

Abstract
Brain edema and the associated increase in intracranial pressure are potentially lethal complications of acute liver failure (ALF). Astrocyte swelling (cytotoxic edema) represents a significant component of the brain edema in ALF, and elevated blood and brain ammonia levels have been strongly implicated in its formation. We earlier showed in cultured astrocytes that oxidative stress (OS) and the mitochondrial permeability transition (mPT) play major roles in the mechanism of ammonia-induced astrocyte swelling. Glutamine, a byproduct of ammonia metabolism, has also been shown to induce OS, the mPT, and astrocyte swelling. Such effects of glutamine were suggested to be mediated by its hydrolysis in mitochondria, potentially yielding high levels of ammonia in this organelle and leading to OS and the mPT. L-histidine, an inhibitor of mitochondrial glutamine transport, was recently shown to mitigate OS, mPT, and cell swelling in cultured astrocytes treated with ammonia. The present study examined whether L-histidine similarly abolishes OS, the mPT, and brain edema in a rat model of ALF. Treatment of rats with thioacetamide caused a significant degree of brain edema, which was associated with induction of OS and the mPT. These changes were completely abolished by L-histidine, supporting a key role of mitochondrial glutamine transport and hydrolysis in the mechanism of the brain edema associated with ALF.
Acute liver failure (ALF) is a life-threatening condition accounting for approximately 80% mortality in these patients.1,2 Such high mortality is in part attributable to the development of severe brain edema, leading to increased intracranial pressure and brain herniation. Currently, there is no effective treatment for the brain edema in ALF other than an emergency liver transplantation.2,3 Of several factors implicated in the development of ALF, ammonia is generally considered an important one as its levels in blood and brain correlate well with the degree of encephalopathy and brain edema.4–7
The edema in ALF is believed to be largely “cytotoxic” because of swelling of astrocytes.5,8,9 Although factors responsible for astrocyte swelling in ALF are incompletely understood, pathophysiological levels of ammonia were shown to result in astrocyte swelling in brain slices,10 as well as in cultured astrocytes.11–13 Similarly, astrocyte swelling has been identified in animal models of hyperammonemia,14,15 as well as in experimental and human ALF.5,8,9
Mechanisms of astrocyte swelling/brain edema in ALF are poorly understood. Recently oxidative stress (OS) has been implicated as a major factor in the development of brain edema.16–18 The mitochondrial permeability transition (mPT), often a consequence of OS, has also been shown to result in cell swelling in cultured astrocytes exposed to pathologically relevant concentrations of ammonia,19 as well as in vivo models of ALF.20
Although ammonia continues to be a major toxin in ALF, several lines of evidence indicate that many of the deleterious effects of ammonia, including seizures, coma, altered vascular response to CO2, and brain edema, are a consequence of glutamine, a byproduct of ammonia metabolism.21–24 High levels of glutamine in brain and cerebrospinal fluid have been identified in ALF.25–27 The production of glutamine results from the action of glutamine synthetase, an enzyme that in brain is largely localized to astrocytes,28 which likely explains the involvement of astrocytes in ALF.
Recent studies using cultured astrocytes documented that many of the deleterious effects caused by ammonia are also a consequence of glutamine. Among these include OS and the mPT,29–31 processes known to contribute to astrocyte swelling (for reviews, see refs18,32). The means by which glutamine exerts OS and the mPT have been postulated to involve the transport of glutamine into mitochondria, and its subsequent hydrolysis in that organelle through the action of phosphate-activated glutaminase (PAG), potentially resulting in the generation of high levels of ammonia in mitochondria.31 Consistent with this view, inhibition of glutamine synthesis or its mitochondrial transport and hydrolysis were found to significantly diminish OS, the mPT and cell swelling in cultured astrocytes.33,34
Although these studies strongly suggest that the transport of glutamine into mitochondria and its subsequent hydrolysis represent critical processes by which glutamine contributes to astrocyte swelling, such a mechanism has not been investigated in vivo. The present study examined whether L-histidine, an inhibitor of mitochondrial glutamine transport, diminishes the severity of OS, the mPT, and the brain edema in an in vivo model of ALF.
Materials and Methods
Induction of ALF
ALF was induced in adult male albino Fisher rats (#344; 200 to 250 g, Charles River Laboratories, Wilmington, MA) by administration of the hepatotoxin thioacetamide (TAA, Sigma-Aldrich, St. Louis, MO) following the method of Hilgier et al.35 This model has been used for more than 25 years and has been validated as morphological, and clinical abnormalities are similar to those observed in humans.35–40 In unpublished observations, we found that this model maintained the integrity of the blood–brain barrier as shown by failure to detect Evans blue in TAA-treated rat brains after intravenous injection. Additionally, TAA has no direct effects on brain as it does not cross the blood–brain barrier.41 In our experience, this model has shown consistency and reproducibility with regard to the clinical course, as well as in the development of brain edema.
TAA (300 mg/kg i.p) was given to animals daily for 3 days. L-histidine (100 mg/kg, Sigma-Aldrich, St. Louis, MO) was dissolved in saline and injected (i.p.) daily 2 hours before each TAA injection. To prevent hypoglycemia and dehydration, rats were given 12.5 ml/kg of fluid therapy (5% dextrose and 0.45% saline with 20 mEq/L of potassium chloride) every 12 hours, s.c. Normal controls received saline (vehicle used for TAA), whereas another group of rats were given L-histidine alone (100 mg/kg) daily for 3 days. TAA-treated rats were clinically monitored, and stages of encephalopathy were graded according to the criteria of Gammal et al39: Grade I, generalized reduction in spontaneous activity; Grade II, mild ataxia; Grade III, lack of spontaneous movement but with intact righting reflexes; Grade IV, loss of righting reflex but intact pain reflex (measured by reaction to tail pinch); Grade V, coma (unresponsive to sensory stimuli) and loss of corneal reflexes. Animals were sacrificed at advanced Grade IV stage, approximately 16 to 24 hours after the third injection of TAA.
Animals were sacrificed by decapitation and blood was obtained from the cervical wound for liver function tests. Blood levels of aspartate and alanine aminotransferases were determined in control and TAA-treated rats 76 h after the initiation of TAA treatment using the Cobas automatic analyzer (Roche Diagnostics, Indianapolis, IN). Cerebral cortex was rapidly dissected and used for determination of protein levels of hemeoxygenase-1 (HO-1), as well as the mPT and brain water content. Livers were excised, fixed in 10% buffered formalin, and processed routinely for histological analysis. All experimental procedures followed guidelines established by National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee.
Brain Ammonia and Glutamine Measurement
Immediately after decapitation, brains were excised and rapidly frozen in liquid nitrogen and stored at −140°C. At the time of analysis, brains were weighed, homogenized in 1 mol/L perchloric acid, and centrifuged at 16,000g for 15 minutes at 4°C. Ammonia and glutamine levels were measured in the supernatant using commercially available ammonia and glutamine assay kits following the manufacturer's protocol (Sigma–Aldrich, St. Louis, MO).
Brain Glutamine Synthetase Measurement
Glutamine synthetase activities were determined from frozen cortical tissue by the methods of Pamiljans et al.42 Briefly, frontal cortical tissue was homogenized in 10 volumes (w/v) of 0.15 mol/L KCl, and a 0.1 ml aliquot of the homogenate was incubated in a reaction buffer containing 10 mmol/L MgCl2, 50 mmol/L l-glutamine, 100 mmol/L imidazole-HCl (pH 7.4), 10 mmol/L β-mercaptoethanol, 50 mmol/L hydroxylamine hydrochloride (pH 7.4), and 10 mmol/L ATP at 37°C for 15 minutes. The reaction was terminated by the addition of a mixture of 0.67 mol/L HCl and 0.2 mol/L trichloroacetic acid. The precipitate was centrifuged, and the absorbance of the supernatant was measured at 530 nm and compared with the absorbance generated by known concentrations of γ-glutamyhydroxamate that were treated with the ferric chloride reagent. Activity was expressed as μmol/mg protein/h.
Brain PAG Measurement
PAG activity was determined by the method of Roberg et al.43 Briefly, frontal cortical tissue was homogenized in 10 volumes (w/v) of buffer containing 250 mmol/L D-mannitol, 70 mmol/L sucrose, 10 mmol/L HEPES, (pH 8.6). Homogenates (100 μl) were incubated at 37°C for 1 hour in 30 mmol/L glutamine, 1 mmol/L potassium EDTA, and 150 mmol/L potassium phosphate buffer. The reaction was terminated by addition of ice-cold 70% ethanol, and the samples were centrifuged for 5 minutes at 12,000g at 4°C. The amount of glutamate produced by the PAG reaction was measured by spectrofluorometry. Briefly, 50 μl of ethanol extract was added to a buffer containing 1.5 mmol/L NAD, 0.23 mmol/L ADP, 0.03% H2O2 in 50 mmol/L Tris-HCl (pH 8). The reaction was started by the addition of five units glutamate dehydrogenase, and 30 minutes after incubation at room temperature the absorbance was measured at 340 nm and compared with the absorbance generated by known concentration of L-glutamate standards. Activity was expressed as nmol/mg protein/min.
Hemeoxygenase-1 Determination
Brain samples from frontal cortex were homogenized in ice-cold lysis buffer (50 mmol/L Tris–HCl pH 7.6, 150 mmol/L NaCl, 50% glycerol 1% NP-40, containing a protease inhibitor cocktail (Roche Applied Science) and centrifuged at 12,000g for 15 minutes. Protein concentrations were measured using the bicinchoninic acid method (Bio-Rad Laboratories, Hercules, CA). Equal amounts of protein were subjected to gel electrophoresis as previously described,44 and the gels were transferred to polyvinylidene difluoride membranes. After blocking with nonfat dry milk, membranes were incubated with rabbit anti-hemeoxygenase-1 (1:1000, Millipore, CA) and mouse anti–α-tubulin antibodies (1:1000, Oncogene, CA). Anti-rabbit and anti-mouse horseradish peroxidase-conjugated secondary antibodies (Vector Laboratories) were used at 1:1000. The peroxidase activity was detected by chemiluminescence using the ECL detection system (Amersham, Arlington Heights, IL). Optical density of the bands was determined with the Chemi-Imager (α Innotech, San Leandro, CA) digital imaging system, and the results were quantified with the Sigma Scan Pro (Jandell Scientific, San Jose, CA) program as a proportion of the signal of a housekeeping protein band (α-tubulin).
Measurement of the mPT
Induction of the mPT was determined by the 2-deoxyglucose (2-DG) entrapment method.45 This method is based on the principle that [3H]−2-DG enters the cell via the glucose transporter present on the plasma membrane, and is then phosphorylated by hexokinase to [3H]−2-deoxyglucose-6-phosphate ([3H]−2-DG-6P) in the cytoplasm. As the inner membrane of normal mitochondria is not permeable to [3H]−2-DG-6P, very little label will be found in mitochondria of normal cells. However, when the mPT is induced, [3H]−2-DG-6P enters mitochondria through the permeability transition pore, which then equilibrates with the cytosolic pool of the isotope. Closure of the permeability transition pore at this stage with the Ca2+ chelator EGTA results in entrapment of [3H]−2-DG-6P within mitochondria. The presence of labeled [3H]−2-DG-6P within mitochondria provides a measure of the mPT.
Rats in each experimental group were injected with 0.2 ml of [3H]−2-DG (20 μCi/ml) 2 hours before sacrifice. The cerebral cortex was homogenized in 0.32 mol/L sucrose containing 1 mmol/L EGTA, 10 mmol/L Tris-HCl, and 2.5 μmol/L cyclosporin A so as to prevent opening of the permeability transition pore. An aliquot (1 ml) of homogenate was saved for measurement of total radioactivity, the remainder was centrifuged at 4000g for 5 minutes, and the supernatant was recentrifuged at 12,000g for 20 minutes. The pellet was washed two times with homogenization buffer and recentrifuged. The final pellet containing mitochondria was suspended in homogenization buffer without cyclosporin A. The radioactivity present in brain homogenates and in the mitochondrial fraction was determined after precipitating these fractions with 5% perchloric acid. The supernatants were collected and neutralized with 3 mol/L potassium bicarbonate and centrifuged at 10,000g for 20 minutes. The final supernatants were transferred to scintillation vials and the radioactivity was determined. The purity of the mitochondrial fraction was assessed by measuring the activity of citrate synthase,46 and the radioactivity present in the mitochondrial fraction was normalized to citrate synthase activity. The results were expressed as the ratio of units of [3H]−2-DG-6P present in the mitochondrial fraction versus homogenate.
Measurement of Brain Edema
Brain water content was determined by the wet/dry weight method. Approximately 10 mg tissue (three to four pieces from each animal) of cerebral cortex was dissected; wet weights of tissue were determined; tissue was dried overnight in an oven at 120°C; and dry weights determined. The difference in wet/dry weights was expressed as percent water content.
Statistical Analysis
Data are represented as mean ± SEM. The data were subjected to analysis of variance followed by Neuman–Keuls posthoc analysis. The mPT and brain water content measurements were repeated at least three times with three rats in each experimental group. Brain ammonia and glutamine levels, as well as glutamine synthetase (GS), PAG, activities and HO-1 protein were determined in triplicate in each sample obtained from 4 separate animals in each experimental group. The statistical analysis showing P < 0.05 was considered significant.
Results
Assessment of ALF
TAA-treated rats appeared normal during the first 40 hours after the first administration of TAA. Between 40 and 60 hours, rats gradually developed Grade I encephalopathy. These symptoms progressively worsened over time, and by approximately 60 to 72 hours the animals evolved to Grade II to III encephalopathy. This was followed by a rapid progression into Grade IV stage (at approximately 80 hours), at which time rats displayed loss of righting reflexes, and unresponsiveness to pain, followed by reduced corneal reflexes. In TAA-treated animals given L-histidine, the progression of symptoms was delayed by approximately 12 hours as compared with rats treated with TAA alone.
The extent of liver injury was assessed by serum alanine aminotransferase and aspartate aminotransferase levels. Alanine aminotransferase activity was increased 60-fold in TAA-treated animals as compared with controls (TAA, 6700 ± 380 U/L versus control, 109 ± 26 U/L). Aspartate aminotransferase activity showed a fivefold increase in TAA-treated animals as compared with controls (TAA 1235 ± 180 U/L versus control 221 ± 45 U/L). Histological assessment of the liver displayed centrolobular necrosis involving approximately 40% of the liver (data not shown). L-histidine treatment did not protect against TAA-induced liver injury as assessed by serum aminotransferase levels as well as by histology (data not shown).
Effect of L-Histidine on Brain Ammonia and Glutamine Levels in Rats with ALF
Rats with ALF resulted in a threefold increase in brain ammonia levels in TAA-treated rats as compared with untreated controls. Similarly, brain glutamine levels were increased 2.5-fold in TAA-treated rats compared with untreated controls (Figure 1, A and B). Administration of L-histidine to TAA-treated rats had no effect on ammonia or glutamine levels as compared with TAA-treated rats alone (Figure 1). Ammonia and glutamine levels in normal rats treated with L-histidine alone were unaffected.
Figure 1.
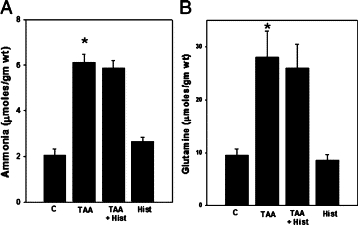
Brain ammonia (A) and glutamine (B) levels in ALF rat. Values were mean ± SEM of triplicate measurements in samples obtained from four separate animals in each experimental group. *P < 0.01 versus control (C). TAA indicates thioacetamide; Hist, L-histidine (100 mg/kg, i.p).
Effect of L-Histidine on Brain GS Activity in Rats with ALF
Rats with ALF showed a 30% (P < 0.05) reduction in GS activity (despite increased glutamine levels), in agreement with an earlier report using another model of ALF47 (Figure 2). L-histidine treatment did not affect the GS activities in rats with ALF. Treatment of normal rats with L-histidine alone had no effect on GS activity.
Figure 2.
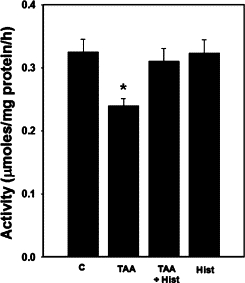
Brain glutamine synthetase activity in ALF rat. Values were mean ± SEM of triplicate measurements in samples obtained from four separate animals in each experimental group. *P < 0.01 versus control (C). There was no statistical difference between TAA versus TAA+ Hist.
Effect of L-Histidine on Brain PAG Activity in Rats with ALF
Rats treated with ALF showed no changes in PAG activity (Figure 3), which is consistent with earlier reports by Albrecht et al48 in rats with ALF induced by TAA. Likewise, L-histidine treatment had no effect on PAG activity in rats with ALF. Treatment of normal rats with L-histidine alone also had no effect on PAG activity.
Figure 3.
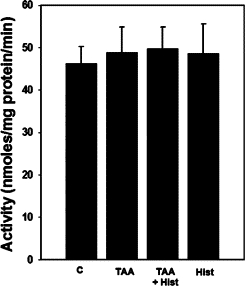
Brain PAG activity in ALF rat. Values were mean ± SEM of triplicate measurements in samples obtained from four separate animals in each experimental group.
Effect of L-Histidine on HO-1 Protein Expression in Rats with ALF
Protein expression of HO-1 in frontal cortex of TAA rats showed a significant increase (34%, P < 0.05). Treatment of TAA rats with L-histidine completely abolished this increase (Figure 4). Treatment of normal rats with L-histidine alone had no effect on HO-1 protein expression (data not shown).
Figure 4.
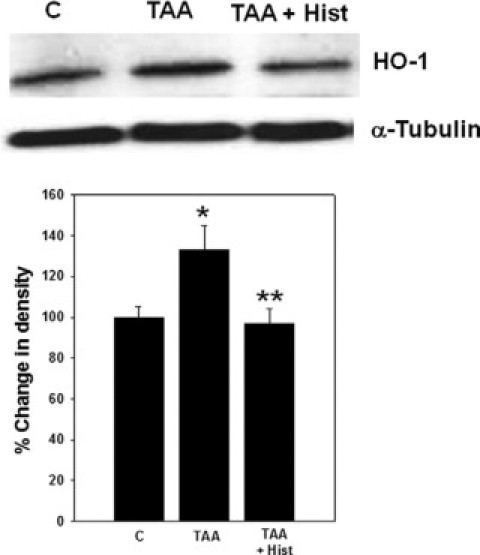
Effect of L-histidine in hemeoxygenase (HO-1) protein expression in rats with ALF. The density of protein bands are expressed as mean ± SEM percentage values of triplicate measurements in samples obtained from four separate animals in each experimental group. *P < 0.05 versus control; **P < 0.01 versus TAA.
Effect of L-Histidine on the Induction of the mPT in Brains of Rats with ALF
ALF resulted in the mPT as demonstrated by a significant increase (80%, P < 0.01) in mitochondrial [3H]−2-DG-6P content. L-histidine (100 mg/kg) completely blocked the induction of the mPT (Figure 5). Lower concentrations of L-histidine (25 and 50 mg/kg) also completely blocked the mPT (data not shown).
Figure 5.
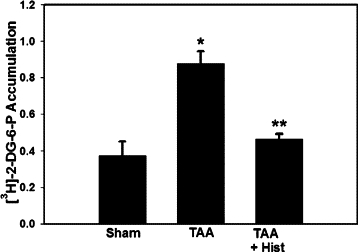
Effect of L-histidine on the mPT in mitochondria from brains of rats with TAA-induced ALF. Values represent the ratio of DPM/unit citrate synthase/DPM/mg homogenate. This ratio was obtained by first normalizing the DPM of [3H]−2-DG-6P in the mitochondrial fraction per unit citrate synthase (a mitochondrial marker), and then divided by [3H]−2-DG-6P DPM values from brain homogenates which were normalized to mg protein. Values in each experimental group represent mean ± SEM (n = 9). *P < 0.05 versus control; **P < 0.01 versus TAA.
Effect of L-Histidine on Brain Edema in Rats with ALF
Rats treated with TAA showed a 2.9% (P < 0.01) increase in brain water content as compared with saline-treated controls (controls and TAA-treated rats were 78.7 ± 0.5% versus 81.05 ± 0.9%, respectively). L-histidine (100 mg/kg) completely inhibited the brain edema in TAA-treated rats (Figure 6). Similar to the mPT, lower doses of L-histidine (25 and 50 mg/kg) also completely blocked the brain edema in TAA rats (data not shown).
Figure 6.
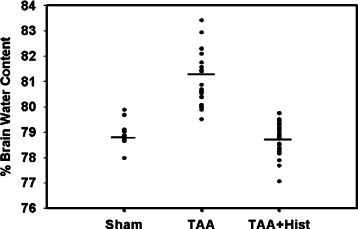
Effect of L-histidine on brain edema in ALF. Values in each experimental group represent percent brain water content of cortical sections (weighing ≈10 mg) obtained from nine rats in each experiments.
Discussion
This study demonstrates that rats with TAA-induced ALF develop brain edema as shown by an increase in brain water content (2.9%). The degree of edema observed in this study is similar to that of other models of ALF induced by hepatic devascularization47,49,50 and by the hepatotoxin galactosamine,51 as well as in hyperammonemic rats.24 Rats with ALF also showed increased protein expression of HO-1, a marker of OS,52–54 in agreement with the findings of Jiang et al.55,56 We also identified the development of the mPT, consistent with our previous studies in ammonia-treated cultured astrocytes.57 The systemic administration of L-histidine completely abolished the HO-1 protein overexpression, induction of the mPT, and brain edema in TAA-treated rats.
Cytotoxic brain edema, principally attributable to astrocyte swelling, is the major neuropathological finding in ALF.5,8,9 Various studies have documented that glutamine, a product of ammonia metabolism, plays a major role in the development of the brain edema in ALF. A strong correlation between blood, brain, and cerebrospinal fluid levels of glutamine and the magnitude of intracranial pressure have been reported in ALF.58,59 Additionally, inhibition of glutamine synthesis by methionine sulfoximine attenuates the brain edema in experimental models of hepatic encephalopathy and hyperammonemia,15,23,24 and reduces cell swelling in cultured astrocytes exposed to ammonia.34,60 For reviews, see refs.31,61
The precise mechanism by which glutamine results in brain edema is not clear. A commonly held view is that glial accumulation of glutamine leads to an osmotic shift of water into astrocytes, resulting in their swelling (osmolyte hypothesis).62 Although this view strongly supports a concordance between cerebral ammonia metabolism and astrocyte swelling, the osmolyte hypothesis appears to be at variance with reports documenting a reduction of brain edema by various modalities in the absence of a commensurate reduction in cerebral glutamine concentration.63–65 Furthermore, a recent study showed an absence of temporal correlation between glutamine concentration and the extent of cell swelling in ammonia-treated cultured astrocytes.66
Another mechanism postulates that glutamine acts to induce OS and the mPT, and that these two factors are ultimately responsible for cell swelling.31 OS is known to contribute to the evolution of astrocyte swelling in culture and in brain slices.44,67–71 OS also has been shown to contribute to brain edema in ALF55 (for reviews see refs16,18). Studies have documented that ammonia and its metabolite glutamine both produce OS.16,30,72 Consistent with the role of OS in ALF, a recent preliminary investigation showed the oxidation and nitration of several proteins in cerebral cortex of rats treated with TAA, and that antioxidants significantly abolished the brain edema in these animals.20
One consequence of OS is induction of the mPT,73 a phenomenon characterized by a sudden increase in the permeability of the inner mitochondrial membrane attributable to the opening of the permeability transition pore. This results in the collapse of inner mitochondrial membrane potential, leading to defective oxidative phosphorylation and bioenergetic failure (for reviews on the mPT, see refs74,75). Ammonia and glutamine both have been shown to induce the mPT in cultured astrocytes,29,57 which was diminished by treatment with antioxidants,29,76 consistent with a role of OS in the induction of the mPT. The mPT has been shown to contribute to astrocyte swelling by ammonia, as cyclosporin A, an inhibitor of the mPT, completely blocked such swelling.19 The present study showing the induction of the mPT in TAA-treated rats suggests that it might also contribute to the brain edema in ALF. Indeed, our preliminary investigations report a significant diminution of brain edema in TAA-treated rats given the mPT inhibitor cyclosporin A.20 The means by which the mPT likely contributes to brain edema is not known. However, it is notable that the mPT also represents an additional source of OS,77,78 a factor that is known to cause brain edema.18 Furthermore, bioenergetic failure, conceivably resulting from the induction of the mPT, may also contribute to brain edema in ALF. While studies document a depletion of ATP levels in cultured astrocytes exposed to ammonia,34 changes in brain ATP levels in ALF are controversial. Nevertheless, alterations in cerebral energy metabolism leading to depletion of brain levels of ATP have been consistently documented in animals models chronic HE and hyperammonemia (for review, see ref79).
It should be noted that the cellular source of mitochondria undergoing the mPT in brains of rats with ALF is not known. Nevertheless, evidence suggests that astrocytes are the principal cells affected in both acute and chronic liver failure, whereas neuronal injury is generally not observed except in the agonal phase of ALF.80 Additionally, in vitro studies document that pathophysiological levels of ammonia results in induction of the mPT in astrocytes but not in neurons.33,57 It is therefore reasonable to assume that the mPT occurred in astrocyte mitochondria in ALF and that it would have contributed to the brain edema.
Although these studies suggest that OS and the mPT likely induced by glutamine (derived from ammonia) represent major factors in the development of astrocyte swelling/brain edema in ALF, the means by which glutamine induces OS and the mPT are not completely clear. It was previously shown that OS and the mPT after treatment of cultured astrocytes with glutamine were diminished by 6-diazo-5-oxo-L-norleucine (DON), an inhibitor of mitochondrial PAG.33 Likewise, DON significantly inhibited cell swelling caused by ammonia in cultured astrocytes.33 It should be emphasized that part of the glutamine synthesized in astrocytes is transported into mitochondria where it is then hydrolyzed to glutamate and ammonia by PAG,81,82 potentially yielding high levels of ammonia in mitochondria that could trigger OS and the mPT. Such OS and the mPT generated in mitochondria would likely contribute to the astrocyte swelling/brain edema observed in hyperammonemic states.31
Although studies in cultured astrocytes support the view that mitochondrial hydrolysis of glutamine (by the action of PAG) represents an important mechanism for astrocyte swelling, the induction of OS and the mPT, it should be noted that the presence of mitochondrial PAG in astrocytes in vivo is debatable. One study reported that in cerebellum PAG was localized in neurons but not in astrocytes.83 However, studies by Subbalakshmi and Murthy84 and Rao and Murthy85 have shown glutaminase activity in astrocytes bulk-isolated from adult rat brain. Similarly, enzyme histochemical studies showed its presence in astrocytes in vivo.86 More definitively, a recent immunohistochemical study documented that astrocytes in vivo do in fact express PAG.87
Consistent with studies on the effect of DON in abolishing OS and the mPT in cultured astrocytes, a recent report documented that L-histidine, a known inhibitor of glutamine transport into brain mitochondria,88–90 also significantly blocked the ammonia-induced cell swelling in cultured astrocytes.34 Likewise, L-histidine abolished OS and the mPT induced by ammonia.34 L-histidine is an essential amino acid that readily crosses the blood–brain barrier.91 AFter systemic administration of L-histidine (1000 mg/kg) to rats, its levels were found to be increased in brain (230%), and such high levels persisted for up to three hours,92 indicating that L-histidine readily enters the brain and its level remains elevated for long periods of time.
The present in vivo study shows that L-histidine completely inhibits HO-1 overexpression (a marker of OS), the mPT, and the brain edema. We examined whether these effects might be attributable to its interference with the brain levels of ammonia and glutamine. However, ammonia and glutamine levels were unaltered, and the activity of glutamine synthetase was not changed in TAA-treated rats given L-histidine (Figures 1 and 2). These studies illustrate that the effect of L-histidine might be mediated by pathways downstream of glutamine synthesis, likely by inhibiting the transport of glutamine into mitochondria and its subsequent hydrolysis.
Similar to studies with GS, we examined for a potential interference of L-histidine on brain PAG activity in TAA-treated rats. PAG activity was found to be unaltered in TAA-treated rat brains, and L-histidine had no effect on PAG activity (Figure 3). The absence of changes in PAG activity is consistent with earlier reports by Albrecht et al.48 These data, however, do not negate the view that mitochondrial hydrolysis of glutamine, yielding high levels of ammonia, likely represent the mechanism for OS, the mPT, and brain edema in ALF. Thus, whereas L-histidine does not interfere with PAG activity, the protective effect of L-histidine is likely mediated by diminishing the transport of glutamine into mitochondria, resulting in a reduction in glutamine hydrolysis, along with a commensurate reduction in ammonia generation in mitochondria.
In addition to its ability to block mitochondrial glutamine transport, L-histidine is known to possess antioxidant properties,93 particularly against singlet oxygen species.94 Studies showing the neuroprotective effects of L-histidine, including a reduction in brain edema in cryogenic injury,95 were suggested to be mediated by reducing OS. It is however not known whether the ability of L-histidine to inhibit OS and brain edema in TAA-treated rats is mediated by its free radical–scavenging effect. Obata et al96 report that only high concentrations of L-histidine (>25 mmol/L) inhibited free radical production in rat striatum caused by 1-methyl-4-phenylpyridinium ion (MPP+). Although brain levels of L-histidine were not measured in our study, a report by Tyfield and Holton97 showed that the systemic administration of L-histidine (500 mg/kg), a fivefold higher concentration than that used in the present study, resulted in brain L-histidine levels of only 1 mmol/L, a concentration insufficient to exert an antioxidant effect.
Whereas our study showed that L-histidine protected rats against TAA-induced brain edema and delayed the clinical progression of ALF, a report examining the role of histidine on brain histamine levels noted that L-histidine (500 mg/kg) did not show improvement in neurological symptoms nor did it influence the timing of coma in rats with ALF induced by hepatic devascularization.98 This study did not assess brain edema. It is possible that the devascularization model, which results in complete liver necrosis leading to a very early onset of coma (14 hours), may be too severe for histidine to show any protective effect. In contrast to their model, rats with ALF induced by TAA showed a lesser degree of hepatocellular necrosis (approximately 40%). Furthermore, the appearance of neurological symptoms, as well as the onset of coma, was significantly delayed (neurological deterioration initiated at approximately 60 hours after first administration of TAA and rats did not become comatose until 80 hours later). It is possible that a lesser severity of liver injury with TAA may explain the ability of L-histidine to provide neuroprotection. It is also possible that the relatively large concentration (500 mg/kg) of L-histidine used by these investigators may have resulted in adverse neurological effects.
In summary, our study demonstrates that L-histidine, an agent known to inhibit mitochondrial glutamine transport, blocks OS, the mPT, and brain edema formation in experimental ALF. These studies suggest that the transport of glutamine into mitochondria, followed by its hydrolysis, potentially yields high levels of ammonia in mitochondria, likely resulting in OS and the mPT, and brain edema. L-histidine may represent a potentially useful therapeutic agent for the brain edema associated with acute liver failure.
Acknowledgements
We thank Dr. Arumugam R. Jayakumar for assistance in these studies.
Footnotes
Supported by The Department of Veterans Affairs Merit review grant and NIH grant DK063311.
References
- 1.Capocaccia L, Angelico M. Fulminant hepatic failure. Clinical features, etiology, epidemiology, and current management. Dig Dis Sci. 1991;36:775–779. doi: 10.1007/BF01311236. [DOI] [PubMed] [Google Scholar]
- 2.Hoofnagle JH, Carithers RL, Jr, Shapiro C, Ascher N. Fulminant hepatic failure: summary of a workshop. Hepatology. 1995;21:240–252. [PubMed] [Google Scholar]
- 3.Bismuth H, Samuel D, Castaing D, Williams R, Pereira SP. Liver transplantation in Europe for patients with acute liver failure. Semin Liver Dis. 1996;16:415–425. doi: 10.1055/s-2007-1007254. [DOI] [PubMed] [Google Scholar]
- 4.Traber PG, Dal Canto M, Ganger DR, Blei AT. Electron microscopic evaluation of brain edema in rabbits with galactosamine-induced fulminant hepatic failure: ultrastructure and integrity of the blood-brain barrier. Hepatology. 1987;7:1272–1277. doi: 10.1002/hep.1840070616. [DOI] [PubMed] [Google Scholar]
- 5.Kato M, Sugihara J, Nakamura T, Muto Y. Electron microscopic study of the blood-brain barrier in rats with brain edema and encephalopathy due to acute hepatic failure. Gastroenterol Jpn. 1989;24:135–142. doi: 10.1007/BF02774187. [DOI] [PubMed] [Google Scholar]
- 6.Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology. 1999;29:648–653. doi: 10.1002/hep.510290309. [DOI] [PubMed] [Google Scholar]
- 7.Ong JP, Aggarwal A, Krieger D, Easley KA, Karafa MT, Van Lente F, Arroliga AC, Mullen KD. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med. 2003;114:188–193. doi: 10.1016/s0002-9343(02)01477-8. [DOI] [PubMed] [Google Scholar]
- 8.Martinez A. Electron microscopy in human hepatic encephalopathy. Acta Neuropathol (Berl) 1968;11:82–86. doi: 10.1007/BF00692797. [DOI] [PubMed] [Google Scholar]
- 9.Traber P, DalCanto M, Ganger D, Blei AT. Effect of body temperature on brain edema and encephalopathy in the rat after hepatic devascularization. Gastroenterology. 1989;96:885–891. [PubMed] [Google Scholar]
- 10.Ganz R, Swain M, Traber P, DalCanto M, Butterworth RF, Blei AT. Ammonia-induced swelling of rat cerebral cortical slices: implications for the pathogenesis of brain edema in acute hepatic failure. Metab Brain Dis. 1989;4:213–223. doi: 10.1007/BF01000297. [DOI] [PubMed] [Google Scholar]
- 11.Norenberg MD, Baker L, Norenberg LO, Blicharska J, Bruce-Gregorios JH, Neary JT. Ammonia-induced astrocyte swelling in primary culture. Neurochem Res. 1991;16:833–836. doi: 10.1007/BF00965694. [DOI] [PubMed] [Google Scholar]
- 12.Olson JE, Evers JA, Holtzman D. Astrocyte volume regulation and ATP and phosphocreatine concentrations after exposure to salicylate, ammonium, and fatty acids. Metab Brain Dis. 1992;7:183–196. doi: 10.1007/BF01000245. [DOI] [PubMed] [Google Scholar]
- 13.Zwingmann C, Flogel U, Pfeuffer J, Leibfritz D. Effects of ammonia exposition on glioma cells: changes in cell volume and organic osmolytes studied by diffusion-weighted and high-resolution NMR spectroscopy. Dev Neurosci. 2000;22:463–471. doi: 10.1159/000017476. [DOI] [PubMed] [Google Scholar]
- 14.Norenberg MD. A light and electron microscopic study of experimental portal-systemic (ammonia) encephalopathy. Progression and reversal of the disorder. Lab Invest. 1977;36:618–627. [PubMed] [Google Scholar]
- 15.Willard-Mack CL, Koehler RC, Hirata T, Cork LC, Takahashi H, Traystman RJ, Brusilow SW. Inhibition of glutamine synthetase reduces ammonia-induced astrocyte swelling in rat. Neuroscience. 1996;71:589–599. doi: 10.1016/0306-4522(95)00462-9. [DOI] [PubMed] [Google Scholar]
- 16.Norenberg MD. Oxidative and nitrosative stress in ammonia neurotoxicity. Hepatology. 2003;37:245–248. doi: 10.1053/jhep.2003.50087. [DOI] [PubMed] [Google Scholar]
- 17.Norenberg MD, Jayakumar AR, Rama Rao KV. Oxidative stress in the pathogenesis of hepatic encephalopathy. Metab Brain Dis. 2004;19:313–329. doi: 10.1023/b:mebr.0000043978.91675.79. [DOI] [PubMed] [Google Scholar]
- 18.Norenberg MD, Jayakumar AR, Rama Rao KV, Panickar KS. New concepts in the mechanism of ammonia-induced astrocyte swelling. Metab Brain Dis. 2007;22:219–234. doi: 10.1007/s11011-007-9062-5. [DOI] [PubMed] [Google Scholar]
- 19.Rama Rao KV, Chen M, Simard JM, Norenberg MD. Suppression of ammonia-induced astrocyte swelling by cyclosporin A. J Neurosci Res. 2003;74:891–897. doi: 10.1002/jnr.10755. [DOI] [PubMed] [Google Scholar]
- 20.Norenberg MD, Jayakumar AR, Reddy PV, Moriyama M, Panickar KS. Oxidative/nitrosative stress, the permeability transition and MAP kinases in the mechanism of brain edema in acute liver failure. J Neurochem. 2007;102:25. [Google Scholar]
- 21.Warren KS, Schenker S. Effect of an inhibitor of glutamine synthesis (methionine sulfoximine) on ammonia toxicity and metabolism. J Lab Clin Med. 1964;64:442–449. [PubMed] [Google Scholar]
- 22.Hawkins RA, Jessy J, Mans AM, De Joseph MR. Effect of reducing brain glutamine synthesis on metabolic symptoms of hepatic encephalopathy. J Neurochem. 1993;60:1000–1006. doi: 10.1111/j.1471-4159.1993.tb03247.x. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi H, Koehler RC, Brusilow SW, Traystman RJ. Inhibition of brain glutamine accumulation prevents cerebral edema in hyperammonemic rats. Am J Physiol. 1991;261:H825–H829. doi: 10.1152/ajpheart.1991.261.3.H825. [DOI] [PubMed] [Google Scholar]
- 24.Blei AT, Olafsson S, Therrien G, Butterworth RF. Ammonia-induced brain edema and intracranial hypertension in rats after portacaval anastomosis. Hepatology. 1994;19:1437–1444. [PubMed] [Google Scholar]
- 25.Hourani BT, Hamlin EM, Reynolds TB. Cerebrospinal fluid glutamine as a measure of hepatic encephalopathy. Arch Intern Med. 1971;127:1033–1036. [PubMed] [Google Scholar]
- 26.Record CO, Buxton B, Chase RA, Curzon G, Murray-Lyon IM, Williams R. Plasma and brain amino acids in fulminant hepatic failure and their relationship to hepatic encephalopathy. Eur J Clin Invest. 1976;6:387–394. doi: 10.1111/j.1365-2362.1976.tb00533.x. [DOI] [PubMed] [Google Scholar]
- 27.McConnell JR, Antonson DL, Ong CS, Chu WK, Fox IJ, Heffron TG, Langnas AN, Shaw BW., Jr Proton spectroscopy of brain glutamine in acute liver failure. Hepatology. 1995;22:69–74. [PubMed] [Google Scholar]
- 28.Norenberg MD, Martinez-Hernandez A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979;161:303–310. doi: 10.1016/0006-8993(79)90071-4. [DOI] [PubMed] [Google Scholar]
- 29.Rama Rao KV, Jayakumar AR, Norenberg MD. Induction of the mitochondrial permeability transition in cultured astrocytes by glutamine. Neurochem Int. 2003;43:517–523. doi: 10.1016/s0197-0186(03)00042-1. [DOI] [PubMed] [Google Scholar]
- 30.Jayakumar AR, Rama Rao KV, Schousboe A, Norenberg MD. Glutamine-induced free radical production in cultured astrocytes. Glia. 2004;46:296–301. doi: 10.1002/glia.20003. [DOI] [PubMed] [Google Scholar]
- 31.Albrecht J, Norenberg MD. Glutamine: a Trojan horse in ammonia neurotoxicity. Hepatology. 2006;44:788–794. doi: 10.1002/hep.21357. [DOI] [PubMed] [Google Scholar]
- 32.Norenberg MD, Rao KV, Jayakumar AR. Mechanisms of ammonia-induced astrocyte swelling. Metab Brain Dis. 2005;20:303–318. doi: 10.1007/s11011-005-7911-7. [DOI] [PubMed] [Google Scholar]
- 33.Rama Rao KV, Jayakumar AR, Norenberg MD. Differential response of glutamine in cultured neurons and astrocytes. J Neurosci Res. 2005;79:193–199. doi: 10.1002/jnr.20295. [DOI] [PubMed] [Google Scholar]
- 34.Pichili VB, Rao KV, Jayakumar AR, Norenberg MD. Inhibition of glutamine transport into mitochondria protects astrocytes from ammonia toxicity. Glia. 2007;55:801–809. doi: 10.1002/glia.20499. [DOI] [PubMed] [Google Scholar]
- 35.Hilgier W, Albrecht J, Krasnicka Z. Thioacetamide-induced hepatic encephalopathy in the rat. I Preliminary morphological and biochemical observations. Neuropatol Pol. 1983;21:487–494. [PubMed] [Google Scholar]
- 36.Krasnicka Z, Albrecht J, Gajkowska B, Mossakowski MJ. Thioacetamide-induced hepatic encephalopathy in the rat. II Cytochemical and ultrastructural studies on astrocytes cultured in vitro. Neuropatol Pol. 1983;21:495–510. [PubMed] [Google Scholar]
- 37.Gammal SH, Jones EA. Hepatic encephalopathy. Med Clin North Am. 1989;73:793–813. doi: 10.1016/s0025-7125(16)30639-3. [DOI] [PubMed] [Google Scholar]
- 38.Zimmermann C, Ferenci P, Pifl C, Yurdaydin C, Ebner J, Lassmann H, Roth E, Hortnagl H. Hepatic encephalopathy in thioacetamide-induced acute liver failure in rats: characterization of an improved model and study of amino acid-ergic neurotransmission. Hepatology. 1989;9:594–601. doi: 10.1002/hep.1840090414. [DOI] [PubMed] [Google Scholar]
- 39.Gammal SH, Basile AS, Geller D, Skolnick P, Jones EA. Reversal of the behavioral and electrophysiological abnormalities of an animal model of hepatic encephalopathy by benzodiazepine receptor ligands. Hepatology. 1990;11:371–378. doi: 10.1002/hep.1840110307. [DOI] [PubMed] [Google Scholar]
- 40.Larsen FS, Adel Hansen B, Pott F, Ejlersen E, Secher NH, Paulson OB, Knudsen GM. Dissociated cerebral vasoparalysis in acute liver failure. A hypothesis of gradual cerebral hyperaemia. J Hepatol. 1996;25:145–151. doi: 10.1016/s0168-8278(96)80066-3. [DOI] [PubMed] [Google Scholar]
- 41.Hilgier W, Zitting A, Albrecht J. The brain octopamine and phenylethanolamine content in rats in thioacetamide-induced hepatogenic encephalopathy. Acta Neurol Scand. 1985;71:195–198. doi: 10.1111/j.1600-0404.1985.tb03188.x. [DOI] [PubMed] [Google Scholar]
- 42.Pamiljans V, Krishnaswamy PR, Dumville G, Meister A. Studies on the mechanism of glutamine synthesis; isolation and properties of the enzyme from sheep brain. Biochemistry. 1962;1:153–158. doi: 10.1021/bi00907a023. [DOI] [PubMed] [Google Scholar]
- 43.Roberg B, Torgner IA, Laake J, Takumi Y, Ottersen OP, Kvamme E. Properties and submitochondrial localization of pig and rat renal phosphate-activated glutaminase. Am J Physiol Cell Physiol. 2000;279:C648–C657. doi: 10.1152/ajpcell.2000.279.3.C648. [DOI] [PubMed] [Google Scholar]
- 44.Jayakumar AR, Panickar KS, Murthy Ch R, Norenberg MD. Oxidative stress and mitogen-activated protein kinase phosphorylation mediate ammonia-induced cell swelling and glutamate uptake inhibition in cultured astrocytes. J Neurosci. 2006;26:4774–4784. doi: 10.1523/JNEUROSCI.0120-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kerr PM, Suleiman MS, Halestrap AP. Reversal of permeability transition during recovery of hearts from ischemia and its enhancement by pyruvate. Am J Physiol. 1999;276:H496–H502. doi: 10.1152/ajpheart.1999.276.2.H496. [DOI] [PubMed] [Google Scholar]
- 46.Shepherd D, Garland PB. The kinetic properties of citrate synthase from rat liver mitochondria. Biochem J. 1969;114:597–610. doi: 10.1042/bj1140597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chatauret N, Desjardins P, Zwingmann C, Rose C, Rao KV, Butterworth RF. Direct molecular and spectroscopic evidence for increased ammonia removal capacity of skeletal muscle in acute liver failure. J Hepatol. 2006;44:1083–1088. doi: 10.1016/j.jhep.2005.11.048. [DOI] [PubMed] [Google Scholar]
- 48.Albrecht J, Hilgier W, Faff L. Rat cerebral mitochondrial glutaminase activity is unaffected by moderate hyperammonemia in two models. Acta Neurobiol Exp (Wars) 1996;56:545–548. doi: 10.55782/ane-1996-1158. [DOI] [PubMed] [Google Scholar]
- 49.Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology. 1999;30:636–640. doi: 10.1002/hep.510300311. [DOI] [PubMed] [Google Scholar]
- 50.Butterworth RF. Mild hypothermia prevents cerebral edema in acute liver failure. J Hepatobiliary Pancreat Surg. 2001;8:16–19. doi: 10.1007/s005340170046. [DOI] [PubMed] [Google Scholar]
- 51.Dixit V, Chang TM. Brain edema and the blood brain barrier in galactosamine-induced fulminant hepatic failure rats. An animal model for evaluation of liver support systems. ASAIO Trans. 1990;36:21–27. [PubMed] [Google Scholar]
- 52.Paschen W, Uto A, Djuricic B, Schmitt J. Hemeoxygenase expression after reversible ischemia of rat brain. Neurosci Lett. 1994;180:5–8. doi: 10.1016/0304-3940(94)90900-8. [DOI] [PubMed] [Google Scholar]
- 53.Matz PG, Massa SM, Weinstein PR, Turner C, Panter SS, Sharp FR. Focal hyperexpression of hemeoxygenase-1 protein and messenger RNA in rat brain caused by cellular stress following subarachnoid injections of lysed blood. J Neurosurg. 1996;85:892–900. doi: 10.3171/jns.1996.85.5.0892. [DOI] [PubMed] [Google Scholar]
- 54.Pai AB, Boyd AV, McQuade CR, Harford A, Norenberg JP, Zager PG. Comparison of oxidative stress markers after intravenous administration of iron dextran, sodium ferric gluconate, and iron sucrose in patients undergoing hemodialysis. Pharmacotherapy. 2007;27:343–350. doi: 10.1592/phco.27.3.343. [DOI] [PubMed] [Google Scholar]
- 55.Jiang W, Desjardins P, Butterworth RF. Hypothermia attenuates oxidative/nitrosative stress, encephalopathy and brain edema in acute (ischemic) liver failure. Neurochem Int. 2009;55:124–128. doi: 10.1016/j.neuint.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 56.Jiang W, Desjardins P, Butterworth RF. Minocycline attenuates oxidative/nitrosative stress and cerebral complications of acute liver failure in rats. Neurochem Int. 2009;55:601–605. doi: 10.1016/j.neuint.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 57.Bai G, Rama Rao KV, Murthy CR, Panickar KS, Jayakumar AR, Norenberg MD. Ammonia induces the mitochondrial permeability transition in primary cultures of rat astrocytes. J Neurosci Res. 2001;66:981–991. doi: 10.1002/jnr.10056. [DOI] [PubMed] [Google Scholar]
- 58.Master S, Gottstein J, Blei AT. Cerebral blood flow and the development of ammonia-induced brain edema in rats after portacaval anastomosis. Hepatology. 1999;30:876–880. doi: 10.1002/hep.510300428. [DOI] [PubMed] [Google Scholar]
- 59.Tofteng F, Hauerberg J, Hansen BA, Pedersen CB, Jorgensen L, Larsen FS. Persistent arterial hyperammonemia increases the concentration of glutamine and alanine in the brain and correlates with intracranial pressure in patients with fulminant hepatic failure. J Cereb Blood Flow Metab. 2006;26:21–27. doi: 10.1038/sj.jcbfm.9600168. [DOI] [PubMed] [Google Scholar]
- 60.Norenberg MD, Bender AS. Astrocyte swelling in liver failure: role of glutamine and benzodiazepines. Acta Neurochir Suppl (Wien) 1994;60:24–27. doi: 10.1007/978-3-7091-9334-1_6. [DOI] [PubMed] [Google Scholar]
- 61.Albrecht J, Dolinska M. Glutamine as a pathogenic factor in hepatic encephalopathy. J Neurosci Res. 2001;65:1–5. doi: 10.1002/jnr.1121. [DOI] [PubMed] [Google Scholar]
- 62.Brusilow SW. Hyperammonemic encephalopathy. Medicine (Baltimore) 2002;81:240–249. doi: 10.1097/00005792-200205000-00007. [DOI] [PubMed] [Google Scholar]
- 63.Cordoba J, Crespin J, Gottstein J, Blei AT. Mild hypothermia modifies ammonia-induced brain edema in rats after portacaval anastomosis. Gastroenterology. 1999;116:686–693. doi: 10.1016/s0016-5085(99)70191-5. [DOI] [PubMed] [Google Scholar]
- 64.Chatauret N, Zwingmann C, Rose C, Leibfritz D, Butterworth RF. Effects of hypothermia on brain glucose metabolism in acute liver failure: a H/C-nuclear magnetic resonance study. Gastroenterology. 2003;125:815–824. doi: 10.1016/s0016-5085(03)01054-0. [DOI] [PubMed] [Google Scholar]
- 65.Zwingmann C, Chatauret N, Rose C, Leibfritz D, Butterworth RF. Selective alterations of brain osmolytes in acute liver failure: protective effect of mild hypothermia. Brain Res. 2004;999:118–123. doi: 10.1016/j.brainres.2003.11.048. [DOI] [PubMed] [Google Scholar]
- 66.Jayakumar AR, Rao KV, Murthy Ch R, Norenberg MD. Glutamine in the mechanism of ammonia-induced astrocyte swelling. Neurochem Int. 2006;48:623–628. doi: 10.1016/j.neuint.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 67.Chan PH, Longar S, Chen S, Yu AC, Hillered L, Chu L, Imaizumi S, Pereira B, Moore K, Woolworth V, Fishman R. The role of arachidonic acid and oxygen radical metabolites in the pathogenesis of vasogenic brain edema and astrocytic swelling. Ann NY Acad Sci. 1989;559:237–247. doi: 10.1111/j.1749-6632.1989.tb22612.x. [DOI] [PubMed] [Google Scholar]
- 68.Staub F, Winkler A, Peters J, Kempski O, Baethmann A. Mechanisms of glial swelling by arachidonic acid. Acta Neurochir Suppl (Wien) 1994;60:20–23. doi: 10.1007/978-3-7091-9334-1_5. [DOI] [PubMed] [Google Scholar]
- 69.Chen CJ, Liao SL, Kuo JS. Gliotoxic action of glutamate on cultured astrocytes. J Neurochem. 2000;75:1557–1565. doi: 10.1046/j.1471-4159.2000.0751557.x. [DOI] [PubMed] [Google Scholar]
- 70.Chan PH, Yurko M, Fishman RA. Phospholipid degradation and cellular edema induced by free radicals in brain cortical slices. J Neurochem. 1982;38:525–531. doi: 10.1111/j.1471-4159.1982.tb08659.x. [DOI] [PubMed] [Google Scholar]
- 71.Brahma B, Forman RE, Stewart EE, Nicholson C, Rice ME. Ascorbate inhibits edema in brain slices. J Neurochem. 2000;74:1263–1270. doi: 10.1046/j.1471-4159.2000.741263.x. [DOI] [PubMed] [Google Scholar]
- 72.Murthy CR, Rama Rao KV, Bai G, Norenberg MD. Ammonia-induced production of free radicals in primary cultures of rat astrocytes. J Neurosci Res. 2001;66:282–288. doi: 10.1002/jnr.1222. [DOI] [PubMed] [Google Scholar]
- 73.Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans. 2006;34:232–237. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- 74.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 75.Norenberg MD, Rao KV. The mitochondrial permeability transition in neurologic disease. Neurochem Int. 2007;50:983–997. doi: 10.1016/j.neuint.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rama Rao KV, Jayakumar AR, Norenberg MD. Role of oxidative stress in the ammonia-induced mitochondrial permeability transition in cultured astrocytes. Neurochem Int. 2005;47:31–38. doi: 10.1016/j.neuint.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 77.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Votyakova TV, Reynolds IJ. Ca2+-induced permeabilization promotes free radical release from rat brain mitochondria with partially inhibited complex I. J Neurochem. 2005;93:526–537. doi: 10.1111/j.1471-4159.2005.03042.x. [DOI] [PubMed] [Google Scholar]
- 79.Rao KV, Norenberg MD. Cerebral energy metabolism in hepatic encephalopathy and hyperammonemia. Metab Brain Dis. 2001;16:67–78. doi: 10.1023/a:1011666612822. [DOI] [PubMed] [Google Scholar]
- 80.Norenberg M. Astrocytes and ammonia in hepatic encephalopathy. In: deVellis J., editor. Human Press; Totowa NJ: 2001. pp. 477–496. [Google Scholar]
- 81.Schousboe A, Hertz L, Svenneby G, Kvamme E. Phosphate activated glutaminase activity and glutamine uptake in primary cultures of astrocytes. J Neurochem. 1979;32:943–950. doi: 10.1111/j.1471-4159.1979.tb04579.x. [DOI] [PubMed] [Google Scholar]
- 82.Sonnewald U, Westergaard N, Jones P, Taylor A, Bachelard HS, Schousboe A. Metabolism of [U-13C5] glutamine in cultured astrocytes studied by NMR spectroscopy: first evidence of astrocytic pyruvate recycling. J Neurochem. 1996;67:2566–2572. doi: 10.1046/j.1471-4159.1996.67062566.x. [DOI] [PubMed] [Google Scholar]
- 83.Laake JH, Takumi Y, Eidet J, Torgner IA, Roberg B, Kvamme E, Ottersen OP. Postembedding immunogold labelling reveals subcellular localization and pathway-specific enrichment of phosphate activated glutaminase in rat cerebellum. Neuroscience. 1999;88:1137–1151. doi: 10.1016/s0306-4522(98)00298-x. [DOI] [PubMed] [Google Scholar]
- 84.Subbalakshmi GY, Murthy CR. Differential response of enzymes of glutamate metabolism in neuronal perikarya and synaptosomes in acute hyperammonemia in rat. Neurosci Lett. 1985;59:121–126. doi: 10.1016/0304-3940(85)90225-3. [DOI] [PubMed] [Google Scholar]
- 85.Rao VL, Murthy CR, Butterworth RF. Glutamatergic synaptic dysfunction in hyperammonemic syndromes. Metab Brain Dis. 1992;7:1–20. doi: 10.1007/BF01000437. [DOI] [PubMed] [Google Scholar]
- 86.Wurdig S, Kugler P. Histochemistry of glutamate metabolizing enzymes in the rat cerebellar cortex. Neurosci Lett. 1991;130:165–168. doi: 10.1016/0304-3940(91)90388-a. [DOI] [PubMed] [Google Scholar]
- 87.Olalla L, Gutierrez A, Jimenez AJ, Lopez-Tellez JF, Khan ZU, Perez J, Alonso FJ, de la Rosa V, Campos-Sandoval JA, Segura JA, Aledo JC, Marquez J. Expression of the scaffolding PDZ protein glutaminase-interacting protein in mammalian brain. J Neurosci Res. 2008;86:281–292. doi: 10.1002/jnr.21505. [DOI] [PubMed] [Google Scholar]
- 88.Zieminska E, Dolinska M, Lazarewicz JW, Albrecht J. Induction of permeability transition and swelling of rat brain mitochondria by glutamine. Neurotoxicology. 2000;21:295–300. [PubMed] [Google Scholar]
- 89.Albrecht J, Dolinska M, Hilgier W, Lipkowski AW, Nowacki J. Modulation of glutamine uptake and phosphate-activated glutaminase activity in rat brain mitochondria by amino acids and their synthetic analogues. Neurochem Int. 2000;36:341–347. doi: 10.1016/s0197-0186(99)00142-4. [DOI] [PubMed] [Google Scholar]
- 90.Hilgier W, Puka M, Albrecht J. Characteristics of large neutral amino acid-induced release of preloaded L-glutamine from rat cerebral capillaries in vitro: effects of ammonia, hepatic encephalopathy, and gamma-glutamyl transpeptidase inhibitors. J Neurosci Res. 1992;32:221–226. doi: 10.1002/jnr.490320211. [DOI] [PubMed] [Google Scholar]
- 91.Oldendorf WH. Stereospecificity of blood-brain barrier permeability to amino acids. Am J Physiol. 1973;224:967–969. doi: 10.1152/ajplegacy.1973.224.4.967. [DOI] [PubMed] [Google Scholar]
- 92.Irisawa Y, Adachi N, Liu K, Arai T, Nagaro T. Alleviation of ischemia-induced brain edema by activation of the central histaminergic system in rats. J Pharmacol Sci. 2008;108:112–123. doi: 10.1254/jphs.08114fp. [DOI] [PubMed] [Google Scholar]
- 93.Wade AM TH. Antioxidant characteristics of L-histidine. J Nutr Biochem. 1998;9:308–315. [Google Scholar]
- 94.Ishibashi T, Lee CI, Okabe E. Skeletal sarcoplasmic reticulum dysfunction induced by reactive oxygen intermediates derived from photoactivated rose bengal. J Pharmacol Exp Ther. 1996;277:350–358. [PubMed] [Google Scholar]
- 95.Ikeda Y, Mochizuki Y, Matsumoto H, Nakamura Y, Dohi K, Jimbo H, Shimazu M, Hayashi M, Matsumoto K. L-histidine but not D-histidine attenuates brain edema following cryogenic injury in rats. Acta Neurochir Suppl. 2000;76:195–197. doi: 10.1007/978-3-7091-6346-7_39. [DOI] [PubMed] [Google Scholar]
- 96.Obata T, Kubota S, Yamanaka Y. Protective effect of histidine on para-nonylphenol-enhanced hydroxyl free radical generation induced by 1-methyl-4-phenylpyridinium ion (MPP+) in rat striatum. Biochim Biophys Acta. 2001;1568:171–175. doi: 10.1016/s0304-4165(01)00214-8. [DOI] [PubMed] [Google Scholar]
- 97.Tyfield LA, Holton JB. The effect of high concentrations of histidine on the level of other amino acids in plasma and brain of the mature rat. J Neurochem. 1976;26:101–105. [PubMed] [Google Scholar]
- 98.Lozeva-Thomas V, Ahonen P, Chatauret N, Tuomisto L, Butterworth RF. Brain histamine in experimental acute liver failure: effects of L-histidine loading. Inflamm Res. 2004;53 Suppl 1:S55–S56. doi: 10.1007/s00011-003-0326-1. [DOI] [PubMed] [Google Scholar]