

Abstract
Local estrogen production plays a key role in proliferative endometrial disorders, such as endometrial hyperplasia and cancer. Hydroxysteroid (17β) dehydrogenase 1 (HSD17B1) is an enzyme that catalyzes with high efficiency the conversion of weakly active estrone into highly potent estradiol. Here we report that female transgenic mice expressing human HSD17B1 invariably develop endometrial hyperplasia in adulthood. These mice also fail to ovulate and have enhanced peripheral conversion of estrone into estradiol in a variety of target tissues, including the uterus. As in humans, endometrial hyperplasia in HSD17B1 transgenic female mice was reversible on ovulation induction, which triggers a rise in circulating progesterone levels, and in response to exogenous progestins. Strikingly, a treatment with an HSD17B1 inhibitor failed to restore ovulation yet completely reversed the hyperplastic morphology of epithelial cells in the glandular compartment, although less so in the luminal epithelium. The data indicate that human HSD17B1 expression enhances endometrial estrogen production, and consequently, estrogen-dependent proliferation. Therefore, HSD17B1 is a promising new therapeutic target in the management of estrogen-dependent endometrial diseases.
Estrogen-dependent uterine disorders, such as endometriosis, endometrial hyperplasia, and cancer, are very prevalent during reproductive years. For example endometriosis affects up to 10% of women and is a major cause of pelvic pain and infertility.1,2 Endometrial carcinoma is the most common invasive cancer of the female genital tract and ranks as the fourth most common malignancy in women in Western Europe and in the United States.3–5 Estrogen-dependent endometrial carcinoma (endometrial carcinoma type I, hereafter referred to as endometrial carcinoma) generally arises from atypical endometrial hyperplasia, whereas hyperplasia without atypia is less likely to become malignant.5,6 Patients with typical hyperplasia are usually managed conservatively with progestins, but hysterectomy remains the treatment of choice for atypical hyperplasia and endometrial carcinoma.3,6 Obesity and anovulation, often associated with polycystic ovary syndrome, are major risk factors for endometrial hyperplasia during reproductive years, as is unopposed estrogen replacement therapy in postmenopausal women.7–10
Hydroxysteroid (17β) dehydrogenases (HSD17Bs) catalyze the conversion of low active 17-ketosteroids and the highly active 17β-hydroxysteroids.11–13 Most of HSD17Bs belong to the ketosteroid reductase family of enzymes, which are also known as short-chain alcohol dehydrogenases/reductases.14 We have previously shown that human HSD17B1 catalyzes the reduction of estrone (E1), which has low biological activity, to highly potent estradiol (E2) in cultured cells and in vivo.15–18 Besides estrogens, androgens are also, albeit less efficient, substrates for human HSD17B1 in vivo.17,19 These studies are in line with the essential role of the enzyme in ovarian E2 biosynthesis. Notably, human HSD17B1 has a wider tissue-distribution than the murine ortholog, which is mainly expressed in the ovaries. We, thus, hypothesized that human HSD17B1 expression enhances estrogen action at target tissues by increasing the E2/E1-ratio, and providing increased concentration of highly active ligand for estrogen receptors (ESR1 and ESR2). In humans, HSD17B1 has been detected in various sex steroid-responsive tissues and linked to various estrogen-dependent diseases, such as breast cancer16,20,21 and endometriosis.2,22 However, HSD17B1 expression in cycling endometrium or in other endometrial disorders, like endometrial cancer, is still controversial with some studies reporting HSD17B1 expression and/or activity,2,4,23–29 whereas others found no evidence of expression at mRNA or protein level.30–32 The human endometrium also reportedly expresses several other HSD17Bs, including types 2, 4, 5, 7, and 12.4,26,33–36 Thus the availability of E2 at a tissue or cellular level is likely to be tightly regulated by the relative activities of various enzymes with opposing functions (activation or inactivation of estrogens). HSD17B2 in particular opposes the activity of HSD17B1, while the role of the other HSD17Bs in regulating local estrogen metabolism remains poorly understood.
Because of its putative role in E2 biosynthesis in ovaries and peripheral target tissues, HSD17B1 is considered a promising drug target for estrogen-dependent diseases. Several preclinical studies indicate that HSD17B1 inhibitors are indeed capable of modulating estrogen responses in vitro and in vivo.16,18,37–39 In the present study, we report that expression of human HSD17B1 in a transgenic mouse model (HSD17B1TG mice)17 not only induces anovulation, but also estrogen-dependent endometrial hyperplasia. Moreover, we demonstrate that this endometrial phenotype can be reversed using novel HSD17B1 inhibitors.
Materials and Methods
Mice
Mice were housed under controlled environmental conditions (12 hours light/12 hours darkness, at 21 ± 1°C) at the Central Animal Laboratory of University of Turku. Soy-free SDS RM3 (Special Diet Service; Witham Essex, UK) and tap water were available ad libitum. To obtain tissue samples, adult mice were terminally anesthetized with 600 to 1000 μl 2.5% tribromoethanol (Avertin; Sigma-Aldrich, St. Louis, MO or Alfa Aesar, Karlsruhe, Germany)40 injection, i.p., and blood was withdrawn from the heart followed by euthanasia by cervical dislocation. Tissues were weighed and frozen at −80°C or processed for histology as described below. Animal experiments were approved by the Finnish Animal Ethics Committee, and the institutional policies on animal experimentation fully meet the requirements as defined in the NIH Guide on animal experimentation. Generation, maintenance, and genotyping of HSD17B1TG mice have been previously described.17,19
Mouse Histology
For histological analysis of the mouse uterus, one of the uterine horns was dissected into five pieces (first piece above the cervix, fifth below the oviduct) before fixation, whereas the ovaries were fixed as whole. Tissues were fixed in 4% paraformaldehyde at room temperature for 15 to 20 hours. After fixation, tissues were dehydrated and paraffin-embedded. Sections, cut at 5 μm thickness at the same level of the uterus, were stained with H&E for microscopic analysis. Normal endometrial histology was defined as normal luminal epithelium and small, round, regular-shaped glands. Hyperplasia was defined as enlarged endometrial glands growing inwards resulting in the formations subglands. Nuclear atypia was defined as abnormally disorganized epithelial cells with round, enlarged nuclei with a granular appearance.
Immunohistochemistry for PCNA
To analyze the rate of proliferation in the uterus, immunohistochemistry was performed with proliferating cell nuclear antigen, NCL-PCNA (n = 6 to 7). After deparaffinization and rehydration, the sections were boiled in 0.01 mol/L sodium citrate (pH 6.0) for 15 minutes and cooled slowly to room temperature. The sections were then exposed to 1% hydrogen peroxide for 20 minutes and incubated overnight with NCL-PCNA mouse monoclonal (clone PC10) antibody (dilution 1:500; Novocastra Laboratories Ltd, Newcastle on Tyne, UK). The sections were afterward incubated with labeled polymer-horseradish peroxidase anti-mouse (DakoCytomation Envision+ System-HRP; Dako, Carpinteria, CA), and the color reaction was performed with 3,3′-diaminobenzidine (liquid DAB+; Dako). The sections were counterstained with hematoxylin and dehydrated in ethanol and xylene before mounting with pertex mounting solution (Histolab, Gothenburg, Sweden).
Uterus Weight Test
Mice were ear-marked at the age of 12 days (d), genotyped and divided into experimental groups (n = 4 to 8) receiving either placebo (corn oil; Sigma-Aldrich), 1 μg/kg/d E1 (Sigma-Aldrich), or 50 μg/kg/d E2 (Sigma-Aldrich) in a 50 μl volume i.p. Dosing was performed once a day from the age of 15 days to 19 days. Mice were terminally anesthetized with 250 to 500 μl 2.5% tribromoethanol followed by euthanasia by cervical dislocation at the age of 20 days, the uterus was dissected and weight was recorded.
Determination of HSD17B1 Activity in Vivo
Radioactive [3H]-E1 (Perkin Elmer, Waltham, MA; Sigma-Aldrich) dissolved in ethanol:saline (20:80) was slowly injected i.v. (61 μg/kg, 2.5 μl/g, 555,000 cpm/μl, ∼1.6 Mbq/mouse, n = 3–8). Adult mice were terminally anesthetized with 600 to 1000 μl 2.5% tribromoethanol injection i.p. and after 15 minutes from the substrate injection, blood was withdrawn from the heart followed by euthanasia by cervical dislocation. Tissues were dissected, placed immediately in liquid nitrogen and stored at −80°C. Tissues were thereafter homogenized by Ultra-Turrax in 500 μl ice cold 50 mmol/L Tris-HCl buffer (pH 7.4). Isopropylether extraction was then performed by adding 2 ml isopropylether to 500 μl homogenate or 150 μl serum. After mixing, samples were centrifuged at 700 rpm for 10 minutes at room temperature, and extraction was repeated for the organic phase. The organic phase was evaporated under nitrogen flow, dissolved in acetonitrile-water (48:52) and centrifuged at 700 rpm for 10 minutes at room temperature. Finally, 50 μl of samples were applied for HPLC (Waters 2695, Waters Corporation, Milford, MA) connected with an online β-counter.
Superovulation Treatment
HSD17B1TG females (n = 4) were treated with 5 IU of pregnant mare serum gonadotropin (PMSG; Sigma- Aldrich), provided i.p., in 100 μl in PBS. After 49 hours, mice were injected with 5 IU of hCG (Schering-Plough, Kenilworth, NJ), i.p., in 100 μl PBS. The length of mouse estrous cycle typically varies from 4 to 7 days41 and 2 superovulation treatments were performed in a period of 8 days to mimic normal estrous cycle. At day 9 mice were euthanized as described in Mice section, and ovarian and uterine samples were dissected for histological analyses.
Progestin Treatment
Medroxyprogesterone acetate (MPA) was delivered for 2 weeks by a subcutaneous pellet that released the hormone 16 mg/kg/d (Innovative Research of America, Sarasota, FL). Adult HSD17B1TG mice at age of 4 to 5 months were anesthetized with 450 to 800 μl 2.5% tribromoethanol, i.p., and the MPA (n = 6) or placebo (n = 7) pellets were inserted subcutaneously. Anesthetic analgesia was achieved by giving 0.15 mg/kg buprenorphine (Temgesic; Schering-Plough) preoperatively. Postoperative analgesia was obtained by injecting 0.1 mg/kg buprenorphine s.c. daily for 3 postoperative days. Two weeks later, mice were euthanized as described in Mice section, and ovarian and uterine samples were dissected for histological analyses.
Inhibitor Treatment
Ten mg/kg/d of a specific HSD17B1 inhibitor (compound 49 in reference 43),42,43 effectively shown to decrease HSD17B1-mediated estrogen-dependent tumor growth in vivo,42 was delivered in dimethyl sulfoxide/propanediol (1:1, Merck, Whitehouse Station, NJ) to mice (n = 6 to 7) for 6 weeks by subcutaneous minipumps (Alzet #2004, Cupertino, CA). To insert minipumps, the mice were anesthetized with 450 to 800 μl 2.5% tribromoethanol and 0.15 mg/kg buprenorphine, i.p. Postoperative analgesia was obtained by providing 0.1 mg/kg buprenorphine s.c. daily for 3 postoperative days. Additional s.c. analgesia was provided when needed with 0.1 mg/kg buprenorphine or 5 mg/kg carprofen (Rimadyl, Pfizer, NY). After a 3-week dosing period, minipumps were substituted with new ones as described above. To obtain samples, mice were euthanized at the age of 5.5 months as described in Mice section.
Statistics
Statistical analyses were performed using SigmaStat 3.1 program (SYSTAT Software Inc., CA) using following tests: Student's t-test or Mann-Whitney test when applicable to compare two groups and one way analysis of variance or Kruskal-Wallis one way analysis of variance of ranks when applicable to analyze many groups. Significance was set as P < 0.05, and mean values ± SEM are presented.
Results
Endometrial Hyperplasia in HSD17B1TG Mice
We recently generated several transgenic (TG) mouse lines that express human HSD17B1 at various levels.17 Histological analysis revealed that HSD17B1TG females invariably suffer from endometrial hyperplasia at the age of 4 months (Figure 1, A−D). The prevalence of endometrial hyperplasia and anovulation was 100% in HSD17B1TG animals and hyperplasias with and without nuclear atypia were detected (Table 1). The proliferation of the hyperplastic glands was confirmed by immunostaining for Pcna expression (Figure 1, E−G). While approximately 25% of animals developed atypical hyperplasia, progression to endometrial carcinomas was not observed by the age of 12 months. These mice also presented from early adulthood with anovulation, characterized by inefficient luteinization of ovarian follicles (Figure 1, H−I). At 1 month of age, before the onset of puberty, the endometrium was entirely normal on histology. Although our analyses were performed predominantly in a mouse line that strongly expresses human HSD17B1 (line 013), endometrial hyperplasia and anovulation were also observed in low-expressing HSD17B1TG lines (data not shown).
Figure 1.
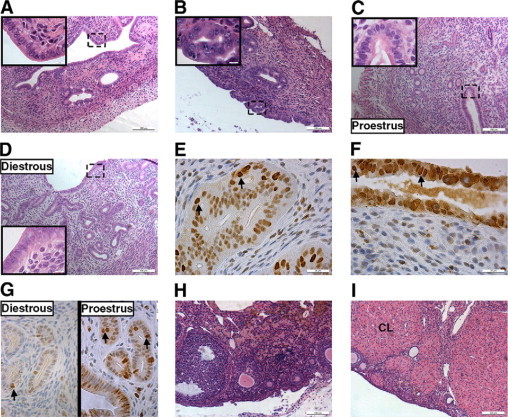
Endometrial hyperplasia in HSD17B1TG females. Endometrial hyperplasia was observed in HSD17B1TG females at the age of 4 months: hyperplastic glands growing inwards and normal luminal epithelium (A), hyperplastic glands growing inwards and nuclear atypia of the luminal epithelium (B), wild-type endometrium at proestrous, which corresponds to the human proliferative phase (C), wild-type endometrium at diestrous, which corresponds to the human secretory phase (D). The sites of the inserts are shown with dash-lined boxes. The expression of a proliferation marker PCNA in hyperplasia without atypia (E), atypical hyperplasia (F), and normal wild-type (G) at proestrous and diestrous. Examples of PCNA-positive cells are indicated by arrows. HSD17B1TG females presented with anovulatory ovaries lacking CLs (H), whereas CLs were abundant in the ovaries of the wild-type littermates (I). CL = corpus luteum.
Table 1.
Prevalence of Endometrial Hyperplasia in HSD17B1TG Mice
| Age (months) | No hyperplasia | Hyperplasia without atypia | Hyperplasia with atypia | |
|---|---|---|---|---|
| TG | 1 (n = 5) | 100% | 0% | 0% |
| 4 (n = 8) | 0% | 75% | 25% | |
| Wild-type | 1 (n = 4) | 100% | 0% | 0% |
| 4 (n = 6) | 100% | 0% | 0% |
HSD17B1 Enhances Uterine Estrogen Action in Vivo
To examine if HSD17B1 modulates estrogen responses in the uterus, immature (age of 15 days) HSD17B1TG females and wild-type littermates were treated for 5 days with vehicle (placebo), 1 μg/kg/d of E1, or 50 μg/kg/d of E2. Uterine weight, which serves as a classical bioassay for estrogen action in this tissue, increased in both E1-treated wild-type and HSD17B1TG animals (Figure 2, A−C). However, the relative increase in weight was four times higher in HSD17B1TG animals (Table 2). As expected, E2 also markedly stimulated uterine growth, and the magnitude of this response was identical between wild-type and HSD17B1TG mice. Notably, uterine weight was modestly higher in placebo-treated HSD17B1TG mice, possibly reflecting more efficient conversion of endogenous E1 to E2. To test this formally, we analyzed the reductive HSD17B capacity, ie, conversion of E1 substrate to E2, in the uterus and other tissues of wild-type and HSD17B1TG mice in vivo. Tissue samples were collected 15 minutes after i.v. injection of radioactive E1 (61 μg/kg). Compared with wild-type animals, the E2/E1 ratio in HSD17B1TG mice was significantly higher in all tissues examined, except for the ovaries (Figure 3). As mentioned, the ovaries of wild-type mice express high levels of Hsd17b1.
Figure 2.
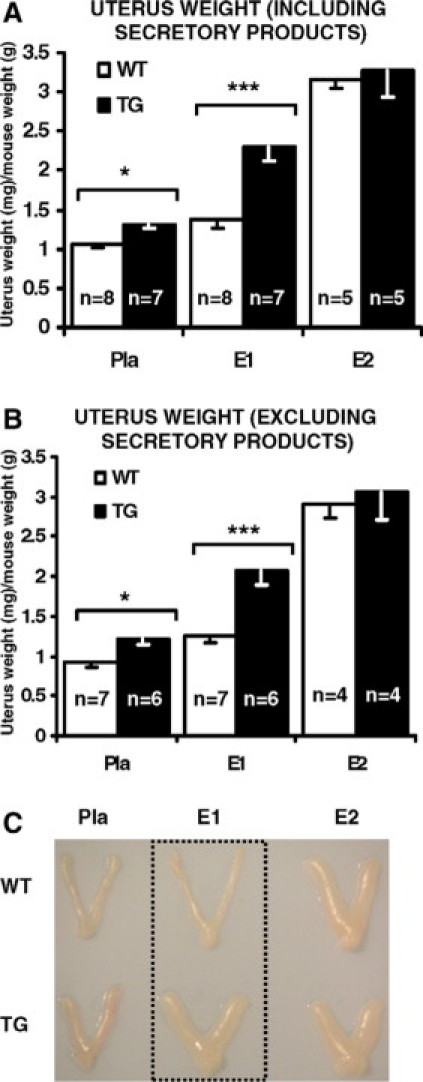
Human HSD17B1 expression enhances uterine response to weak estrogen estrone. Estrogen action was enhanced in the presence of E1 in HSD17B1TG uterus as indicated by the fact that the uterus weight of HSD17B1TG females increased more with a small dose of E1 than the weight of wild-type uterus. The different growth response was observed in the weights both by including (A) and excluding (B) the uterine secretory products, ie, full and empty uterus, respectively. Macroscopic images (C) of wild-type and HSD17B1TG uteri treated with placebo, and with equal dose of E1 (1 μg/kg/d) and E2 (50 μg/kg/d). E1 = estrone, E2 = estradiol, Pla = placebo, *P < 0.05, ***P < 0.001.
Table 2.
Uterus Weight and Magnitude of E1-Induced Uterine Growth
| Uterus weight (mg) |
|||
|---|---|---|---|
| Placebo | E1 | E1-induced growth (mg) | |
| TG (n = 7) | 11.1 (±0.39) | 19.5 (±0.78)*** | 8.4 |
| Wild-type (n = 8) | 9.8 (±0.38) | 11.8 (±0.79) | 2 |
P < 0.001 between E1 treated TG and wild-type uterus weight containing secretory products. Average uterus weight (mg) ± SEM and the magnitude of E1-induced uterine growth (mg) are presented.
Figure 3.
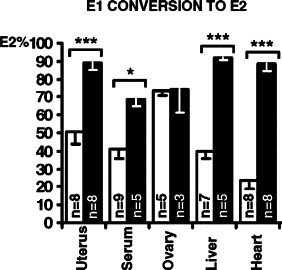
Human HSD17B1 induces estradiol production from estrone in vivo. Increased conversion of E1 to E2 was observed in HSD17B1TG mice in all extra-gonadal tissues studied, as compared with wild-type littermate tissues. E1 = estrone, E2 = estradiol, white bars represent wild-type mice, black bars represent HSD17B1TG mice, *P < 0.05, ***P < 0.001.
HSD17B1TG Mice Serve as a Model for Human Endometrial Hyperplasia
We reasoned that endometrial hyperplasia in HSD17B1TG mice could, as is the case in humans, be a consequence of persistent anovulation, which abolishes cyclic progesterone secretion and results in unopposed estrogen action in target tissues like the uterus. To test this conjecture, animals were treated with exogenous gonadotropins to induce ovulation and consequent luteinization of ovarian follicles. Two PMSG/hCG injections were given at a 4-day interval to mimic the normal estrous cycle in mice. Induction of ovulation was sufficient to reverse endometrial hyperplasia in HSD17B1TG mice (Figure 4, A−B, Table 3), indicating that the endometrial phenotype was indeed a consequence of ovarian dysfunction. Treatment with a high dose of progestin (MPA) for 2 weeks also reversed the endometrial hyperplasia but, as anticipated, did not induce ovulation (Figure 4, C−D, Table 3). To test if HSD17B1 could serve as a drug target for the treatment of endometrial hyperplasia, HSD17B1TG females were treated with a specific HSD17B1 inhibitor for 6 weeks. Similarly to MPA, the HSD17B1 inhibitor completely reversed the hyperplastic morphology of the glandular endometrial compartment without inducing luteinization of ovarian follicles in HSD17B1TG animals (Figure 5, A−F, Table 3). The endometrial luminal epithelial cells, however, remained prolonged proestrous-like in 50% of animals treated with this inhibitor (Figure 5, B−C) and this was often associated with focal endometrial inflammation, characterized by the accumulation of eosinophilic granulocytes and other leukocytes (data not shown). However, significant difference in the expression of PCNA was not found between inhibitor- and placebo-treated groups as analyzed by immunohistochemistry (data not shown). The data indicate that endometrial hyperplasia in HSD17B1TG mice is the sum of the effects of ovarian dysfunction and enhanced local E2 production in response to endometrial HSD17B1 expression.
Figure 4.
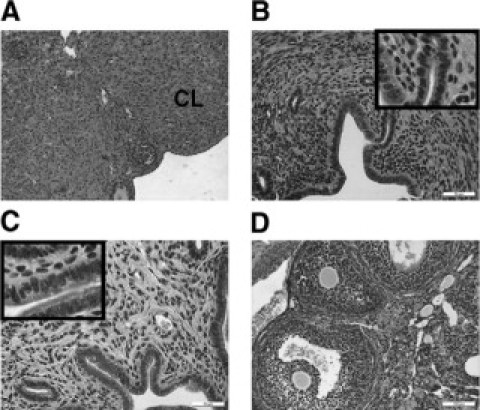
Treatment of endometrial hyperplasia of HSD17B1TG females with progestins. Multiple CLs were induced in adult HSD17B1TG ovaries by gonadotophin (PMSG/hCG) treatment (A). The gonadotrophin treatment also reversed endometrial hyperplasia in HSD17B1TG mice (B). Treating HSD17B1TG mice with progestin (MPA) also reversed endometrial hyperplasia (C), but the ovary remained anovulatory (D). CL = corpus luteum.
Table 3.
Endometrial Hyperplasia After Different Treatments In HSD17B1TG Mice
| Treatment | No hyperplasia | Hyperplasia without atypia | Hyperplasia with atypia |
|---|---|---|---|
| PMSG/hCG | 100% | 0% | 0% |
| MPA (n = 5) | 100% | 0% | 0% |
| HSD17B1 inhibitor (n = 6) | 100% | 0% | 0% |
| Placebo (n = 7) | 0% | 57.1% | 42.9% |
PMSG/hCG = pregnant mare serum gonadotropin, hCG = human chorion gonadotropin, MPA = medroxyprogesterone acetate.
Figure 5.
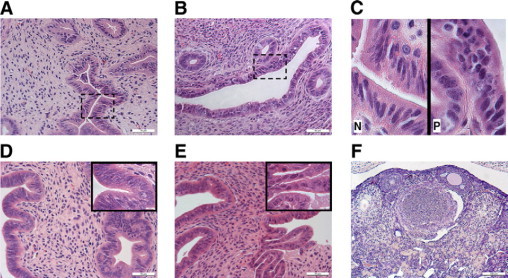
Treatment of endometrial hyperplasia of HSD17B1TG females with an HSD17B1 inhibitor. The treatment with an HSD17B1 inhibitor reversed the hyperplastic glandular morphology in adult TG females to normal (A, B), while the luminal epithelium appeared as normal or prolonged proestrous-like (C). The sites of the inserts in C are indicated with dash-lines boxes in A and B. Several stages of hyperplasia were observed in placebo-treated HSD17B1TG females: hyperplasia without atypia (D) and hyperplasia with atypia (E). The inhibitor did not induce ovulation in the HSD17B1TG ovary (F). N = normal, P = prolonged proestrous.
Discussion
Endometrial hyperplasia is a precursor of endometrial carcinoma and is caused by imbalanced actions of estrogens and progestins. Thus, both increased estrogen and decreased progestin concentrations can induce endometrial hyperplasia and cancer. This type of hormonal imbalance could be caused, for example, during estrogen replacement therapy or tamoxifen treatment, which both increase the risk for endometrial carcinoma.7,9,10 Endometrial cancer is more prevalent after menopause, but obese women with increased estrogen production in the adipose tissue and polycystic ovary syndrome patients with anovulatory menstrual cycles are at increased risk already during reproductive years.8 The correlation between serum E2 levels and the risk of endometrial cancer is controversial44–46 and increased intratissular E2 concentrations are considered a more definite risk factor.44,46 Progestins antagonize estrogen-mediated cell proliferation in the endometrium47,48 and are, therefore, widely included in hormone replacement therapy to decrease the risk of endometrial hyperplasia and cancer.49,50 The endocrinology of the mouse endometrium closely resembles that of humans. As in humans, endometrial hyperplasia and carcinoma can be induced by continuous estrogen exposure in rodents.51 In line with this, mice lacking Esr1 are resistant and the uterus remains hypoplastic,52 whereas progesterone receptor knockout mice respond to combined estrogen and progestin stimulation by developing abnormally enlarged uteri and endometrial hyperplasia, indicative of unopposed estrogen stimulation.53
Several human HSD17Bs are capable of converting E1 to E2, including HSD17B1,15–17 HSD17B7,33 and HSD17B12,35 but the role of each of these enzymes in extra-gonadal E2 formation remains still unclear. Although HSD17B1 expression in various peripheral tissues is low, its catalytic efficacy is markedly higher than that of HSD17B7 or HSD17B12,35 suggesting a central role for this enzyme in peripheral E2 formation. We recently reported that HSD17B1 expression increases estrogen responses in target cells in the presence of E1 in vivo.16,42 However, we also recently demonstrated that the enzyme is not fully specific for estrogenic substrates, as previously thought, but rather it also activates androgens.17,19 The enhanced sex steroid action resulting in estrogenization or androgenization depends on the availability of substrates. Thus, during fetal life there are androgenic precursors available for masculinization of HSD17B1TG females, while after the onset of puberty there is plenty of 17-keto estrogen (E1) available to be further activated to E2 by HSD17B1. The observation that a small dose of E1 suffices to markedly increase uterine weight of immature HSD17B1TG mice further demonstrates the ability of HSD17B1 to enhance estrogen action in target tissues. Increased local E2 production in response to E1 was apparent in all TG tissues with the exception of the ovaries, which in mice express high levels of endogenous Hsd17b1.54 Our data suggest that HSD17B1 plays a major role in determining the gradient between the E2 concentrations in serum and peripheral tissues, as has been reported for postmenopausal breast cancer,55 a tissue with abundant HSD17B1 expression.56,57
We have shown that overexpression of human HSD17B1 in mice enhances estrogen action in the uterus and, in combination with persistent anovulation, that this invariably causes endometrial hyperplasia with and without atypia. However, endometrial carcinomas were not observed in HSD17B1TG mice, indicating that other mechanisms, such as phosphatase and tensin homolog inactivation, loss of forkhead box O subclass transcription factor 1, and hyperactivity of phosphoinositide-3 kinase pathway, are key to endometrial carcinogenesis.58 Endometrial hyperplasia in HSD17B1TG mice closely resembled human disease and was efficiently reversed on normalizing the estrogen/progestin ratio in response to either ovulation induction or exogenous progestins. Treatment with an HSD17B1 inhibitor also restored endometrial glandular morphology, but incompletely blocked proliferation of luminal epithelial cells. Targeting activity with this compound was insufficient to induce ovulation. One explanation for this could be a failure in the programming of the hypothalamus-pituitary-gonadal axis, known to be induced by abnormal concentrations of estrogen and androgens during development and after birth,59 and consequent lack of luteinizing hormone surge, which would normally induce ovulation. The compound also often caused an endometrial inflammatory response for reasons as yet unclear.
Expression of HSD17B1 in human endometrium and endometrial cancer is contentious, with some studies reporting expression at mRNA or protein level and others not.2,4,22–32,60–62 Also other estrogen-metabolizing enzymes, for example HSD17B7, HSD17B12, HSD17B5, HSD17B2, HSD17B4, HSD17B8, aromatase, steroid sulfatase, and estrogen sulfotransferase have been detected in the endometrium under different pathological conditions, such as endometrial cancer,4,36,63 endometriosis,2,36,64 and polycystic ovary syndrome.65,66 Thus, the combination of the activities of these various steroid-metabolizing enzymes ultimately determines the hormonal status of the endometrium. Studies have indicated that oxidative activity is higher in the endometrium than reductive activity, and therefore, estrogen inactivation is higher than synthesis,67 and the current data suggest that HSD17B1 is not the main enzyme in the estrogen production of the normal and malignant endometrium, but nevertheless, the enzyme is expressed in both tissues, and significant reductive activity has been detected in the human endometrium.67,68 Therefore, the inhibition of HSD17B1-induced reductive activity would further decrease the estrogen synthesis, and thus, decrease local estrogen concentration, and consequently, estrogen-dependent proliferation. Furthermore, due to the inevitable role of HSD17B1 in ovarian E2 biosynthesis, HSD17B1 inhibitors could also decrease estrogen action via decreasing both local and circulating E2 concentration when used in premenopausal patients.
Acknowledgements
Ms. Erja Mäntysalo, Ms. Taija Leinonen, Ms. Erica Nyman, Ms. Jonna Palmu, Ms. Anu Salminen, Ms. Taina Kirjonen, Ms. Hannele Rekola, Ms. Riikka Kytömaa, and Mr. Jussi Mantere are acknowledged for technical help.
Footnotes
Supported by Hormos Medical Ltd. subsidiary of QuatRx Pharmaceuticals, The Academy of Finland (Projects 77920, 211480 and 121527), the Sigrid Juselius Foundation, the Turku Graduate School of Biomedical Sciences, the Finnish Cultural Foundation, and the Finnish Union of Experts in Science. Hormos Medical Ltd. and Solvay Pharmaceuticals are acknowledged for providing the HSD17B1 inhibitors.
References
- 1.Eskenazi B, Warner ML. Epidemiology of endometriosis. Obstet Gynecol Clin North Am. 1997;24:235–258. doi: 10.1016/s0889-8545(05)70302-8. [DOI] [PubMed] [Google Scholar]
- 2.Dassen H, Punyadeera C, Kamps R, Delvoux B, Van Langendonckt A, Donnez J, Husen B, Thole H, Dunselman G, Groothuis P. Estrogen metabolizing enzymes in endometrium and endometriosis. Hum Reprod. 2007;22:3148–3158. doi: 10.1093/humrep/dem310. [DOI] [PubMed] [Google Scholar]
- 3.Kurman R. Blaustein's pathology of the female genital tract. Springer-Verlag; New York: 2002. pp. 467–559. [Google Scholar]
- 4.Smuc T, Rupreht R, Sinkovec J, Adamski J, Rizner TL. Expression analysis of estrogen-metabolizing enzymes in human endometrial cancer. Mol Cell Endocrinol. 2006;248:114–117. doi: 10.1016/j.mce.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 5.Horn LC, Meinel A, Handzel R, Einenkel J. Histopathology of endometrial hyperplasia and endometrial carcinoma: an update. Ann Diagn Pathol. 2007;11:297–311. doi: 10.1016/j.anndiagpath.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 6.Lax SF. Molecular genetic changes in epithelial, stromal and mixed neoplasms of the endometrium. Pathology. 2007;39:46–54. doi: 10.1080/00313020601146822. [DOI] [PubMed] [Google Scholar]
- 7.Kelsey JL, LiVolsi VA, Holford TR, Fischer DB, Mostow ED, Schwartz PE, O'Connor T, White C. A case-control study of cancer of the endometrium. Am J Epidemiol. 1982;116:333–342. doi: 10.1093/oxfordjournals.aje.a113417. [DOI] [PubMed] [Google Scholar]
- 8.Gallup DG, Stock RJ. Adenocarcinoma of the endometrium in women 40 years of age or younger. Obstet Gynecol. 1984;64:417–420. [PubMed] [Google Scholar]
- 9.Sherman ME. Theories of endometrial carcinogenesis: a multidisciplinary approach. Mod Pathol. 2000;13:295–308. doi: 10.1038/modpathol.3880051. [DOI] [PubMed] [Google Scholar]
- 10.Schneider HP. HRT and cancer risks. Maturitas. 2002;43 Suppl 1:S35–S52. doi: 10.1016/s0378-5122(02)00147-0. [DOI] [PubMed] [Google Scholar]
- 11.Mindnich R, Moller G, Adamski J. The role of 17 beta-hydroxysteroid dehydrogenases. Mol Cell Endocrinol. 2004;218:7–20. doi: 10.1016/j.mce.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 12.Lukacik P, Kavanagh KL, Oppermann U. Structure and function of human 17beta-hydroxysteroid dehydrogenases. Mol Cell Endocrinol. 2006;248:61–71. doi: 10.1016/j.mce.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Moeller G, Adamski J. Integrated view on 17beta-hydroxysteroid dehydrogenases. Mol Cell Endocrinol. 2009;301:7–19. doi: 10.1016/j.mce.2008.10.040. [DOI] [PubMed] [Google Scholar]
- 14.Peltoketo H, Luu-The V, Simard J, Adamski J. 17beta-hydroxysteroid dehydrogenase (HSD)/17-ketosteroid reductase (KSR) family; nomenclature and main characteristics of the 17HSD/KSR enzymes. J Mol Endocrinol. 1999;23:1–11. doi: 10.1677/jme.0.0230001. [DOI] [PubMed] [Google Scholar]
- 15.Poutanen M, Miettinen M, Vihko R. Differential estrogen substrate specificities for transiently expressed human placental 17 beta-hydroxysteroid dehydrogenase and an endogenous enzyme expressed in cultured COS-m6 cells. Endocrinology. 1993;133:2639–2644. doi: 10.1210/endo.133.6.8243287. [DOI] [PubMed] [Google Scholar]
- 16.Husen B, Huhtinen K, Saloniemi T, Messinger J, Thole HH, Poutanen M. Human hydroxysteroid (17-beta) dehydrogenase 1 expression enhances estrogen sensitivity of MCF-7 breast cancer cell xenografts. Endocrinology. 2006;147:5333–5339. doi: 10.1210/en.2006-0778. [DOI] [PubMed] [Google Scholar]
- 17.Saloniemi T, Lamminen T, Huhtinen K, Welsh M, Saunders P, Kujari H, Poutanen M. Activation of androgens by hydroxysteroid (17beta) dehydrogenase 1 in vivo as a cause of prenatal masculinization and ovarian benign serous cystadenomas. Mol Endocrinol. 2007;21:2627–2636. doi: 10.1210/me.2007-0144. [DOI] [PubMed] [Google Scholar]
- 18.Lamminen T, Saloniemi T, Huhtinen K, Koskimies P, Messinger J, Husen B, Thole H, Poutanen M. In vivo mouse model for analysis of hydroxysteroid (17beta) dehydrogenase 1 inhibitors. Mol Cell Endocrinol. 2009;301:158–162. doi: 10.1016/j.mce.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 19.Saloniemi T, Welsh M, Lamminen T, Saunders P, Makela S, Streng T, Poutanen M. Human HSD17B1 expression masculinizes transgenic female mice. Mol Cell Endocrinol. 2009;301:163–168. doi: 10.1016/j.mce.2008.10.047. [DOI] [PubMed] [Google Scholar]
- 20.Poutanen M, Isomaa V, Lehto VP, Vihko R. Immunological analysis of 17 beta-hydroxysteroid dehydrogenase in benign and malignant human breast tissue. Int J Cancer. 1992;50:386–390. doi: 10.1002/ijc.2910500310. [DOI] [PubMed] [Google Scholar]
- 21.Gunnarsson C, Jerevall PL, Hammar K, Olsson B, Nordenskjold B, Jansson A, Stal O. Amplification of HSD17B1 has prognostic significance in postmenopausal breast cancer. Breast Cancer Res Treat. 2008;108:35–41. doi: 10.1007/s10549-007-9579-7. [DOI] [PubMed] [Google Scholar]
- 22.Einspanier A, Lieder K, Bruns A, Husen B, Thole H, Simon C. Induction of endometriosis in the marmoset monkey (Callithrix jacchus) Mol Hum Reprod. 2006;12:291–299. doi: 10.1093/molehr/gal031. [DOI] [PubMed] [Google Scholar]
- 23.Maentausta O, Sormunen R, Isomaa V, Lehto VP, Jouppila P, Vihko R. Immunohistochemical localization of 17 beta-hydroxysteroid dehydrogenase in the human endometrium during the menstrual cycle. Lab Invest. 1991;65:582–587. [PubMed] [Google Scholar]
- 24.Maentausta O, Boman K, Isomaa V, Stendahl U, Backstrom T, Vihko R. Immunohistochemical study of the human 17 beta-hydroxysteroid dehydrogenase and steroid receptors in endometrial adenocarcinoma. Cancer. 1992;70:1551–1555. doi: 10.1002/1097-0142(19920915)70:6<1551::aid-cncr2820700618>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 25.Martel C, Rheaume E, Takahashi M, Trudel C, Couet J, Luu-The V, Simard J, Labrie F. Distribution of 17 beta-hydroxysteroid dehydrogenase gene expression and activity in rat and human tissues. J Steroid Biochem Mol Biol. 1992;41:597–603. doi: 10.1016/0960-0760(92)90390-5. [DOI] [PubMed] [Google Scholar]
- 26.Miettinen MM, Mustonen MV, Poutanen MH, Isomaa VV, Vihko RK. Human 17 beta-hydroxysteroid dehydrogenase type 1 and type 2 isoenzymes have opposite activities in cultured cells and characteristic cell- and tissue-specific expression. Biochem J. 1996;314:839–845. doi: 10.1042/bj3140839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeitoun K, Takayama K, Sasano H, Suzuki T, Moghrabi N, Andersson S, Johns A, Meng L, Putman M, Carr B, Bulun SE. Deficient 17beta-hydroxysteroid dehydrogenase type 2 expression in endometriosis: failure to metabolize 17beta-estradiol. J Clin Endocrinol Metab. 1998;83:4474–4480. doi: 10.1210/jcem.83.12.5301. [DOI] [PubMed] [Google Scholar]
- 28.Kasai T, Shozu M, Murakami K, Segawa T, Shinohara K, Nomura K, Inoue M. Increased expression of type I 17beta-hydroxysteroid dehydrogenase enhances in situ production of estradiol in uterine leiomyoma. J Clin Endocrinol Metab. 2004;89:5661–5668. doi: 10.1210/jc.2003-032085. [DOI] [PubMed] [Google Scholar]
- 29.Fechner S, Husen B, Thole H, Schmidt M, Gashaw I, Kimmig R, Winterhager E, Grummer R. Expression and regulation of estrogen-converting enzymes in ectopic human endometrial tissue. Fertil Steril. 2007;88:1029–1038. doi: 10.1016/j.fertnstert.2006.11.153. [DOI] [PubMed] [Google Scholar]
- 30.Marovitz W, Loucopoulos A, Satyaswaroop PG, Gurpide E, Todd R, Naftolin F. Apparent immunologic nonidentity of human placental and endometrial 17beta-estradiol dehydrogenase. Am J Obstet Gynecol. 1980;138:643–647. doi: 10.1016/0002-9378(80)90081-2. [DOI] [PubMed] [Google Scholar]
- 31.Casey ML, MacDonald PC, Andersson S. 17 beta-Hydroxysteroid dehydrogenase type 2: chromosomal assignment and progestin regulation of gene expression in human endometrium. J Clin Invest. 1994;94:2135–2141. doi: 10.1172/JCI117569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Utsunomiya H, Suzuki T, Kaneko C, Takeyama J, Nakamura J, Kimura K, Yoshihama M, Harada N, Ito K, Konno R, Sato S, Okamura K, Sasano H. The analyses of 17beta-hydroxysteroid dehydrogenase isozymes in human endometrial hyperplasia and carcinoma. J Clin Endocrinol Metab. 2001;86:3436–3443. doi: 10.1210/jcem.86.7.7661. [DOI] [PubMed] [Google Scholar]
- 33.Torn S, Nokelainen P, Kurkela R, Pulkka A, Menjivar M, Ghosh S, Coca-Prados M, Peltoketo H, Isomaa V, Vihko P. Production, purification, and functional analysis of recombinant human and mouse 17beta-hydroxysteroid dehydrogenase type 7. Biochem Biophys Res Commun. 2003;305:37–45. doi: 10.1016/s0006-291x(03)00694-6. [DOI] [PubMed] [Google Scholar]
- 34.Ito K, Utsunomiya H, Suzuki T, Saitou S, Akahira J, Okamura K, Yaegashi N, Sasano H. 17Beta-hydroxysteroid dehydrogenases in human endometrium and its disorders. Mol Cell Endocrinol. 2006;248:136–140. doi: 10.1016/j.mce.2005.11.038. [DOI] [PubMed] [Google Scholar]
- 35.Luu-The V, Tremblay P, Labrie F. Characterization of type 12 17beta-hydroxysteroid dehydrogenase, an isoform of type 3 17beta-hydroxysteroid dehydrogenase responsible for estradiol formation in women. Mol Endocrinol. 2006;20:437–443. doi: 10.1210/me.2005-0058. [DOI] [PubMed] [Google Scholar]
- 36.Smuc T, Rizner TL. Aberrant pre-receptor regulation of estrogen and progesterone action in endometrial cancer. Mol Cell Endocrinol. 2009;301:74–82. doi: 10.1016/j.mce.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 37.Laplante Y, Cadot C, Fournier MA, Poirier D. Estradiol and estrone C-16 derivatives as inhibitors of type 1 17beta-hydroxysteroid dehydrogenase: Blocking of ER(+) breast cancer cell proliferation induced by estrone. Bioorg Med Chem. 2008;16:1849–1860. doi: 10.1016/j.bmc.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 38.Day JM, Foster PA, Tutill HJ, Parsons MF, Newman SP, Chander SK, Allan GM, Lawrence HR, Vicker N, Potter BV, Reed MJ, Purohit A. 17beta-hydroxysteroid dehydrogenase Type 1, and not Type 12, is a target for endocrine therapy of hormone-dependent breast cancer. Int J Cancer. 2008;122:1931–1940. doi: 10.1002/ijc.23350. [DOI] [PubMed] [Google Scholar]
- 39.Laplante Y, Rancourt C, Poirier D. Relative involvement of three 17beta-hydroxysteroid dehydrogenases (types 1, 7 and 12) in the formation of estradiol in various breast cancer cell lines using selective inhibitors. Mol Cell Endocrinol. 2009;301:146–153. doi: 10.1016/j.mce.2008.08.026. [DOI] [PubMed] [Google Scholar]
- 40.Weiss J, Zimmermann F. Tribromoethanol (Avertin) as an anaesthetic in mice. Lab Anim. 1999;33:192–193. doi: 10.1258/002367799780578417. [DOI] [PubMed] [Google Scholar]
- 41.Staley K, Scharfman H. A woman's prerogative. Nat Neurosci. 2005;8:697–699. doi: 10.1038/nn0605-697. [DOI] [PubMed] [Google Scholar]
- 42.Husen B, Huhtinen K, Poutanen M, Kangas L, Messinger J, Thole H. Evaluation of inhibitors for 17beta-hydroxysteroid dehydrogenase type 1 in vivo in immunodeficient mice inoculated with MCF-7 cells stably expressing the recombinant human enzyme. Mol Cell Endocrinol. 2006;248:109–113. doi: 10.1016/j.mce.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 43.Messinger J, Husen B, Koskimies P, Hirvela L, Kallio L, Saarenketo P, Thole H. Estrone C15 derivatives–a new class of 17beta-hydroxysteroid dehydrogenase type 1 inhibitors. Mol Cell Endocrinol. 2009;301:216–224. doi: 10.1016/j.mce.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 44.Vermeulen-Meiners C, Poortman J, Haspels AA, Thijssen JH. The endogenous concentration of estradiol and estrone in pathological human postmenopausal endometrium. J Steroid Biochem. 1986;24:1073–1078. doi: 10.1016/0022-4731(86)90362-6. [DOI] [PubMed] [Google Scholar]
- 45.Potischman N, Hoover RN, Brinton LA, Siiteri P, Dorgan JF, Swanson CA, Berman ML, Mortel R, Twiggs LB, Barrett RJ, Wilbanks GD, Persky V, Lurain JR. Case-control study of endogenous steroid hormones and endometrial cancer. J Natl Cancer Inst. 1996;88:1127–1135. doi: 10.1093/jnci/88.16.1127. [DOI] [PubMed] [Google Scholar]
- 46.Berstein LM, Tchernobrovkina AE, Gamajunova VB, Kovalevskij AJ, Vasilyev DA, Chepik OF, Turkevitch EA, Tsyrlina EV, Maximov SJ, Ashrafian LA, Thijssen JH. Tumor estrogen content and clinico-morphological and endocrine features of endometrial cancer. J Cancer Res Clin Oncol. 2003;129:245–249. doi: 10.1007/s00432-003-0427-9. [DOI] [PubMed] [Google Scholar]
- 47.Graham JD, Clarke CL. Physiological action of progesterone in target tissues. Endocr Rev. 1997;18:502–519. doi: 10.1210/edrv.18.4.0308. [DOI] [PubMed] [Google Scholar]
- 48.Wheeler DT, Bristow RE, Kurman RJ. Histologic alterations in endometrial hyperplasia and well-differentiated carcinoma treated with progestins. Am J Surg Pathol. 2007;31:988–998. doi: 10.1097/PAS.0b013e31802d68ce. [DOI] [PubMed] [Google Scholar]
- 49.Beresford SA, Weiss NS, Voigt LF, McKnight B. Risk of endometrial cancer in relation to use of oestrogen combined with cyclic progestagen therapy in postmenopausal women. Lancet. 1997;349:458–461. doi: 10.1016/S0140-6736(96)07365-5. [DOI] [PubMed] [Google Scholar]
- 50.Feeley KM, Wells M. Hormone replacement therapy and the endometrium. J Clin Pathol. 2001;54:435–440. doi: 10.1136/jcp.54.6.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gunin AG, Kapitova IN, Suslonova NV. Effects of histone deacetylase inhibitors on estradiol-induced proliferation and hyperplasia formation in the mouse uterus. J Endocrinol. 2005;185:539–549. doi: 10.1677/joe.1.06118. [DOI] [PubMed] [Google Scholar]
- 52.Couse JF, Korach KS. Contrasting phenotypes in reproductive tissues of female estrogen receptor null mice. Ann NY Acad Sci. 2001;948:1–8. doi: 10.1111/j.1749-6632.2001.tb03981.x. [DOI] [PubMed] [Google Scholar]
- 53.Lydon JP, DeMayo FJ, Funk CR, Mani SK, Hughes AR, Montgomery CA, Jr, Shyamala G, Conneely OM, O'Malley BW. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9:2266–2278. doi: 10.1101/gad.9.18.2266. [DOI] [PubMed] [Google Scholar]
- 54.Nokelainen P, Puranen T, Peltoketo H, Orava M, Vihko P, Vihko R. Molecular cloning of mouse 17 beta-hydroxysteroid dehydrogenase type 1 and characterization of enzyme activity. Eur J Biochem. 1996;236:482–490. doi: 10.1111/j.1432-1033.1996.00482.x. [DOI] [PubMed] [Google Scholar]
- 55.Pasqualini JR, Chetrite G, Blacker C, Feinstein MC, Delalonde L, Talbi M, Maloche C. Concentrations of estrone, estradiol, and estrone sulfate and evaluation of sulfatase and aromatase activities in pre- and postmenopausal breast cancer patients. J Clin Endocrinol Metab. 1996;81:1460–1464. doi: 10.1210/jcem.81.4.8636351. [DOI] [PubMed] [Google Scholar]
- 56.Miyoshi Y, Ando A, Shiba E, Taguchi T, Tamaki Y, Noguchi S. Involvement of up-regulation of 17beta-hydroxysteroid dehydrogenase type 1 in maintenance of intratumoral high estradiol levels in postmenopausal breast cancers. Int J Cancer. 2001;94:685–689. doi: 10.1002/ijc.1525. [DOI] [PubMed] [Google Scholar]
- 57.Shibuya R, Suzuki T, Miki Y, Yoshida K, Moriya T, Ono K, Akahira J, Ishida T, Hirakawa H, Evans DB, Sasano H. Intratumoral concentration of sex steroids and expression of sex steroid-producing enzymes in ductal carcinoma in situ of human breast. Endocr Relat Cancer. 2008;15:113–124. doi: 10.1677/ERC-07-0092. [DOI] [PubMed] [Google Scholar]
- 58.Goto T, Takano M, Albergaria A, Briese J, Pomeranz KM, Cloke B, Fusi L, Feroze-Zaidi F, Maywald N, Sajin M, Dina RE, Ishihara O, Takeda S, Lam EW, Bamberger AM, Ghaem-Maghami S, Brosens JJ. Mechanism and functional consequences of loss of FOXO1 expression in endometrioid endometrial cancer cells. Oncogene. 2008;27:9–19. doi: 10.1038/sj.onc.1210626. [DOI] [PubMed] [Google Scholar]
- 59.Robinson J. Prenatal programming of the female reproductive neuroendocrine system by androgens. Reproduction. 2006;132:539–547. doi: 10.1530/rep.1.00064. [DOI] [PubMed] [Google Scholar]
- 60.Maentausta O, Peltoketo H, Isomaa V, Jouppila P, Vihko R. Immunological measurement of human 17 beta-hydroxysteroid dehydrogenase. J Steroid Biochem. 1990;36:673–680. doi: 10.1016/0022-4731(90)90187-w. [DOI] [PubMed] [Google Scholar]
- 61.Maentausta O, Menjivar M, Vihko R. Time-resolved immunofluorometric assay of 17 beta-hydroxysteroid dehydrogenase in plasma. Clin Chem. 1991;37:1412–1415. [PubMed] [Google Scholar]
- 62.Utsunomiya H, Suzuki T, Ito K, Moriya T, Konno R, Sato S, Yaegashi N, Okamura K, Sasano H. The correlation between the response to progestogen treatment and the expression of progesterone receptor B and 17beta-hydroxysteroid dehydrogenase type 2 in human endometrial carcinoma. Clin Endocrinol (Oxf) 2003;58:696–703. doi: 10.1046/j.1365-2265.2003.01766.x. [DOI] [PubMed] [Google Scholar]
- 63.Rizner TL, Smuc T, Rupreht R, Sinkovec J, Penning TM. AKR1C1 and AKR1C3 may determine progesterone and estrogen ratios in endometrial cancer. Mol Cell Endocrinol. 2006;248:126–135. doi: 10.1016/j.mce.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 64.Smuc T, Pucelj MR, Sinkovec J, Husen B, Thole H, Lanisnik Rizner T. Expression analysis of the genes involved in estradiol and progesterone action in human ovarian endometriosis. Gynecol Endocrinol. 2007;23:105–111. doi: 10.1080/09513590601152219. [DOI] [PubMed] [Google Scholar]
- 65.Leon L, Bacallao K, Gabler F, Romero C, Valladares L, Vega M. Activities of steroid metabolic enzymes in secretory endometria from untreated women with polycystic ovary syndrome. Steroids. 2008;73:88–95. doi: 10.1016/j.steroids.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 66.Bacallao K, Leon L, Gabler F, Soto E, Romero C, Valladares L, Vega M. In situ estrogen metabolism in proliferative endometria from untreated women with polycystic ovarian syndrome with and without endometrial hyperplasia. J Steroid Biochem Mol Biol. 2008;110:163–169. doi: 10.1016/j.jsbmb.2008.03.031. [DOI] [PubMed] [Google Scholar]
- 67.Delvoux B, Groothuis P, D'Hooghe T, Kyama C, Dunselman G, Romano A. Increased production of 17beta-estradiol in endometriosis lesions is the result of impaired metabolism. J Clin Endocrinol Metab. 2009;94:876–883. doi: 10.1210/jc.2008-2218. [DOI] [PubMed] [Google Scholar]
- 68.Delvoux B, Husen B, Aldenhoff Y, Koole L, Dunselman G, Thole H, Groothuis P. A sensitive HPLC method for the assessment of metabolic conversion of estrogens. J Steroid Biochem Mol Biol. 2007;104:246–251. doi: 10.1016/j.jsbmb.2007.03.006. [DOI] [PubMed] [Google Scholar]