrosis. Eur J Gastroenterol Hepatol. 2003;15:1073–1078. doi: 10.1097/00042737-200310000-00002. [DOI] [PubMed] [Google Scholar]
- 47.Strausbaugh SD, Davis PB. Cystic fibrosis: a review of epidemiology and pathobiology. Clin Chest Med. 2007;28:279–288. doi: 10.1016/j.ccm.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 48.Craig JM, Haddad H, Scwachman H. The pathological changes in the liver in cystic fibrosis of the pancreas. AM J Dis Child. 1957;93:357–369. doi: 10.1001/archpedi.1957.02060040359002. [DOI] [PubMed] [Google Scholar]
- 49.Porta EA, Stein AA, Patterson P. Ultrastructural changes of the pancreas and liver in cystic fibrosis. Am J Clin Pathol. 1964;42:451–465. doi: 10.1093/ajcp/42.5.451. [DOI] [PubMed] [Google Scholar]
- 50.Feranchak AP. In: Cystic fibrosis liver disease. Liver Disease in Children. ed 3. Suchy F., Sokol R., Balistreri W., editors. Cambridge University Press; New York, NY: 2007. pp. 572–594. [Google Scholar]
- 51.Chaudry G, Navarro OM, Levine DS, Oudijhane K. Abdominal manifestations of cystic fibrosis in children. Pediatr Radiol. 2006;36:233–240. doi: 10.1007/s00247-005-0049-2. [DOI] [PubMed] [Google Scholar]
- 52.Oppenheimer EH, Esterly JR. Pathology of cystic fibrosis review of the literature and comparison with 146 autopsied cases. Perspect Pediatr Pathol. 1975;2:241–278. [PubMed] [Google Scholar]
- 53.Esterly JR, Oppenheimer EH. Observations in cystic fibrosis of the pancreas. I. The gallbladder. Bull Johns Hopkins Hosp. 1962;110:247–255. [PubMed] [Google Scholar]
- 54.Agrons GA, Corse WR, Markowitz RI, Suarez ES, Perry DR. Gastrointestinal manifestations of cystic fibrosis: radiologic-pathologic correlation. Radiographics. 1996;16:871–893. doi: 10.1148/radiographics.16.4.8835977. [DOI] [PubMed] [Google Scholar]
- 55.Imrie JR, Fagan DG, Sturgess JM. Quantitative evaluation of the development of the exocrine pancreas in cystic fibrosis and control infants. Am J Pathol. 1979;95:697–708. [PMC free article] [PubMed] [Google Scholar]
- 56.Blackman SM, Deering-Brose R, McWilliams R, Naughton K, Coleman B, Lai T, Algire M, Beck S, Hoover-Fong J, Hamosh A, Fallin MD, West K, Arking DE, Chakravarti A, Cutler DJ, Cutting GR. Relative contribution of genetic and nongenetic modifiers to intestinal obstruction in cystic fibrosis. Gastroenterology. 2006;131:1030–1039. doi: 10.1053/j.gastro.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Drumm ML, Konstan MW, Schluchter MD, Handler A, Pace R, Zou F, Zariwala M, Fargo D, Xu A, Dunn JM, Darrah RJ, Dorfman R, Sandford AJ, Corey M, Zielenski J, Durie P, Goddard K, Yankaskas JR, Wright FA, Knowles MR, Gene Modifier Study Group Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353:1443–1453. doi: 10.1056/NEJMoa051469. [DOI] [PubMed] [Google Scholar]
- 58.Accurso FJ, Sontag MK. Gene modifiers in cystic fibrosis. J Clin Invest. 2008;118:839–841. doi: 10.1172/JCI35138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gu Y, Harley IT, Henderson LB, Aronow BJ, Vietor I, Huber LA, Harley JB, Kilpatrick JR, Langefeld CD, Williams AH, Jegga AG, Chen J, Wills-Karp M, Arshad SH, Ewart SL, Thio CL, Flick LM, Filippi MD, Grimes HL, Drumm ML, Cutting GR, Knowles MR, Karp CL. Identification of IFRD1 as a modifier gene for cystic fibrosis lung disease. Nature. 2009;458:1039–1042. doi: 10.1038/nature07811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grubb BR, Boucher RC. Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol Rev. 1999;79:S193–S214. doi: 10.1152/physrev.1999.79.1.S193. [DOI] [PubMed] [Google Scholar]
- 61.Bartoszewski R, Rab A, Jurkuvenaite A, Mazur M, Wakefield J, Collawn JF, Bebok Z. Activation of the unfolded protein response by deltaF508 CFTR. Am J Respir Cell Mol Biol. 2008;39:448–457. doi: 10.1165/rcmb.2008-0065OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Durie PR, Kent G, Philips MJ, Ackerley CA. Characteristic multiorgan pathology of cystic fibrosis in a long-living cystic fibrosis transmembrane regulator knockout murine model. Am J Pathol. 2004;164:1481–1493. doi: 10.1016/S0002-9440(10)63234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blyth DI, Pedrick MS, Savage TJ, Bright H, Beesley JE, Sanjar S. Induction, duration, and resolution of airway goblet cell hyperplasia in a murine model of atopic asthma: effect of concurrent infection with respiratory syncytial virus and response to dexamethasone. Am J Respir Cell Mol Biol. 1998;19:38–54. doi: 10.1165/ajrcmb.19.1.2930. [DOI] [PubMed] [Google Scholar]
- 64.Döring G, Gulbins E. Cystic fibrosis and innate immunity: how chloride channel mutations provoke lung disease. Cell Microbiol. 2009;11:208–216. doi: 10.1111/j.1462-5822.2008.01271.x. [DOI] [PubMed] [Google Scholar]
- 65.Chang HJ, Suh JI, Kwon SY. Gallstone formation and gallbladder mucosal changes in mice fed a lithogenic diet. J Korean Med Sci. 1999;14:286–292. doi: 10.3346/jkms.1999.14.3.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robert R, Norez C, Becq F. Disruption of CFTR chloride channel alters mechanical properties and cAMP-dependent Cl- transport of mouse aortic smooth muscle cells. J Physiol. 2005;568:483–495. doi: 10.1113/jphysiol.2005.085019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vandebrouck C, Melin P, Norez C, Robert R, Guibert C, Mettey Y, Becq F. Evidence that CFTR is expressed in rat tracheal smooth muscle cells and contributes to bronchodilation. Respir Res. 2006;7:113. doi: 10.1186/1465-9921-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Michoud MC, Robert R, Hassan M, Moynihan B, Haston C, Govindaraju V, Ferraro P, Hanrahan JW, Martin JG. Role of the cystic fibrosis transmembrane conductance channel in human airway smooth muscle. Am J Respir Cell Mol Biol. 2009;40:217–222. doi: 10.1165/rcmb.2006-0444OC. [DOI] [PubMed] [Google Scholar]
- 69.Duenhoelter JH, Pritchard JA. Fetal respiration. A review Am J Obstet Gynecol. 1977;129:326–338. doi: 10.1016/0002-9378(77)90793-1. [DOI] [PubMed] [Google Scholar]
- 70.Robinson M, Bye PT. Mucociliary clearance in cystic fibrosis. Pediatr Pulmonol. 2002;33:293–306. doi: 10.1002/ppul.10079. [DOI] [PubMed] [Google Scholar]
- 71.Inglis SK, Wilson SM. Cystic fibrosis and airway submucosal glands. Pediatr Pulmonol. 2005;40:279–284. doi: 10.1002/ppul.20183. [DOI] [PubMed] [Google Scholar]
- 72.Elizur A, Cannon CL, Ferkol TW. Airway inflammation in cystic fibrosis. Chest. 2008;133:489–495. doi: 10.1378/chest.07-1631. [DOI] [PubMed] [Google Scholar]
- 73.Dubin PJ, McAllister F, Kolls JK. Is cystic fibrosis a TH17 disease? Inflamm Res. 2007;56:221–227. doi: 10.1007/s00011-007-6187-2. [DOI] [PubMed] [Google Scholar]