

Abstract
Mutation and loss of function in p53 are common features among human breast cancers. Here we use BALB/c-Trp53+/− mice as a model to examine the sequence of events leading to mammary tumors. Mammary gland proliferation rates were similar in both BALB/c-Trp53+/− mice and wild-type controls. In addition, sporadic mammary hyperplasias were rare in BALB/c-Trp53+/− mice and not detectably different from those of wild-type controls. Among the 28 mammary tumors collected from BALB/c-Trp53+/− mice, loss of heterozygosity for Trp53 was detected in more than 90% of invasive mammary tumors. Transplantation of Trp53+/− ductal hyperplasias also indicated an association between loss of the wild-type allele of Trp53 and progression to invasive carcinomas. Therefore, loss of p53 function seems to be a rate-limiting step in progression. Moreover, expression of biomarkers such as estrogen receptor α, progesterone receptor, Her2/Neu, and activated Notch1 varied among mammary tumors, suggesting that multiple oncogenic lesions collaborate with loss of p53 function. Expression of biomarkers was retained when tumor fragments were transplanted to syngeneic hosts. Tumors expressing solely luminal or basal keratins were also observed (27 and 11%, respectively), but the largest class of tumors expressed both luminal and basal keratins (62%). Overall, this panel of transplantable tumors provides a resource for detailed evaluation of the cell lineages undergoing transformation and preclinical testing of therapeutic agents targeting a variety of oncogenic pathways including cancer stem cells.
Compromised function of the p53 tumor suppressor pathway remains among the most common alterations found in cancers.1,2 Although disruption of p53 function predisposes to a broad spectrum of malignancies, the breast epithelium seems exquisitely sensitive to proper p53 function. Polymorphisms in MDM2, CHK2, and ATM alter the stability and activity of p53 and have been linked to breast cancer risk in women. The activity of p53 has also been shown to be responsive to hormones in rodent models.3–5 Exogenous estrogen and progesterone are sufficient to render the mammary epithelium resistant to carcinogen-induced tumors, mimicking the protective effect afforded by a full-term pregnancy,6 and the p53 pathway participates in the hormone-induced protection.7,8 Furthermore, breast cancer is the most prevalent tumor among women with Li-Fraumeni syndrome, which is most commonly associated with heterozygous mutations in TP53.9–11 Reduced dosage of the p53 gene has been associated with haploinsufficiency with respect to levels of p53 protein and activity, cell cycle arrest, apoptosis, and homology-directed DNA repair.12–14 Therefore, the level of p53 activity is a critical regulator of tumor suppressor pathways and breast cancer risk.
The mechanisms by which p53 suppresses tumors are diverse. The roles of p53 in mediating cell cycle arrest and apoptosis have been described extensively.15 More recently, the activities of p53 have been expanded to include regulation of DNA repair, senescence, autophagy, cellular metabolism, and microRNA processing.16–21 In addition, loss of p53 was shown to permit expansion of the pool of pluripotent embryonic stem cells22,23 and cancer stem cells.23–25 The activities of p53 that are critical for suppression of tumors vary among tissues. In the thymus, the proapoptotic activity of p53 was necessary to suppress lymphomas, whereas the cell cycle checkpoint function was dispensable.26 In contrast, senescence is the principal pathway leading to regression of liver tumors after restoration of p53 function27 and seems to be the prominent pathway in sarcomas as well.28 Among the known breast cancer susceptibility genes there is a convergence of function highlighting the central role of homology-directed repair of DNA double-strand breaks in breast cancer risk.29 Thus, fidelity of double-strand break repair may be a critical pathway controlled by p53 in breast tissue.
The sequential changes that occur in normal tissue and in premalignant mammary lesions as a consequence of heterozygous mutations in TP53 provide clues to the mechanisms that initiate the carcinogenic cascade as well as the cellular origins of breast cancer. Although it is difficult to monitor sequential changes in human breast cancers, spontaneous mammary tumors are common in BALB/c-Trp53+/− female mice,30,31 providing a model to examine the sequence of phenotypic and genetic alterations during mammary tumorigenesis. Using this model, we demonstrate that heterozygosity for Trp53 does not increase proliferation of the epithelium or the incidence of precancerous lesions. However, loss of the wild-type allele of Trp53 was associated with the transition from hyperplastic to invasive phenotypes. In a set of 28 spontaneous tumors, histological phenotypes and expression of oncogenes were heterogeneous. The majority of tumors expressed markers of both luminal and basal epithelia (62%), suggesting that progenitor cells are the most common origin. However, significant numbers of tumors expressed purely luminal keratins (27%) or basal keratins (11%). Although distinct patterns of keratins were observed, stem cell markers were expressed similarly in tumors with only keratins associated with luminal cells (K8/18) as well as tumors expressing both luminal and basal cell keratins (K8/18 and K5/6). Therefore, it seems that tumors arise most frequently from progenitor cells, which then commit toward more differentiated lineages during progression. Lineage decisions were not associated with specific activation of either Notch1 or Her2. Because tumor phenotypes were stable after transplantation, this panel of tumors can be used to speed preclinical testing of chemotherapeutic agents.
Materials and Methods
Mice
BALB/c-Trp53+/− mice were generated as described previously,32 by backcrossing (C57BL/6 × 129/Sv) Trp53−/− mice onto the BALB/cMed strain for 11 generations. Mice were genotyped by multiplex PCR as described previously.33 BALB/c-Trp53+/+ and Trp53+/− mice were monitored weekly for tumor development or morbidity and were palpated for mammary tumors. Ninety-seven virgin female BALB/c-Trp53+/− mice and 30 Trp53+/+ mice were included in the study.
Estrous Stage Determination
Stages of estrus were determined by cytological evaluation of vaginal smears. Vaginal smears of adult female nulliparous mice aged 35 to 37 weeks were taken daily. Once the estrous stages were determined, mice were injected intraperitoneally with 30 μg of 5-bromo-2-deoxyuridine (BrdU) (Sigma-Aldrich, St. Louis, MO)/g b.wt. and sacrificed 2 hours later. Mammary glands were isolated and processed for immunohistological analysis.
Mammary Gland Whole Mounts
Mammary glands were removed and spread on glass slides and fixed in Carnoy's fixative (60% ethanol, 30% chloroform, and 10% glacial acetic acid) for 3 hours. The glands were washed in 70% ethanol for 15 minutes followed by a brief rinse in distilled water. The tissues were then stained in carmine alum solution (1 g of carmine and 2.5 g of aluminum potassium sulfate/500 ml of water) at 4°C overnight. The tissue was dehydrated by soaking it in a series of solutions with increasing concentrations of ethanol (70 to 100%), washed in xylene twice, and mounted on a slide.
Isolation and Culture of Transplantable Mammary Tumor Cells
For transplantable tumor fragments, each mammary tumor from BALB/c-Trp53+/− mice was minced and frozen in Dulbecco's modified Eagle's medium (DMEM):F12 with 10% fetal bovine serum, 7% dimethyl sulfoxide following a general cell-freezing method and kept in liquid nitrogen. To isolate tumor cells, mammary tumors were removed from mice, rinsed in PBS, minced, and incubated 3 hours at 37°C in medium containing DMEM:F12 supplemented with 25 mmol/L HEPES, 1.2 g/L NaHCO3, 10 μg/ml insulin, 5 ng/ml epidermal growth factor, 2% adult bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B, and 0.2% collagenase type 3 (Worthington 4182, 223 U/mg, Worthington Biochemicals, Freehold, NJ). After dissociation and centrifugation, cells were grown in DMEM:F12 containing 2% adult bovine serum, 10 μg/ml insulin, 5 ng/ml epidermal growth factor, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B. The cells were expanded for six passages before freezing in liquid nitrogen.
Transplantation of Mammary Hyperplasias, Tumor Fragments, and Tumor Cell Lines
Procedures for transplantation of tissues into cleared mammary fat pads have been described previously.34 In brief, 21- to 24-day old BALB/c wild-type females were used as transplant recipients. The endogenous mammary epithelium was surgically removed from the fourth inguinal glands to provide a cleared mammary fat pad. Fragments of PH1b and PH2 hyperplastic outgrowth lines35 or spontaneous mammary tumors were inserted into the cleared mammary fat pads. Tumor cell lines were injected (5 × 105 cells in 10 μl of saline) into each cleared fat pad. Mice were palpated 3 times/week to monitor for tumor development. To test hormone dependence, tumor fragments were transplanted into recipients, which were then ovariectomized when tumors were palpable.
Histology and Immunohistochemistry
All collected tissues were fixed overnight at 4°C in 10% neutral-buffered formalin and then were stored in 70% ethanol until they were embedded in paraffin. Sections (4-μm-thick) were deparaffinized in xylene and then were hydrated through a series of graded ethanol. The sections used for histopathological evaluation were stained with H&E. Immunohistochemical staining was performed using an EnVision+ System/HRP kit (DakoCytomation, Carpenteria, CA) after antigen retrieval. The antibodies used were anti-estrogen receptor (ER) α (1:200, MC 20, Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-progesterone receptor (PR) (1:50, C-19, Santa Cruz Biotechnology, Inc.), and anti-mouse cytokeratin 6 (1:5000, PRB-169P, Covance, Berkeley, CA). BrdU staining was performed using a BrdU staining kit (Invitrogen, Carlsbad, CA). Immunofluorescence staining followed published procedures.36 In brief, hydrated slides were boiled in sodium citrate (10 mmol/L) for 15 minutes. After the slides cooled, they were incubated in blocking buffer (5% bovine serum albumin-0.5% Tween 20 in PBS) for 1 hour under room temperature. Then the slides were incubated in primary antibodies, anti-cytokeratin 5 (1:8000, PRB-160P, Covance), and anti-cytokeratin 8/18 (1:400, GP11, Progen Biotechnick, Heidelberg, Germany) in a humid chamber at room temperature overnight. After a series of washing steps, Texas Red- or Cy2-conjugated secondary antibodies (1:200, Jackson ImmunoResearch Laboratories, West Grove, PA) and 4,6-diamidino-2-phenylindole (1 μg/ml, Sigma-Aldrich) was added to the samples for 1 hour under room temperature. Slides were mounted in mounting media (2.5% Dabco-50 mmol/L Tris-HCl, pH 8.0–90% glycerol) and sealed with clear nail polish.
Trp53 Genotyping and Loss of Heterozygosity
Loss of heterozygosity (LOH) at Trp53 in hyperplastic outgrowths and tumors was determined by Southern blotting as described previously.37 The epithelium in hyperplastic outgrowths (PH1b and PH2) was enriched by digestion in DMEM:F12 supplemented with 25 mmol/L HEPES, 1.2 g/L NaHCO3, 10 μg/ml insulin, 5 ng/ml epidermal growth factor, 2% adult bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B, and 0.2% collagenase type 3 (223 U/mg), as described for cell lines, followed by centrifugation. Genomic DNA was extracted from the enriched epithelial pellet. For tumors, DNA was extracted directly from frozen tissue. Genomic DNA was digested with StuI and EcoRI. Southern blots were hybridized with a probe spanning exons 7 to 9 of the Trp53 gene. The intensity of the wild-type and null bands was quantified using a PhosphorImager (Cyclone, Packard Bioscience, Boston MA) and OptiQuant software. The ratios of wild-type/null band hybridization values were calculated. Loss of the wild-type allele was defined as the ratio of wild-type/null alleles <0.5.
Western Blot
Flash-frozen tissues were homogenized in buffer (50 mmol/L Tris, 150 mmol/L NaCl, 1% Triton X-100, 1 mmol/L sodium vanadate, 10 mmol/L sodium fluoride, 10 mmol/L β-glycerol phosphate, and 1× protease inhibitor [P8340, Sigma-Aldrich]) using 200 μl of buffer/50 mg of tissue. The protein concentration was determined using BCA reagent (Pierce Chemical, Rockford, IL). Lysates (50 μg) were separated by 6% gel electrophoresis and electrophoretically transferred to nitrocellulose membrane. The membrane was incubated with anti-Her2/Neu (1:250, C20, Santa Cruz Biotechnology, Inc.), anti-phospho Her2/Neu (Tyr1248, 1:1000, Stressgen Bioreagents, Ann Arbor, MI), anti-Notch1 (1:1000, mN1A, eBioscience, San Diego, CA), or anti-β-actin (1:1000, A3853, Sigma-Aldrich), followed by incubation with horseradish peroxidase-conjugated secondary antibodies, and developed using an enhanced chemiluminescence solution (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Isolation of Mammary Gland Organoids
Isolation of mammary gland organoids (enrichment of epithelium cells) was performed essentially as described previously.38 In brief, mammary glands were removed from mice, rinsed in PBS, minced, and incubated on a shaker for 1 hour at 37°C in medium containing DMEM:F12 supplemented with 25 mmol/L HEPES, 1.2 g/L NaHCO3, 5% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mg/ml collagenase type 3 (223 U/mg), and 100 U/ml hyaluronidase (H3506, Sigma-Aldrich). After enzyme digestion the fat layer was decanted, and the remaining organoids were centrifuged and washed three times with PBS. The organoid pellets were subject to RNA extraction immediately after the washes.
RNA Isolation and PCR Array Analysis
Total RNA from mammary gland organoid and tumor samples was extracted by QIAzol Lysis Reagent (Qiagen, Valencia, CA) and further purified using an RNeasy Mini Kit (Qiagen). PCR array analysis was performed by using 1 μg of total RNA of each sample and the RT2 Profiler PCR Array for mouse stem cells (SABiosciences, Frederick, MD) according to the recommendations of the manufacturer. Expression levels on each array plate were normalized using two housekeeping genes: Hprt1 and Hsp90ab1. Tumor samples were compared with mammary gland organoids from age-matched BALB/c-Trp53+/− mice. Analysis of ΔCt and fold changes was conducted using the online data analysis tool supported by the array manufacturer. Genes were considered to be differentially expressed if the fold change between tumor and mammary epithelial organoids was >2.5. Genes that are differentially expressed between the “mixed keratin” tumors (V06 and V22) and “luminal keratin” tumors (V07 and V14) were selected if the ratio differed by more than fourfold.
Results
Analysis of Preneoplastic Changes in BALB/c-Trp53+/− Mammary Tissues
To examine the events during mammary tumorigenesis, tissues were collected from BALB/c-Trp53+/+ and BALB/c-Trp53+/− nulliparous female mice at 8-week intervals up to 52 weeks. The overall ductal structure showed no difference in branching or alveologenesis between the two groups (Figure 1A). Proliferation rates were also similar for both genotypes at 36 weeks (Figure 1B). Sporadic hyperplastic foci were seen in BALB/c-Trp53+/− mice (Figure 1C), but the incidence was too low to compare between the groups. Thus, heterozygosity for Trp53 did not increase proliferation or the appearance of precancerous lesions.
Figure 1.
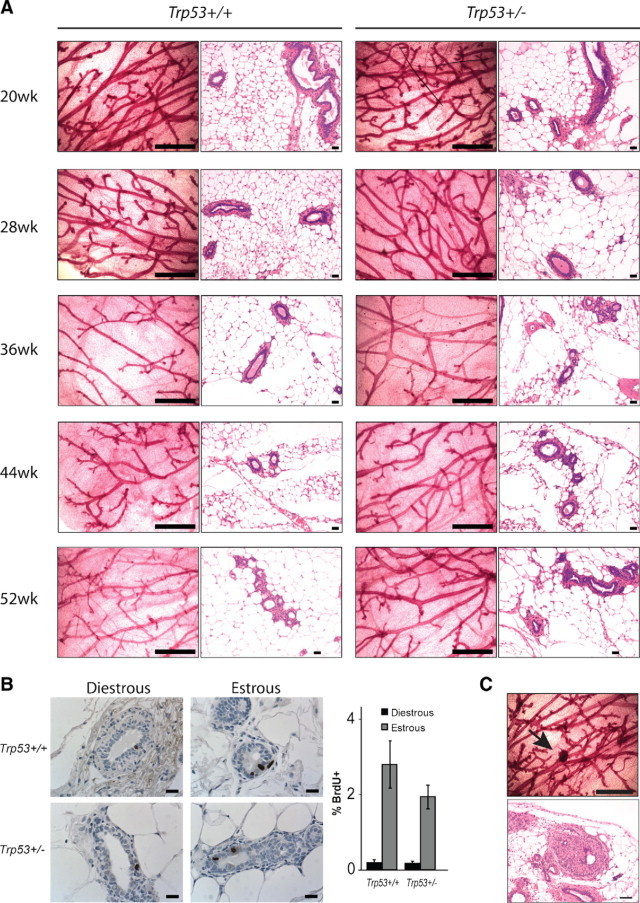
Mammary gland morphology in BALB/c-Trp53+/+ and -Trp53+/− mice. A: Whole mount and H&E staining of mammary glands from 20- to 52-week-old mice. B: Proliferation of mammary epithelial cells was determined at different estrous phases by BrdU incorporation. The histogram shows the average percentage of BrdU-positive cells. No significance was detected between the Trp53 genotypes in either stage. At least 2500 epithelial cells were counted per slide for a minimum of three mice per genotype. C: Hyperplastic foci appeared at low incidence in whole mounts (upper panels, arrow) and with H&E (lower panels) from Trp53+/− mice. Scale bars: 1 mm (whole mounts); 40 μm (H&E staining); 20 μm (BrdU staining).
LOH for Trp53 is observed frequently in breast cancers and is also observed in 90% of spontaneous mammary tumors from BALB/c-Trp53+/− mice.37 In this panel, well differentiated lesions such as mammary intraepithelial neoplasias (MINs) retained the wild-type allele of Trp53, whereas poorly differentiated adenocarcinomas had nearly complete LOH.37 To explore the timing of Trp53 LOH, two transplantable hyperplastic lines (PH1b and PH2) derived from BALB/c-Trp53+/− mice35 were transplanted into cleared mammary fat pads in BALB/c-Trp53+/+ hosts. The PH outgrowths displayed abnormal ductal morphologies in the recipient glands and later developed into invasive carcinomas with a latency of 16 to 26 weeks. Despite the Trp53 heterozygous status of the PH lines, eight of nine of the resulting tumors lost the wild-type allele of Trp53 (Figure 2). These results suggested that complete loss of Trp53 is not necessary to establish premalignant lesions such as hyperplasias or MINs, but LOH is strongly associated with the transition into invasive tumor.
Figure 2.
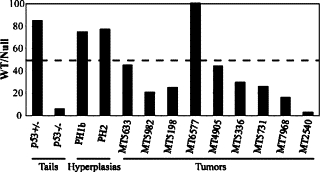
Loss of heterozygosity during mammary tumorigenesis in BALB/c-Trp53+/− mice. The proportions of wild-type (WT) and null (NULL) alleles in tumors were determined by Southern blot hybridization and were compared with the ratios for tail DNAs from mice that were homozygous for the wild-type allele or heterozygous (Trp53+/+ and Trp53+/−, respectively). Reductions in the wild-type allele below 50% (indicated by the dashed line) were considered significant loss of heterozygosity. The transplanted ductal hyperplasias (PH1b and PH2) used for transplantation retained the wild-type allele at levels equivalent to tail DNA controls. However, tumors developing from the hyperplasias showed severe loss of heterozygosity in all cases except for MT6577.
Pathological and Genetic Alterations Associated with BALB/c-Trp53+/− Mammary Tumors
We next explored whether mammary tumorigenesis in BALB/c-Trp53+/− mice follows a restricted set of pathological or molecular pathways. The histological features of the total 28 spontaneous mammary lesions collected ranged from intraepithelial neoplasia to invasive carcinomas (Figure 3, A–H). Because collections were focused on large palpable lesions, the majority of tumors were poorly differentiated invasive ductal adenocarcinomas. A minority of tumors (2 of 28) underwent transdifferentiation into adenosquamous carcinoma or had focal squamous differentiation. A complete summary of tumor characteristics is provided in Table 1.
Figure 3.
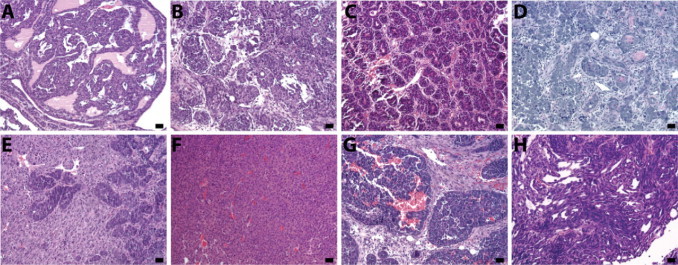
Histological characterization of BALB/c-Trp53+/− mammary lesions. Mammary lesions from BALB/c-Trp53+/− mice showed diverse histological features including intraepithelial neoplasia (MIN, V05, A) ductal adenocarcinoma (V03, B) adenocarcinoma with acinar morphology (V23, C) adenocarcinoma with squamous differentiation (V01, D) carcinosarcoma (V08, E) solid sheets of tumors with little stroma (V26, F) adenocarcinoma with strong stromal reaction (V13, G) and adenocarcinoma with spindle cell feature (V28, H). Scale bars = 20 μm.
Table 1.
Features of Spontaneous Mammary Tumors from BALB/c-Trp53+/− Mice
| Sample | Tumor type | Latency (wk) | LOH % WT/Null | PR | ER | Her2/Neu | K8/18 | K5 | K6 |
|---|---|---|---|---|---|---|---|---|---|
| V01 | Adenocarcinoma | 30 | 12.6 | − | − | − | − | + | + |
| V02 | Adenocarcinoma | 30 | Lost null signal | − | − | − | + | + | + |
| V03 | Adenocarcinoma | 40.7 | 5.9 | + | + | − | + | + | + |
| V04 | Adenocarcinoma | 46.7 | 8.8 | − | − | − | + | + | + |
| V05 | MIN | 47.1 | 64.9 | + | + | − | + | + | + |
| V06 | Adenocarcinoma | 46.3 | 1.4 | − | − | − | + | + | + |
| V07 | Adenocarcinoma | 48.9 | 7.7 | − | − | NA | + | − | − |
| V08 | carcinosarcoma | 49.1 | NA | − | − | NA | + | + | + |
| V09 | Adenocarcinoma | 42 | 10.2 | + | + | − | + | + | + |
| V10 | Adenocarcinoma | 42 | 4.5 | − | − | + | + | + | + |
| V11 | Adenocarcinoma | 53.9 | 46 | − | − | NA | + | − | − |
| V12 | Adenocarcinoma | 53.9 | 28.2 | + | + | NA | NA | NA | NA |
| V13 | Adenocarcinoma | 52 | NA | − | − | NA | − | + | + |
| V14 | Adenocarcinoma | 39.7 | 8.2 | − | − | + | + | − | − |
| V15 | Adenocarcinoma | 50.4 | 54.8 | − | − | − | + | − | − |
| V16 | Adenocarcinoma | 50.4 | NA | − | − | NA | + | + | + |
| V17 | Adenocarcinoma | 50.9 | 8.2 | − | − | + | + | + | + |
| V18 | Adenocarcinoma | 47.4 | 0 | − | − | + | + | + | + |
| V19 | Adenosquamous carcinoma | 50.1 | 28.6 | − | − | − | + | + | + |
| V20 | Adenocarcinoma | 52.9 | 40 | − | − | + | − | + | + |
| V21 | Adenocarcinoma | 47.1 | 12.8 | − | − | + | + | + | + |
| V22 | Adenocarcinoma | 48.1 | 29.7 | − | − | − | + | + | + |
| V23 | Adenocarcinoma | 47.9 | 8.7 | − | − | − | + | − | − |
| V24 | Adenocarcinoma | 50.4 | 15.5 | − | − | − | − | − | + |
| V25 | Adenosquamous carcinoma | 48.6 | 34.1 | − | − | + | + | + | + |
| V26 | Adenocarcinoma | 40.7 | 7.4 | − | − | + | + | − | − |
| V27 | Adenocarcinoma | 49.1 | 44.7 | − | − | − | + | + | + |
| V28 | Adenocarcinoma | 48.7 | 29.7 | − | − | − | + | − | − |
NA, not available.
Deregulation of ERα and PR expression was observed during progression of the mammary gland lesions in BALB/c-Trp53+/− mice. Individual ERα- and PR-positive cells are distributed in normal ducts throughout Trp53+/− mammary glands (Figure 4, A–C). An increase in clusters of ERα- and PR-positive cells was detected in ductal hyperplasias from BALB/c-Trp53+/− mice (Figure 4, D–F). Expression of ERα and PR was retained in the majority of early lesions (MINs; Figure 4, G–I) but lost in most invasive carcinomas (Figure 4, M–O). In this panel, 3 of 27 malignant tumors were ERα+/PR+ (Figure 4, J–L). The expression of ovarian steroid receptors during tumorigenesis in BALB/c-Trp53+/− mammary tissues mimics the pattern during progression of human breast cancers.39
Figure 4.
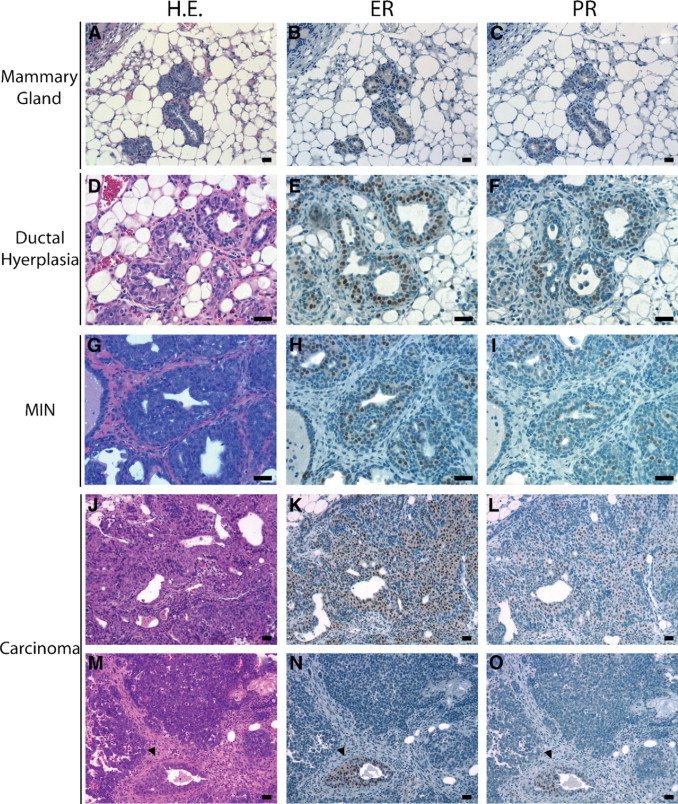
Expression of estrogen and progesterone receptors in mammary tissues and tumors. Consecutive sections were stained with either H&E (H.E.) or antibodies detecting estrogen receptor α or progesterone receptor. Preinvasive lesions (hyperplasias and MIN) retained expression of the steroid hormone receptors, whereas expression in tumors was variable. A–C: Mammary duct. D–F: Ductal hyperplasia. G–I: Mammary intraepithelial neoplasia (V05). J–L: ERα+/PR+ mammary adenocarcinoma (V09); M–O: ERα−/PR− mammary adenocarcinoma (V13). Arrowheads indicate entrapped mammary duct. Scale bars = 20 μm.
Selected oncogenic alterations may collaborate with loss of p53 to stimulate progression of tumors in specific cellular compartments, yielding tumors with distinct phenotypes. Her2/Neu expression was examined because Her2/Neu is commonly overexpressed in human breast cancers, often in conjunction with mutations in p53.40 Similar to levels in breast cancers, levels of Her2/Neu protein were elevated in 36% (8 of 22) of the BALB/c-Trp53+/− mammary tumors. The Her2/Neu protein seemed to be active because it was phosphorylated (Figure 5). Although protein levels were elevated, they were not associated with amplification of the Her2/Neu gene as determined by Southern blot (data not shown). Recent findings have suggested that p53 regulates Notch1 receptor activities, although the effect seems to differ among tissues.41,42 Activated Notch1 expression was identified in BALB/c-Trp53+/− mammary tumor (Figure 5). Levels of activated Notch1 varied among BALB/c-Trp53+/− mammary tumors but were independent of histopathological types or expression of other biomarkers. These results reveal the heterogeneous molecular alterations among mammary tumors from BALB/c-Trp53+/− mice.
Figure 5.
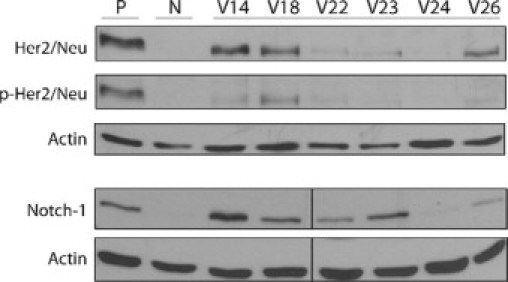
Expression of Her2/Neu and Notch1 oncogenes in mammary tumors. Western analysis using anti-Her2/Neu, anti-phospho-Her2/Neu (p-Her2/Neu) antibodies, and antibody against intracellular domain of Notch1 were used to examine expression in tumors. Nulliparous mammary tissue (N) from Trp53+/− mice was used for comparison. Lysates from A431 cells and mouse splenocytes cells (stimulated with CD3 and CD28) were used as positive controls (P) for Her2/Neu and Notch1, respectively.
Although all mammary tumors exhibited highly invasive phenotypes, distant metastases were not detected during the observation period up to 14 months. The metastatic potential of these mammary tumors could be masked, as mice were sacrificed because of large mammary tumors or coincident lymphomas. To further address the metastatic potential of the mammary tumors, tumor fragments from eight of the primary BALB/c-Trp53+/− mammary tumors (V01, V02, V06, V07, V09, V14, V20, and V22) were transplanted into mammary fat pads that had been cleared of endogenous epithelium in BALB/c-Trp53+/+ hosts. The tumor outgrowths were resected to allow longer periods of observation. The tumor outgrowths retained the histological features of the primary tumors. In the case of the V09 tumor, both primary tumors and outgrowths after transplantation expressed ERα (Figure 6, A–D). Although the tumor transplants expressed ERα at levels greater than those of the endogenous epithelium, their growth was unaffected by ovariectomy (Figure 6E) and no metastases were observed. Among tumors that resembled the “triple-negative” breast cancers, none of the four tumor outgrowth lines formed metastases (V01, V02, V06, and V22). Two outgrowth lines, V07 (ERα−/PR−) and V20 (ERα−/PR−, Her2/Neu+), formed metastases in the lungs (Figure 6F).
Figure 6.
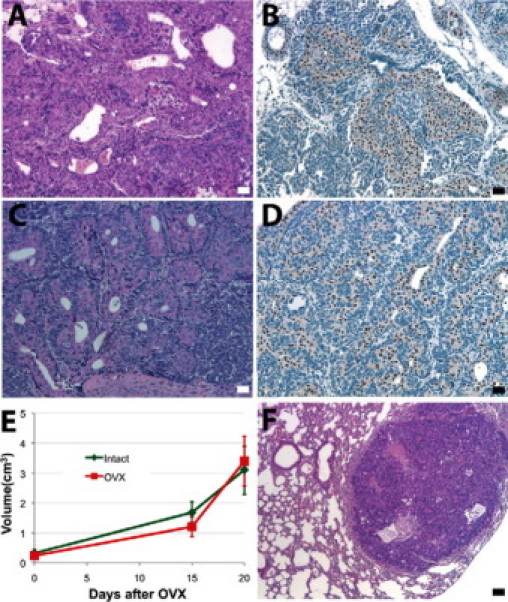
Outgrowths from transplanted tumor fragments. ERα+ adenocarcinoma V09 (A and B) fragments were transplanted into mammary fat pads that had been cleared of endogenous epithelium in BALB/c-Trp53+/+ hosts. The tumor outgrowth in recipient BALB/c wild-type mouse remained ERα+ adenocarcinoma (C and D). Half of the recipient mice were ovariectomized (OVX) when tumors were palpable. Tumor growth (E) was compared between the ovary intact group and OVX group. Metastasis was seen in mice bearing transplants of tumor fragment from the V07 tumor (F). Scale bars = 20 μm.
Origins of Mammary Tumors
To investigate cellular origins, tumors from BALB/c-Trp53+/− mice were stained for markers of basal and luminal epithelial lineages (cytokeratins 5 or 8/18, respectively). A mixture of cells expressing either K5 or K8/18 (Figure 7A) was observed in 62% of the tumors, but coexpression of K5 and K8/18 in individual cells was rare. Both of the K5-positive cells and K8/18-positive cells were pleomorphic and disorganized and were found in large quantities within each tumor. Only luminal cytokeratins were detected in 26% of tumors (Figure 7D), whereas 11% of the tumors expressed only K5-positive cells (Figure 7G). Tumors were also stained with cytokeratin 6 (K6) because it was found to be preferentially expressed in mammary stem/progenitor cells43 and expanded in mammary tumors originating from progenitor cells.44–46 None of the K8/18 only-positive tumors contained K6-positive cells (Figure 7, E–F), whereas all of the double-positive tumors and K5-positive tumors showed K6-positive cells (Figure 7, B and C, H and I). As the majority of tumors contained both luminal and basal cell lineages, it seems that tumors most often originate within the population of bipotent progenitors.
Figure 7.
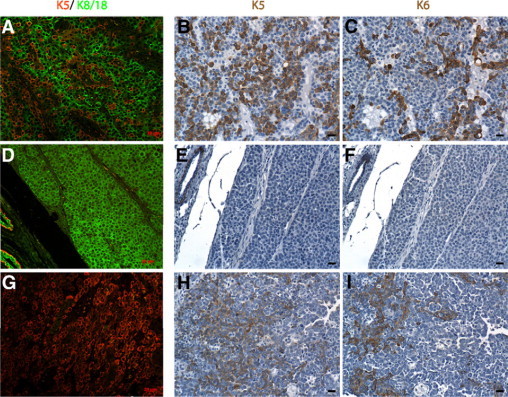
Heterogeneous patterns of keratin expression in mammary tumors from BALB/c-Trp53+/− mice. Mammary ducts from BALB/c-Trp53+/− mice expressed cytokeratins normally with K5 localized to the basal/myoepithelium and K8/18 restricted to the luminal epithelium. A: Most of the mammary tumors express a mixture of cells expressing K8/18 (green) with K5 (red). Although both cell populations were similarly abundant, cells expressing both luminal and basal keratins were rare. A smaller number of tumors have only K8/18-expressing cells (D) or only K5-expressing cells (G). Serial sections were also stained for K6, confirming the expanded populations of cells with basal-like cytokeratins among tumors composed of mixed populations of cells (A and C) and tumors expressing only K5 (G and I). In contrast, K8/18 only-expressing tumors contain no K6-expressing cells (D and F). Although basal-like tumors contained cells that were positive for K5 and K6, individual cells do not necessarily express both (see B versus C, E versus F, H versus I). Scale bars = 20 μm.
Stem cell-related genes were shown to be enriched in aggressive breast cancers and have been suggested to reflect the origins of cancers within the progenitor cells of the breast epithelium.47 Therefore, expression of a panel of 84 stem cell-related genes was profiled in tumors that expressed either mixed luminal and basal keratins (V06 and V22) and tumors expressing only luminal keratins (V07 and V14). Distinct differences were observed for nine genes (Figure 8A, top panel). N-cadherin (Cdh2) was elevated 22-fold in the mixed keratin tumors compared with normal mammary epithelial organoids, whereas the luminal-only keratin tumors showed a fourfold decrease compared with the normal mammary epithelial organoids. Expression of Krt15 was also specifically elevated in the mixed keratin tumors along with cyclins D2 and E1 (Ccnd2 and Ccne1). However, the overwhelming majority of genes showed similar patterns in both the tumors with mixed keratins as well as the tumors with luminal-only keratins (Figure 8A, bottom panel). Although it is not surprising that both sets of tumors had elevations in proliferation-associated genes (Figure 8, A–I) compared with the normal mammary epithelium, levels of cytokines and growth factors were decreased (Figure 8A, II). Both Notch and Wnt pathways have been implicated in maintenance of mammary stem cells.48–50 In both sets of tumors, ligands and receptors for Notch and Wnt were increased, indicating that signaling through these pathways is increased dramatically compared with the normal mammary epithelium (Figure 8A, V and VI). Therefore, there is broad overlap in the signaling pathways found in the tumors, suggesting a common origin of the tumors regardless of the pattern of keratins expressed.
Figure 8.
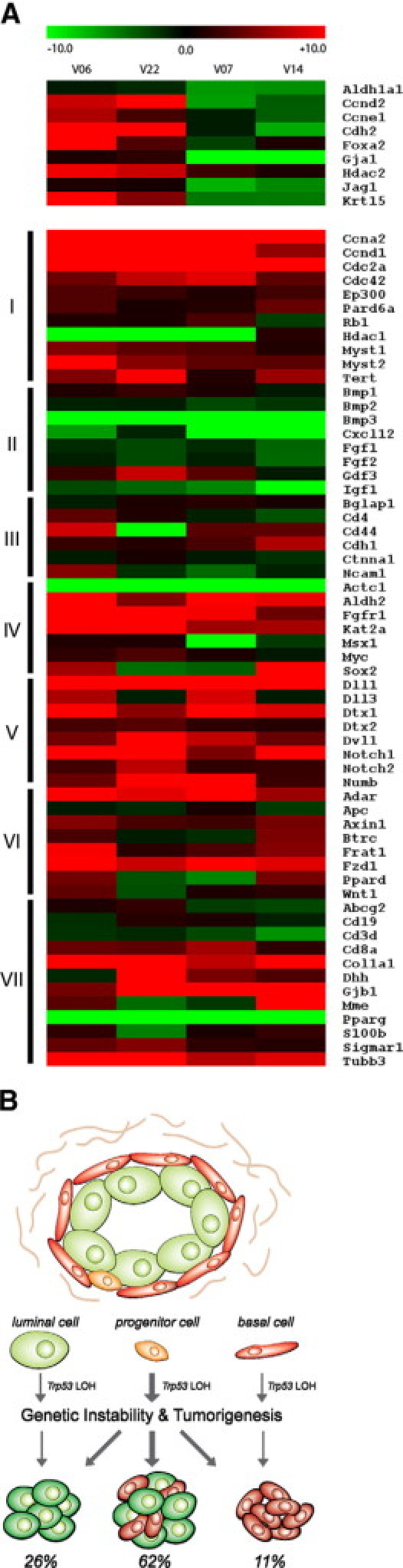
Origin of mammary tumors in BALB/c-Trp53+/− mice. A: Expression of stem cell-related genes in tumor samples differing in keratins expressed. Expression of stem cell-related genes was examined in tumors expressing mixed keratins (V06 and V22) and tumors with luminal-only keratins (V07 and V14). The expression of each gene is expressed as a ratio compared with the levels in normal mammary epithelium obtained by enzymatic digestion. A set of 9 genes are differently expressed between the two groups (top panel). However, 69 genes showed similar patterns of expression between the groups (bottom panel). The genes are grouped according to functions or pathways: I, cell cycle regulators, chromosome modulators, and cell division-related genes; II, cytokines and growth factors; III, cell adhesion molecule; IV, embryonic stem cell-related genes; V, notch pathway-related genes; VI, Wnt pathway related genes; and VII, tissue-specific stem cell markers. B: Cellular origins of mammary tumors in BALB/c-Trp53+/− mice. Mammary glands in BALB/c-Trp53+/− mice display normal ductal and alveolar structures composed of luminal (green) and basal cells (red). Progenitor cells (orange) with a potential to differentiate into both epithelial lineages also reside in the normal structure. Loss of the wild-type allele of Trp53 in any of these cells would abolish the rate-limiting step in tumorigenesis, resulting in invasive mammary tumors. Because the majority of mammary tumors contain tumor cells expressing luminal and basal markers, a clonal origin within bipotent cells is suggested as the most common pathway. For tumors expressing only luminal or basal markers, there could be two possibilities. They may originate from bipotent progenitors and later partially differentiate into tumors with single lineage cells or they could originate within the lineage-restricted cells. The overall similarity of the stem cell gene expression pattern of tumors suggested the former path.
Discussion
Although the p53 tumor suppressor pathway is commonly disrupted in a variety of cancers, breast tissue seems to be uniquely sensitive to the proper functioning of this pathway to prevent tumors. Heritable mutations in the TP53 gene have been linked to Li-Fraumeni syndrome, and breast cancer is the most common tumor type in women.9,11 Somatic mutation of TP53 is also common in sporadic breast cancers1,2 and may initiate tumorigenesis by pathways similar to those in Li-Fraumeni syndrome. Similar to patients with Li-Fraumeni syndrome, BALB/c-Trp53+/− mice exhibit a prevalence of mammary tumors as well as lymphomas and sarcomas.31 The slow onset of mammary tumors allowed detailed analysis of the cellular targets and molecular events leading to mammary tumors. Although deficiency in p53 has been associated with impaired cell cycle control and apoptosis, there was no detectable difference in proliferation rates within the mammary epithelium of Trp53+/− and Trp53+/+ mice (Figure 1). In contrast, both basal and radiation-induced apoptosis in the mammary epithelium were decreased with Trp53 gene dosage.12 Therefore, haploinsufficiency with respect to apoptosis, not proliferation, is associated with the susceptibility to mammary tumors in BALB/c-Trp53+/− mice. Haploinsufficiency in p53-mediated apoptosis has been observed in the involuting prostate of mice,51 and the pro-apoptotic role of p53 seems to be critical for suppression of lymphomas, whereas cell cycle checkpoint function was dispensable.26 Thus, a decrease in p53 dosage was suggested to be sufficient to initiate breast tumors.52 However, in our study, the diminished spontaneous apoptosis in Trp53+/− mammary epithelium did not lead to a discernible increase in preneoplastic lesions compared with that in the Trp53+/+ mice. The glands were normal with respect to ductal branching and alveologenesis. Therefore, Trp53 haploinsufficiency alone did not permit initiation of preneoplasia.
Although TP53 LOH is frequently observed in breast tumors among patients with Li-Fraumeni syndrome,53 it has been unclear whether loss of the wild-type allele of TP53 initiates preneoplasia or whether it accompanies the transition to invasive cancers. In BALB/c-Trp53+/− mammary tissues, LOH was prevalent in invasive tumors but not hyperplasia or MIN. Although rates of LOH in tumors differ between strains of mice and segregate with the predisposition to mammary tumors,37 LOH at Trp53 was also observed in K14-cre,Trp53fl/+ mice in a mixed 129 × FVB background.54 Therefore, LOH seems to be a rate-limiting step in the genesis of mammary tumors. Because p53 plays an important role in responding to oncogenic stimuli and the DNA damage response, it has been proposed that p53 function limits growth of preneoplastic cells and loss is necessary for progression.55 These results favor a model of carcinogenesis in which activation of oncogenes stimulate preneoplasia in both Trp53+/− and Trp53+/+ tissues. Although progression is limited by p53, the proapoptotic activity and double-strand break repair pathways are impaired in Trp53+/− tissues, allowing illegitimate mitotic recombination,37 which can abolish the wild-type allele of Trp53 and eliminate the rate-limiting step in the tumor progression.
Although the requirement for loss of p53 function seems uniform, the histological appearance of the tumors in p53-deficient mice suggests divergent cellular origins. Oncogene-specific tumor phenotypes have been reported in transgenic mice expressing Her2/Neu or Ras,56–58 and, thus, the heterogeneity of the tumors may reflect the collaborating oncogenes. On the other hand, the tumor phenotypes may depend on the cellular origins. Selective deletion of Trp53 in basal or luminal compartments using the Cre recombinase under control of K14, MMTV, or WAP promoters resulted in heterogeneous tumor phenotypes,54,59,60 suggesting that tumors can arise from both luminal and basal cells. However, the origins were not definitive because Cre activity was evident in both the luminal and basal cells in these experiments. A WAP-Cre that is restricted to the luminal epithelium and is pregnancy-dependent to drive activation of a point mutant of Trp53 (R270H) yielded tumors that were positive as well as negative for ERα,61 suggesting that the luminal lineage can yield both ERα-positive and -negative tumors. Using more extensive immunophenotyping, we found that 62% of spontaneous mammary tumors in BALB/c-Trp53+/− mice expressed both luminal and basal keratins (K8/18 and either K5 or K6) suggesting origin of the tumors within bipotent progenitors (summarized in Figure 8B). A smaller fraction of tumors expressed only luminal or basal keratins. There was no association between the oncogenes expressed and tumor phenotypes because Her2/Neu and Notch1 were detected in tumors with either luminal or basal keratins, whereas the ERα-positive tumors were positive for both luminal and basal keratins. Although tumor V09 expresses high levels of ERα and PR and the phenotype was stable when tumor fragments were transplanted, its growth was estrogen-independent (Figure 6). This observation underscores the challenge of predicting origins or behavior of tumors on the basis of a limited number of biomarkers.
As the majority of tumors expressed both luminal and basal keratins, it would seem that tumors initiate most often within bipotent progenitor cells. It is possible that progenitor cells may be most susceptible to transformation, but decreased p53 function may also lead to expansion of the progenitor cell pool. The p53 protein plays a fundamental role in restricting the pool of stem cells in embryonic stem cells22,23,62 as well as in adult tissues including neuronal63,64 and hematopoietic systems.65,66 Loss of p53 allows expansion of cancer stem cells either through asymmetric division or by promoting phenotypic plasticity and the acquisition of stem cell characteristics.24,25 Mammary tumors from BALB/c-Trp53+/− mice showed evidence of activated Wnt and Notch signaling. Aberrant signaling in these pathways could result in stem cell expansion and tumorigenesis in mammary glands.48–50 However, it remains possible that the tumors initiate within lineage-restricted cells and acquire the stem cell signature during progression.
Although the tumors seem to initiate most often in bipotent progenitors, the tumors express a diversity of histological features and biomarkers. This diversity recapitulates many aspects of human breast cancer. The panel of 28 transplantable tumors provides a relevant resource for preclinical testing of therapeutic agents targeting cancer stem cells or oncogenic changes occurring during the progression from locally invasive to metastatic breast cancer.
Acknowledgements
We thank Ms. Brooke Bentley for immunohistochemistry support and Dr. Mary Hagen for her help in array analysis.
Footnotes
Supported by grants from the National Institutes of Health (R01-CA095164, R01-CA105452, and R01-ES015739 to D.J.J.).
Current address of A.C.B.: John Curtin School of Medical Research, Australian National University, Canberra ACT 0200, Australia.
References
- 1.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 2.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed M, Rahman N. ATM and breast cancer susceptibility. Oncogene. 2006;25:5906–5911. doi: 10.1038/sj.onc.1209873. [DOI] [PubMed] [Google Scholar]
- 4.Bond GL, Hirshfield KM, Kirchhoff T, Alexe G, Bond EE, Robins H, Bartel F, Taubert H, Wuerl P, Hait W, Toppmeyer D, Offit K, Levine AJ. MDM2 SNP309 accelerates tumor formation in a gender-specific and hormone-dependent manner. Cancer Res. 2006;66:5104–5110. doi: 10.1158/0008-5472.CAN-06-0180. [DOI] [PubMed] [Google Scholar]
- 5.Meijers-Heijboer H, Van Den Ouweland A, Klijn J, Wasielewski M, De Snoo A, Oldenburg R, Hollestelle A, Houben M, Crepin E, Veghel-Plandsoen M, Elstrodt F, Van Duijn C, Bartels C, Meijers C, Schutte M, McGuffog L, Thompson D, Easton DF, Sodha N, Seal S, Barfoot R, Mangion J, Chang-Claude J, Eccles D, Eeles R, Evans DG, Houlston R, Murday V, Narod S, Peretz T, Peto J, Phelan C, Zhang HX, Szabo C, Devilee P, Goldgar D, Futreal PA, Nathanson KL, Weber BL, Rahman N, Stratton MR. Low-penetrance susceptibility to breast cancer due to CHEK2*1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002;31:55–59. doi: 10.1038/ng879. [DOI] [PubMed] [Google Scholar]
- 6.Sivaraman L, Stephens LC, Markaverich BM, Clark JA, Krnacik S, Conneely OM, O'Malley BW, Medina D. Hormone-induced refractoriness to mammary carcinogenesis in Wistar-Furth rats. Carcinogenesis. 1998;19:1573–1581. doi: 10.1093/carcin/19.9.1573. [DOI] [PubMed] [Google Scholar]
- 7.Jerry DJ, Kittrell FS, Kuperwasser C, Laucirica R, Dickinson ES, Bonilla PJ, Butel JS, Medina D. A mammary-specific model demonstrates the role of the p53 tumor suppressor gene in tumor development. Oncogene. 2000;19:1052–1058. doi: 10.1038/sj.onc.1203270. [DOI] [PubMed] [Google Scholar]
- 8.Medina D, Kittrell FS. p53 function is required for hormone-mediated protection of mouse mammary tumorigenesis. Cancer Res. 2003;63:6140–6143. [PubMed] [Google Scholar]
- 9.Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, Eden OB, Varley JM. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene. 2001;20:4621–4628. doi: 10.1038/sj.onc.1204621. [DOI] [PubMed] [Google Scholar]
- 10.Kleihues P, Schauble B, zur Hausen A, Esteve J, Ohgaki H. Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol. 1997;150:1–13. [PMC free article] [PubMed] [Google Scholar]
- 11.Nichols KE, Malkin D, Garber JE, Fraumeni JF, Jr, Li FP. Germ-line p53 mutations predispose to a wide spectrum of early-onset cancers. Cancer Epidemiol Biomarkers Prev. 2001;10:83–87. [PubMed] [Google Scholar]
- 12.Dunphy KA, Blackburn AC, Yan H, O'Connell LR, Jerry DJ. Estrogen and progesterone induce persistent increases in p53-dependent apoptosis and suppress mammary tumors in BALB/c-Trp53+/− mice. Breast Cancer Res. 2008;10:R43. doi: 10.1186/bcr2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu X, Lozano G, Donehower LA. Activities of wildtype and mutant p53 in suppression of homologous recombination as measured by a retroviral vector system. Mutat Res. 2003;522:69–83. doi: 10.1016/s0027-5107(02)00261-0. [DOI] [PubMed] [Google Scholar]
- 14.Lynch CJ, Milner J. Loss of one p53 allele results in four-fold reduction of p53 mRNA and protein: a basis for p53 haplo-insufficiency. Oncogene. 2006;25:3463–3470. doi: 10.1038/sj.onc.1209387. [DOI] [PubMed] [Google Scholar]
- 15.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 16.Bensaad K, Vousden KH. p53: new roles in metabolism. Trends Cell Biol. 2007;17:286–291. doi: 10.1016/j.tcb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Bertrand P, Saintigny Y, Lopez BS. p53's double life: transactivation-independent repression of homologous recombination. Trends Genet. 2004;20:235–243. doi: 10.1016/j.tig.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 18.Gatz SA, Wiesmuller L. p53 in recombination and repair. Cell Death Differ. 2006;13:1003–1016. doi: 10.1038/sj.cdd.4401903. [DOI] [PubMed] [Google Scholar]
- 19.Sugrue MM, Shin DY, Lee SW, Aaronson SA. Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc Natl Acad Sci USA. 1997;94:9648–9653. doi: 10.1073/pnas.94.18.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 22.Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, Xu Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7:165–171. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 23.Qin H, Yu T, Qing T, Liu Y, Zhao Y, Cai J, Li J, Song Z, Qu X, Zhou P, Wu J, Ding M, Deng H. Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J Biol Chem. 2007;282:5842–5852. doi: 10.1074/jbc.M610464200. [DOI] [PubMed] [Google Scholar]
- 24.Godar S, Ince TA, Bell GW, Feldser D, Donaher JL, Bergh J, Liu A, Miu K, Watnick RS, Reinhardt F, McAllister SS, Jacks T, Weinberg RA. Growth-inhibitory and tumor-suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang M, Behbod F, Atkinson RL, Landis MD, Kittrell F, Edwards D, Medina D, Tsimelzon A, Hilsenbeck S, Green JE, Michalowska AM, Rosen JM. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 2008;68:4674–4682. doi: 10.1158/0008-5472.CAN-07-6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM, Lowe SW. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell. 2002;1:289–298. doi: 10.1016/s1535-6108(02)00047-8. [DOI] [PubMed] [Google Scholar]
- 27.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 29.Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell. 2007;11:103–105. doi: 10.1016/j.ccr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 30.Blackburn AC, Hill LZ, Roberts AL, Wang J, Aud D, Jung J, Nikolcheva T, Allard J, Peltz G, Otis CN, Cao QJ, Ricketts RS, Naber SP, Mollenhauer J, Poustka A, Malamud D, Jerry DJ. Genetic mapping in mice identifies DMBT1 as a candidate modifier of mammary tumors and breast cancer risk. Am J Pathol. 2007;170:2030–2041. doi: 10.2353/ajpath.2007.060512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuperwasser C, Hurlbut GD, Kittrell FS, Dickinson ES, Laucirica R, Medina D, Naber SP, Jerry DJ. Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice—a model for Li-Fraumeni syndrome. Am J Pathol. 2000;157:2151–2159. doi: 10.1016/S0002-9440(10)64853-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jerry DJ, Kuperwasser C, Downing SR, Pinkas J, He C, Dickinson E, Marconi S, Naber SP. Delayed involution of the mammary epithelium in BALB/c-p53null mice. Oncogene. 1998;17:2305–2312. doi: 10.1038/sj.onc.1202157. [DOI] [PubMed] [Google Scholar]
- 33.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 34.Medina D. The mammary gland: a unique organ for the study of development and tumorigenesis. J Mammary Gland Biol Neoplasia. 1996;1:5–19. doi: 10.1007/BF02096299. [DOI] [PubMed] [Google Scholar]
- 35.Medina D, Kittrell FS, Shepard A, Stephens LC, Jiang C, Lu J, Allred DC, McCarthy M, Ullrich RL. Biological and genetic properties of the p53 null preneoplastic mammary epithelium. FASEB J. 2002;16:881–883. doi: 10.1096/fj.01-0885fje. [DOI] [PubMed] [Google Scholar]
- 36.Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S, Chandrasekharan S, Backlund MG, Yin Y, Khramtsov AI, Bastein R, Quackenbush J, Glazer RI, Brown PH, Green JE, Kopelovich L, Furth PA, Palazzo JP, Olopade OI, Bernard PS, Churchill GA, Van DT, Perou CM. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blackburn AC, Mclary SC, Naeem R, Luszcz J, Stockton DW, Donehower LA, Mohammed M, Mailhes JB, Soferr T, Naber SP, Otis CN, Jerry DJ. Loss of heterozygosity occurs via mitotic recombination in Trp53+/− mice and associates with mammary tumor susceptibility of the BALB/c strain. Cancer Res. 2004;64:5140–5147. doi: 10.1158/0008-5472.CAN-03-3435. [DOI] [PubMed] [Google Scholar]
- 38.Gomm JJ, Browne PJ, Coope RC, Liu QY, Buluwela L, Coombes RC. Isolation of pure populations of epithelial and myoepithelial cells from the normal human mammary gland using immunomagnetic separation with Dynabeads. Anal Biochem. 1995;226:91–99. doi: 10.1006/abio.1995.1196. [DOI] [PubMed] [Google Scholar]
- 39.Shoker BS, Jarvis C, Sibson DR, Walker C, Sloane JP. Oestrogen receptor expression in the normal and pre-cancerous breast. J Pathol. 1999;188:237–244. doi: 10.1002/(SICI)1096-9896(199907)188:3<237::AID-PATH343>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 40.Horak E, Smith K, Bromley L, LeJeune S, Greenall M, Lane D, Harris AL. Mutant p53. EGF receptor and c-erbB-2 expression in human breast cancer. Oncogene. 1991;6:2277–2284. [PubMed] [Google Scholar]
- 41.Laws AM, Osborne BA. p53 regulates thymic Notch1 activation. Eur J Immunol. 2004;34:726–734. doi: 10.1002/eji.200324772. [DOI] [PubMed] [Google Scholar]
- 42.Lefort K, Mandinova A, Ostano P, Kolev V, Calpini V, Kolfschoten I, Devgan V, Lieb J, Raffoul W, Hohl D, Neel V, Garlick J, Chiorino G, Dotto GP. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKα kinases. Genes Dev. 2007;21:562–577. doi: 10.1101/gad.1484707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith GH, Mehrel T, Roop DR. Differential keratin gene expression in developing, differentiating, preneoplastic, and neoplastic mouse mammary epithelium. Cell Growth Differ. 1990;1:161–170. [PubMed] [Google Scholar]
- 44.Li Y, Welm B, Podsypanina K, Huang S, Chamorro M, Zhang X, Rowlands T, Egeblad M, Cowin P, Werb Z, Tan LK, Rosen JM, Varmus HE. Evidence that transgenes encoding components of the Wnt signaling pathway preferentially induce mammary cancers from progenitor cells. Proc Natl Acad Sci USA. 2003;100:15853–15858. doi: 10.1073/pnas.2136825100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Livasy CA, Karaca G, Nanda R, Tretiakova MS, Olopade OI, Moore DT, Perou CM. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod Pathol. 2006;19:264–271. doi: 10.1038/modpathol.3800528. [DOI] [PubMed] [Google Scholar]
- 46.Sotgia F, Williams TM, Cohen AW, Minetti C, Pestell RG, Lisanti MP. Caveolin-1-deficient mice have an increased mammary stem cell population with upregulation of Wnt/β-catenin signaling. Cell Cycle. 2005;4:1808–1816. doi: 10.4161/cc.4.12.2198. [DOI] [PubMed] [Google Scholar]
- 47.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004;6:R605–R615. doi: 10.1186/bcr920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Farnie G, Clarke RB. Mammary stem cells and breast cancer—role of Notch signalling. Stem Cell Rev. 2007;3:169–175. doi: 10.1007/s12015-007-0023-5. [DOI] [PubMed] [Google Scholar]
- 50.Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/β-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci USA. 2007;104:618–623. doi: 10.1073/pnas.0606599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Colombel M, Radvanyi F, Blanche M, Abbou C, Buttyan R, Donehower LA, Chopin D, Thiery JP. Androgen suppressed apoptosis is modified in p53 deficient mice. Oncogene. 1995;10:1269–1274. [PubMed] [Google Scholar]
- 52.Børresen-Dale AL. TP53 and breast cancer. Hum Mutat. 2003;21:292–300. doi: 10.1002/humu.10174. [DOI] [PubMed] [Google Scholar]
- 53.Varley JM, Thorncroft M, McGown G, Appleby J, Kelsey AM, Tricker KJ, Evans DG, Birch JM. A detailed study of loss of heterozygosity on chromosome 17 in tumours from Li-Fraumeni patients carrying a mutation to the TP53 gene. Oncogene. 1997;14:865–871. doi: 10.1038/sj.onc.1201041. [DOI] [PubMed] [Google Scholar]
- 54.Liu X, Holstege H, van der GH, Treur-Mulder M, Zevenhoven J, Velds A, Kerkhoven RM, van Vliet MH, Wessels LF, Peterse JL, Berns A, Jonkers J. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci USA. 2007;104:12111–12116. doi: 10.1073/pnas.0702969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 56.Andrechek ER, Hardy WR, Siegel PM, Rudnicki MA, Cardiff RD, Muller WJ. Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc Natl Acad Sci USA. 2000;97:3444–3449. doi: 10.1073/pnas.050408497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cardiff RD, Anver MR, Gusterson BA, Hennighausen L, Jensen RA, Merino MJ, Rehm S, Russo J, Tavassoli FA, Wakefield LM, Ward JM, Green JE. The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene. 2000;19:968–988. doi: 10.1038/sj.onc.1203277. [DOI] [PubMed] [Google Scholar]
- 58.Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell. 1987;49:465–475. doi: 10.1016/0092-8674(87)90449-1. [DOI] [PubMed] [Google Scholar]
- 59.Derksen PW, Liu X, Saridin F, van der Gulden H, Zevenhoven J, Evers B, van B, Jr, Griffioen AW, Vink J, Krimpenfort P, Peterse JL, Cardiff RD, Berns A, Jonkers J. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell. 2006;10:437–449. doi: 10.1016/j.ccr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 60.Lin SC, Lee KF, Nikitin AY, Hilsenbeck SG, Cardiff RD, Li A, Kang KW, Frank SA, Lee WH, Lee EY. Somatic mutation of p53 leads to estrogen receptor α-positive and -negative mouse mammary tumors with high frequency of metastasis. Cancer Res. 2004;64:3525–3532. doi: 10.1158/0008-5472.CAN-03-3524. [DOI] [PubMed] [Google Scholar]
- 61.Wijnhoven SW, Zwart E, Speksnijder EN, Beems RB, Olive KP, Tuveson DA, Jonkers J, Schaap MM, van den Berg J, Jacks T, van Steeg H, de Vries A. Mice expressing a mammary gland-specific R270H mutation in the p53 tumor suppressor gene mimic human breast cancer development. Cancer Res. 2005;65:8166–8173. doi: 10.1158/0008-5472.CAN-05-1650. [DOI] [PubMed] [Google Scholar]
- 62.Pan G, Thomson JA. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 2007;17:42–49. doi: 10.1038/sj.cr.7310125. [DOI] [PubMed] [Google Scholar]
- 63.Medrano S, Burns-Cusato M, Atienza MB, Rahimi D, Scrable H. Regenerative capacity of neural precursors in the adult mammalian brain is under the control of p53. Neurobiol Aging. 2009;30:483–497. doi: 10.1016/j.neurobiolaging.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meletis K, Wirta V, Hede SM, Nister M, Lundeberg J, Frisen J. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133:363–369. doi: 10.1242/dev.02208. [DOI] [PubMed] [Google Scholar]
- 65.Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5:e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dumble M, Moore L, Chambers SM, Geiger H, Van ZG, Goodell MA, Donehower LA. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood. 2007;109:1736–1742. doi: 10.1182/blood-2006-03-010413. [DOI] [PMC free article] [PubMed] [Google Scholar]