

Abstract
Certain serine proteases are considered to be signaling molecules that act through protease-activated receptors (PARs). Our recent studies have implicated PAR1 and PAR4 (thrombin receptors) and PAR2 (trypsin receptor) in human colon cancer growth. Here we analyzed the expression of KLK4, a member of the kallikrein-related peptidase (KLK) family of serine proteases and explored whether this member can activate PAR1 and PAR2 in human colon cancer cells. Immunohistochemistry showed KLK4 expression in human colon adenocarcinomas and its absence in normal epithelia. KLK4 (1 μmol/L) initiated loss of PAR1 and PAR2 from the HT29 cell surface as well as increased intracellular calcium transients in HT29 cells. This KLK4-induced Ca2+ flux was abrogated after an initial challenge of the cells with TRAP (SFLLR-NH2; 100 μmol/L), which is known to desensitize PAR1 and PAR2. Interestingly, PAR1 blocking antibody, which inhibits cleavage and activation by thrombin, dramatically reduced KLK4-induced Ca2+ influx, but blocking cleavage of PAR2 failed to attenuate the KLK4-induced Ca2+ flux. Consistently, desensitization with AP1 (TFFLR-NH2), targeting PAR1, attenuated most of the Ca2+ flux induced by KLK4. KLK4 also induced a rapid and significant ERK1/2 phosphorylation in HT29 cells. Our results demonstrate, for the first time, that KLK4 is aberrantly expressed in colon cancer and capable of inducing PAR1 signaling in cancer cells. These data suggest that KLK4 signaling via PAR1 may represent a novel pathway in colon tumorigenesis.
The progression from normal colonic mucosa to malignant tumor is a multistep process involving genetic alterations in oncogenes and/or tumor suppressor genes that lead to aberrant growth.1,2 Proteases have long been associated with colon cancer progression because of their ability to degrade extracellular matrices, which facilitates invasion and metastasis.3 However, recent studies have shown that these enzymes target a diversity of substrates and promote some steps of tumor development.4 The traditional view of the role of proteases in tumor growth and progression has therefore significantly changed recently. Besides their contribution to cancer progression by degrading extracellular matrix proteins, it is now clear that a subclass of proteases serve as signaling molecules controlling cell functions through specific membrane receptors, the protease-activated receptors (PARs).5,6 PARs are seven transmembrane-spanning G protein–coupled receptors that are targeted by a variety of serine proteases and also by enzymes from other protease families including matrix metalloprotease-1.6–8 To date four members of the family have been described: PAR1 to PAR4.6,9 Originally, PAR1, PAR3, and PAR4 were described to be activated only by thrombin, whereas PAR2 is activated by trypsin and mast cell tryptase but not by thrombin. Cleavage of PARs by activating serine proteases exposes a new amino terminus that functions as a tethered ligand. This new ligand binds to the core of the receptor and initiates signal transduction resulting in stimulation of phosphoinositide breakdown and cytosolic calcium mobilization.5,6 Short synthetic peptides (activating peptides or APs) corresponding to the newly exposed amino terminus are able to selectively activate a given PAR receptor and mimic cellular effects of the protease.5,6,10
We have previously demonstrated that trypsin acts through PAR2 as a very robust growth factor for human colon cancer cells.11 Moreover, we have shown that the thrombin receptors PAR1 and PAR4 are aberrantly expressed in human colon cancer cells in vivo and in vitro and their activation induces cell proliferation and motility in human colon cancer cells.12,13 The mechanism by which PARs control proliferation involves a series of events that lead to the activation of ERK1/2.14,15 These data support the idea that PARs as well as their activators may be considered as important contributors to the development of human colon cancer. Since their initial characterization, many PAR-activating serine proteases have been reported, including cathepsin G, epithelial trypsin IV, and mesotrypsin.6,9,16–18 The discovery of novel PAR activators has generated additional interest in the possible role of PAR receptors in cancer biology. However, the physiological activators of PARs in colonic tumors have not been characterized yet. Very recently, some members of the tumor-associated serine protease family of kallikrein-related peptidases (KLK) have been shown to function as PAR activators in vitro and in vivo.19 These findings have been partially confirmed in vitro by others who demonstrated the ability of KLK5 and KLK14 to activate PAR2 by analyzing calcium mobilization in cells stably transfected with PAR2,20 whereas in prostate cancer cells, KLK4 and KLK2 were found to initiate cell signaling.21,22
The KLK superfamily consists of fifteen (KLK1–KLK15) trypsin- or chymotrypsin-like serine proteinases.23 KLK proteases, which are secreted into the extracellular space of a wide range of tissues, are involved in a number of physiological processes24 and malignancies including tumor establishment, growth, and/or metastasis.25 Recently, we have structurally and biochemically characterized a series of KLK proteases including KLK4.26 KLK4 is a trypsin-like serine protease displaying arginine/lysine-specific protease activity27 similar to the known PAR activators.6 In this context, the purpose of this study was to determine KLK4 expression in colon cancer tumors in vivo and to examine its ability to modulate PAR activity and signaling in HT29 colon cancer cells in vitro. Our results demonstrate for the first time the aberrant expression of KLK4 in colonic tumors and show its ability to induce PAR1 signaling that promoted ERK1/2-MAP kinase activation in colon cancer cells. These results provide a link between the increased expression of KLK4 and the ectopic expression of PAR1 in colon cancer. Thus, we can speculate that KLK4 is a potential endogenous activator of PAR1.
Materials and Methods
Reagents
The reagents were obtained from the following sources: the activating peptides TFLLR-NH2 (AP1), and SLIGKV-NH2 (AP2) or SFLLRN-NH2 (TRAP, thrombin receptor agonist peptide that activates both PAR1 and PAR2), NeoMPS (Strasbourg, France); 2- furoyl-LIGRLO-NH2 (gift from Dr Hollenberg, Calgary, Canada); highly purified α-thrombin (3000 U/mg), Kordia Laboratory Supplies (Leiden, Netherlands); trypsin (16,000 U/mg), goat anti-mouse IgG coupled to fluorescein, Sigma Chemical Co. (St Louis, MO); Fura-2-AM, Molecular Probes (Leiden, Netherlands). Antibodies were purchased from the following vendors: phospho-specific antibodies to ERK1/2, Cell Signaling Technologies (Beverly, MA); polyclonal anti-ERK1/2 antibodies, Santa Cruz Biotechnology (Santa Cruz, CA); WEDE-15 and SPAN-12 monoclonal antibodies,28 Beckman Coulter (Villepinte, France). All other chemicals were purchased from Interchim (Asnière, France).
Cell Culture
The human colon cancer cell line HT29 was obtained from the American Type Culture Collection (Rockville, MD). Cells underwent routine culture in 25-cm2 plastic flasks (Costar, Cambridge, MA) and were maintained at 37°C in a humidified atmosphere of 5% CO2/air in Dulbecco's modified Eagle's medium, Invitrogen (Cergy Pontoise, France) containing 4.5 g glucose/L, supplemented with 10% fetal calf serum.
Recombinant Kallikrein-Related Peptidase 4
Recombinant KLK4 was expressed in E. coli and purified as described previously.27 Briefly, the bacterial expression plasmid pQE-30 (Qiagen, Hilden, Germany) encodes a synthetic pro-form of KLK protease with an N-terminally located histidine-tag followed by an enterokinase cleavage site. Bacterial cell pellets were solubilized in 6 mol/L guanidinium hydrochlorid, 100 mmol/L NaH2PO4, 8 mmol/L 2-mercaptoethanol, 10 mmol/L Tris/HCl, pH 8.0 and the (nonglycosylated) recombinant protein was purified via its histidine-tag by using nickel-nitrilotriacetic acid agarose affinity chromatography (Qiagen). KLK4 was eluted with 8 mol/L urea, 100 mmol/L NaH2PO4, 8 mmol/L 2-mercaptoethanol, 10 mmol/L Tris/HCl, pH 4.0, after washing by stepwise lowering of the pH from 8.0 to 4.5. The protein was incubated with 10 mmol/L DTT overnight at 25°C and then dialyzed against the 100-fold volume 4 mol/L urea, 50 mmol/L of Tris/HCl, pH 8.0, 100 mmol/L NaCl, and 0.005% Tween-20 for 12 hours. Refolding of KLK4 was performed in 2 mol/L urea, 50 mmol/L Tris/HCl, pH 8.0, 100 mmol/L NaCl, 5 mmol/L reduced glutathione, 0.5 mmol/L oxidized glutathione (Sigma; GSH:GSSG ratio 10:1), 2 mmol/L CaCl2, 0.002% NaN3, 0.005% Tween-20, pH 8.0 in the 100-fold volume of the sample at 4°C for 40 hours and subsequently in the 100-fold sample volume of the same buffer containing 1 mol/L urea, at 4°C for 24 hours. The refolding buffer was twice exchanged with 100 mmol/L NaCl, 50 mmol/L Tris/HCl, 0.002% NaN3 at pH 8.0, incubated for 24 hours each. After refolding, soluble KLK4 was incubated with porcine enterokinase (Genscript, Piscataway, NJ) resulting in removal of the N-terminal histidine tag along with the enterokinase site. Enterokinase was removed using an immobilized antibody (Sigma) and active KLK4 was purified by benzamidine-sepharose affinity chromatography (Amersham Biosciences, Little Chalfont, UK). KLK4 activity was checked against the synthetic substrate Boc-Val-Pro-Arg-AMC (I-1120; Bachem, Germany Bubendorf, Switzerland) and against the macromolecular substrate pro-urokinase (Grünenthal, Aachen, Germany) as previously described.29
Generation of Polyclonal Antibodies Directed to KLK4
Hens were immunized intramuscularly (pectoral muscle) with 20 μg of the recombinant pro-enzyme form of KLK4 (see above) per injection following the protocol by McKiernan et al.30 Antibodies were isolated from egg yolk using a standard step precipitation procedure using increasing concentrations of polyethylene glycol (PEG precipitation). This procedure yielded approximately 95% pure IgY (avian analogue of IgG; SDS–PAGE). Monospecific antibodies were further affinity purified using peptide affinity chromatography using a peptide derived from a flexible surface-associated loop that is specific of KLK4 (aa 109 to 122 according to the chymotrypsin numbering; for an alignment see 31). The region selected is not conserved among members of the KLK family. Purified monospecific polyclonal antibodies (named pAb 617A) were then characterized using ‘one-side ELISA’ methodology in which the immunogen or irrelevant proteins are coupled to the microtiter well (Seiz et al, submitted for publication). Specificity of the antibodies was further verified by Western blot analysis, which demonstrated reactivity toward recombinant pro-KLK4, but no reactivity toward related recombinant pro-enzyme forms of closely related kallikrein-related peptidases, namely KLK2, KLK3, as well as KLK5 to KLK15. As a control for the antibody specificity we analyzed KLK4 expression using immunohistochemistry analysis on a panel of normal and pathological human tissue sections. We found that KLK4 expression pattern (Seiz et al, submitted for publication) is consistent with the pattern reported by others.24,32
Immunohistochemistry
Immunohistochemistry was performed on archival formalin-fixed paraffin-embedded tissue samples from normal colonic mucosa and colonic adenocarcinomas (Pathology Department of Bichat-Claude Bernard Hospital, Paris). Tissues were used in accordance with the requirement of the Human Research Committee of the Bichat-Claude Bernard Hospital and according to the French bioethical law.33 After deparaffinization by xylene and rehydration in serial ethanol dilutions, tissue sections underwent heat-induced antigen retrieval by pressure cooking for 4 minutes in 0.01 mol/L citrate buffer (pH 6.0). Subsequently, endogenous peroxidase was blocked applying 0.5% H2O2 for 10 minutes at room temperature. Sections were then incubated overnight at 4°C with chicken pAb 617A (1:700 dilution), before a rabbit anti-chicken IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) was applied for 30 minutes. In identically treated control sections, primary antibody was either omitted or replaced by green antibody diluent (Dako, Hamburg, Germany) or neutralized with a 20-fold excess of the immunogen. Specific binding of the antibody was detected using the EnVision+ peroxidase polymer system with 3,3′-diaminobenzidine as chromogen (Dako). Sections were counterstained with Mayer's hemalum and coverslipped with Assistant Histokitt.
Immunofluorescence Staining
PAR1 and PAR2 immunofluorescence detection was performed on HT29 cells grown on glass coverslips. The monoclonal PAR2 antibodies mAb 13-8 is directed against 24 aa in the extracellular N-terminal region of PAR2. The amino acid identity between PAR2 and PAR1 in the 24-aa stretch is 17% and is even less for PAR3 and PAR4. Thus it is highly unlikely that mAb 13-8 will recognize any of the other PAR family members. The mAb 13-8 specificity has been tested by flow cytometric analysis on CHO cells stably transfected with human PAR2 compared with parental CHO cells. Moreover, mAb 13-8 blocks specifically trypsin mediated calcium mobilization, whereas thrombin-mediated calcium mobilization is unaffected by the PAR2 antibody (Virca et al, unpublished data).
We also performed immunofluorescence studies after incubation of HT29 cells for various times (5, 10, 15, 30, and 60 minutes) at 37°C with either thrombin (0.01 μmol/L), trypsin (0.01 μmol/L), or KLK4 (1 μmol/L). Cells were washed three times in PBS before being fixed in 2% paraformaldehyde, washed three times in PBS, and then incubated with PBS containing 2% BSA for 15 minutes before application of the primary anti-PAR1 (WEDE-15) or anti-PAR2 (mAb 13-8) antibodies for 2 hours at room temperature at a 1:100 and 1:200 dilution, respectively. Cells were washed in PBS containing 1% BSA and secondary antibody, goat anti-mouse IgG coupled to fluorescein, was applied for 45 minutes at room temperature. The cells were washed again in PBS containing 1% BSA, and finally washed in PBS. Negative controls were obtained by omitting primary antibodies. The cells were then mounted in Vectashield medium (Vector, Peterborough, UK). Images were examined under a fluorescence microscope (Leica DM IRB; magnification ×630).
Intracellular Calcium Measurement
Intracellular calcium concentration was measured using Fura-2/AM. HT29 cells were seeded onto the center of glass coverslips and cultured in Dulbecco's modified Eagle's medium to 80% confluence. Coverslips were then loaded with 5 μmol/L of Fura-2/AM in Na-Hepes-buffered saline (saline pH 7.4, containing 135 mmol/L NaCl, 4.6 mmol/L KCl, 1.2 mmol/L MgCl2, 11 mmol/L HEPES, 11 mmol/L glucose with or without 1.5 mmol/L CaCl2) containing 0.01% pluronic acid for 45 to 60 minutes at 37°C. They were then washed in Na-HEPES buffer and placed at 37°C in a fluorimeter. Cells were treated with KLK4, thrombin, or human trypsin or the PARs agonists peptides TRAP (SFLLR-NH2) and AP1 (TFFLR-NH2), and changes in intracellular Ca2+ were monitored. Fluorescence was measured using a dual-wavelength excitation fluorimeter at 340 and 380 nm for excitation and 510 nm for emission.
Western Blot Analysis
For ERK phosphorylation assays, cells were grown in six-cluster wells (Costar) to 70% confluence and serum-deprived for 48 hours. Quiescent cells were treated with test substances for various time periods as indicated. Cells were lysed with RIPA buffer (1× PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) containing protease inhibitor cocktail (Sigma) and 1 mmol/L sodium orthovanadate for 30 minutes at 4°C, and lysates were centrifuged at 12,000g for 15 minutes. Equal amounts of extracts (50 μg) were separated by SDS–PAGE and transferred onto nitrocellulose membrane. Membranes were incubated in blocking Tris-buffered saline buffer (20 mol/L Tris, 50 mol/L NaCl) containing 5% (w/v) low-fat milk and 0.1% (v/v) Tween 20 and then probed with phospho-specific antibodies to ERK1/2 (1:2000) overnight at 4°C. Subsequently, blots were washed and incubated with the anti–IgG-peroxidase–linked secondary antibody for 1 hour at room temperature before detection by use of a chemiluminescent detection kit (NEN Life Science, Paris, France) and exposure to X-ray film. Membranes were reprobed with a polyclonal anti-ERK1/2 antibody (1:1000) that recognizes total ERK1/2 regardless of its phosphorylation state and served as loading control.
Results
KLK4 Is Expressed in Colon Cancer Tumors in Vivo
We analyzed KLK4 expression in human colonic adenocarcinomas versus normal colonic epithelium by immunohistochemistry using p617A, the KLK4-specific polyclonal antibody (for details see Materials and Methods section). A discrete immunoreactivity is detected in the “normal-appearing” mucosa, from patients with colon cancer, removed far from the neoplastic tissue (Figure 1A). Similarly, in normal colonic samples from control patients, almost no staining was observed (not shown). KLK4 expression is clearly seen in the mild dysplastic mucosa contiguous to a cancerous lesion (Figure 1B), and expression increases in intensity as the dysplasia progresses (data not shown). Staining is localized in the columnar absorptive cells and in goblet cells; however, the intensity of labeling varies among the different patient's adenocarcinomas analyzed as shown in (Figure 1, C–E). KLK4 is observed mainly in the cytoplasm of cancer cells. No specific staining was seen in negative control sections where the primary antibody was omitted (not shown) or preabsorbed with 20-fold excess of recombinant KLK4 (Figure 1F). These observations show that human colonic adenocarcinomas aberrantly express higher levels of KLK4 than normal mucosa and suggest that the also upregulated PARs11–13 may represent potential substrates for KLK4 in this pathology.
Figure 1.
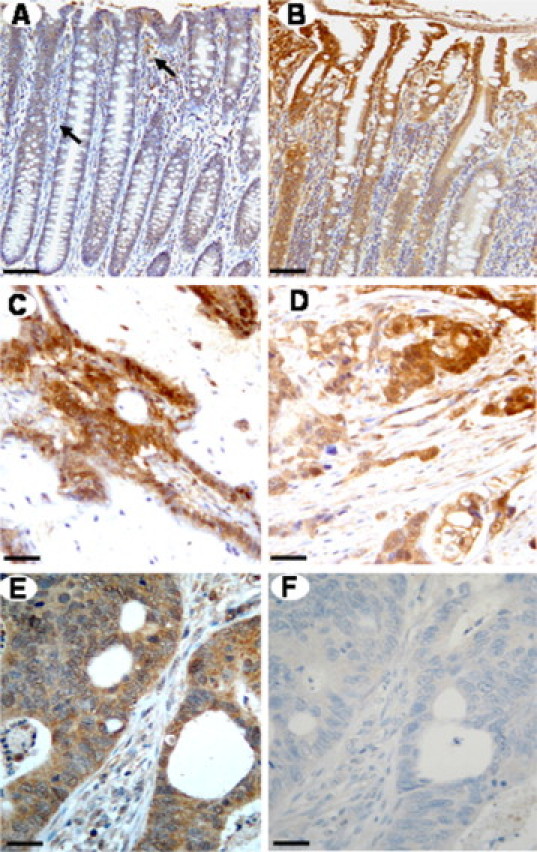
Representative immunostaining of the KLK4 in paraffin sections of normal-appearing colonic mucosa and colonic tissues from patients with adenocarcinoma. A: Faint epithelial KLK4 immunoreactivity in “normal-appearing” mucosa removed far from the cancerous lesion. Positive KLK4 staining is also seen in the lamina propria and in a few macrophages (arrow). B: Immunostaining for KLK4 in the dysplastic colonic mucosa close to the cancerous lesion seen in D. C–E: Moderate to strong positive immunoreactivity in the adenocarcinomas of three different patients. F: In the same carcinoma as in E, no immunoreactivity is detected when the primary antibody is preadsorbed with 20-fold excess of recombinant KLK4. Scale bar = 100 μm (A and B), 200 μm (C and D), or 400 μm (E and F).
KLK4 Induces Loss of PAR1 and PAR2 from the Cell Surface of Colon Cancer–Derived Cell Line HT29
Because PAR4 is expressed at very low levels in colon cancer cell lines in general and specifically in HT29 cells,13 only PAR1 and PAR2 were analyzed in this study. By using immunofluorescence microscopy with antibodies directed against the N-terminal domains of either PAR1 and PAR2, we examined the loss of PAR1 and PAR2 immunoreactivity at the cell surface of KLK4-treated cells, a finding that may reflect receptor internalization.34 As shown in Figure 2, PAR1 and PAR2, which are expressed on HT29 cells,11,12 were readily detected at the plasma membrane of unstimulated HT29 cells. Stimulation of HT29 cells with KLK4 (1 μmol/L) for 5 minutes resulted in a decrease of PAR1 and PAR2 immunoreactivity at the cell surface (Figure 2). Longer stimulations of the cells with thrombin or KLK4 resulted in an increased loss of PAR1 receptor immunoreactivity from the cell surface, with nearly complete loss at 30 to 60 minutes (not shown). In controls and as expected, thrombin and trypsin treatment also caused PAR1 and PAR2 internalization, respectively (Figure 2).5,34 Interestingly, immunostaining of PAR1 was not significantly affected by trypsin treatment (Figure 2). Similarly, thrombin treatment did not affect PAR2 staining. These data indicate that KLK4 mediates both PAR1 and PAR2 N-terminal cleavage and loss of the receptor staining from HT29 cell surface.
Figure 2.
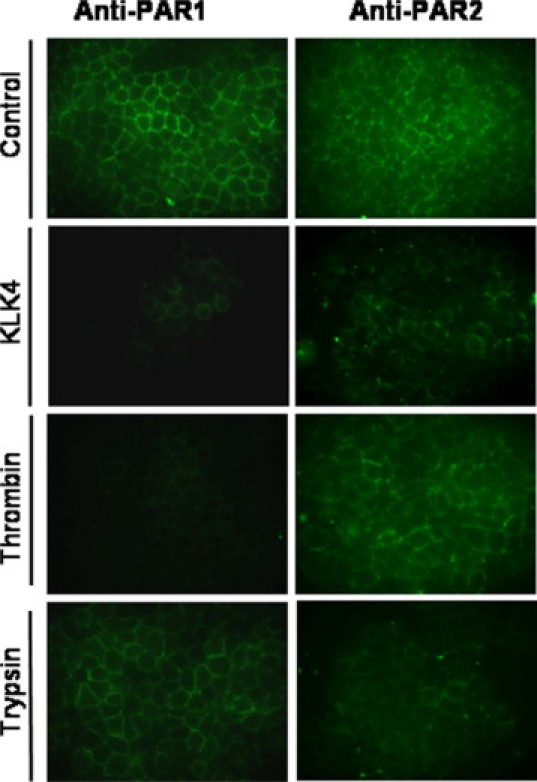
KLK4 induces loss of PAR1 and PAR2 from the surface of colonic cancer HT29 cells. HT29 cells were treated with thrombin (0.01 μmol/L), trypsin (0.01 μmol/L), KLK4 (1 μmol/L; stimulated), or with vehicle (unstimulated) for 5 minutes at 37°C. Cells were fixed using 2% paraformaldehyde and immunostained with PAR1 or PAR2 monoclonal antibodies that recognize epitopes within the N-terminal extracellular domains of PAR1 and PAR2, respectively (×630). Results are representative of two independent experiments.
Calcium Signaling Triggered by KLK4 Is Mediated by PAR1 but Not by PAR2
The immunofluorescence analysis data suggest that the loss of PAR1 and PAR2 seen in KLK4-treated HT29 cells is attributable either to receptor internalization or cleavage upstream or downstream of the activation site, the latter process often referred to as receptor disarming.6 PAR-mediated calcium mobilization requires proteolytic cleavage at a specific activation site.5,6 Therefore, we investigated whether KLK4 can trigger calcium signaling in HT29 cells that constitutively express PAR1 and PAR2.11,12 On cell exposure to 0.5 μmol/L of KLK4, a very small peak of intracellular Ca2+ mobilization was detected (data not shown), whereas, as shown in Figure 3, HT29 cells challenge with 1 μmol/L of KLK4 initiated a significant transient Ca2+ mobilization (Figure 3A). Positive control agonists for PAR1 (thrombin) and PAR2 (trypsin) each induced changes in Ca2+ mobilization in HT29 cells (Figure 3, B and C), confirming that the receptors (PAR1 and PAR2) expressed on HT29 are functional. As a control, no transient Ca2+ mobilization was seen when heat-inactivated KLK4 was added to HT29 cells (data not shown).
Figure 3.
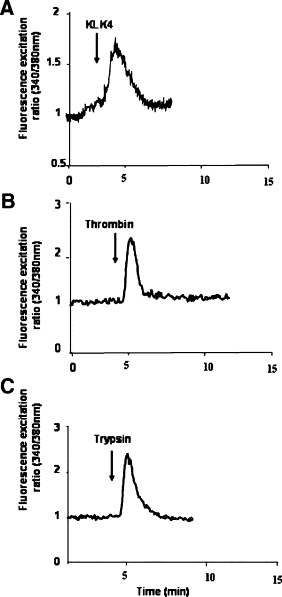
KLK4 induces calcium mobilization in HT29 cells. HT29 cells were loaded for 60 minutes at 37°C using Fura-2/AM. Cells were challenged with (A) KLK4 (1 μmol/L), (B) thrombin (0.01 μmol/L), or (C) trypsin (0.01 μmol/L). Addition of the enzymes is indicated by arrows. These results are representative of three others.
The specificity of the response via PAR1 and for PAR2 was demonstrated by cross desensitization studies using a specific agonist peptide.10,35 As shown in Figure 4A, a challenge of the cells with TRAP (SFLLR-NH2; 100 μmol/L), which is known to desensitize PAR1 and PAR2 but not other PARs,10 abrogated subsequent KLK4-induced Ca2+ flux. Similarly, thrombin and trypsin, the known agonists of PAR1 and PAR2, respectively, had no effect on calcium mobilization after an initial challenge with TRAP (SFLLR-NH2; Figure 4, B and C). To check the integrity of the cells, carbachol (50 μmol/L)36 was subsequently added of to the same cells and gave the expected response (Figure 4, A–C). These experiments suggest that KLK4 signals mainly via PAR1 and/or PAR2.
Figure 4.
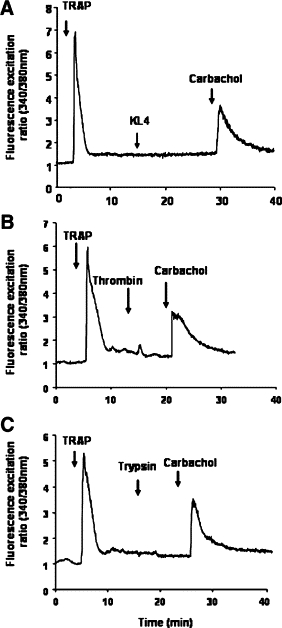
KLK4 signals via PARs in HT29 cells. HT29 cells were loaded for 60 minutes at 37°C using Fura-2/AM. A: Cells were challenged by addition of TRAP (SFLLR-NH2; 100 μmol/L) followed by a second challenge with KLK4 (1 μmol/L). B: The cells were challenged first with the activating peptide TRAP (SFLLR-NH2; 100 μmol/L) followed by a second challenge with thrombin (0.01 μmol/L). C: The cells were challenged first with the activating peptide TRAP (SFLLR-NH2; 100 μmol/L) followed by a second challenge with trypsin (0.01 μmol/L). Note that cells are still responsive to a third challenge with carbachol (50 μmol/L; A–C). All of the compounds were given at the arrows. These results are representative of two others.
To determine which PAR is activated by KLK4, we used monoclonal antibodies directed against the sequence spanning the cleavage sites of PAR1 (SPAN-12) or PAR2 (mAb 13-8) (Virca et al, unpublished results, and this study). As shown in Figure 5A, the SPAN-12 antibody efficiently blocked PAR1 cleavage by thrombin, as very little, if any, Ca2+ influx was detected. Interestingly, KLK4–induced Ca2+ mobilization was also abrogated, whereas trypsin-mediated responses were not significantly affected (Figure 5A). On the other hand, blocking cleavage of PAR2 with the monoclonal antibody antagonist mAb 13-8 completely abrogated trypsin-induced Ca2+ influx, and was without significant effect on the KLK4-induced Ca2+ flux (Figure 5B). Consistent with these findings, in the presence of PAR2 blocking antibody, KLK4 induced Ca2+ mobilization, and subsequent incubation with thrombin led to a dramatically reduced signal indicating that KLK4 desensitized most of PAR1 in HT29 cells (Figure 5C). The KLK4-induced loss of PAR2 immunostaining at HT29 cell surface seen in Figure 2 may be attributable to PAR2 cleavage at a site upstream of the activation site without any further consequences on the functionality of the receptor. In concordance with these data, after complete desensitization of PAR1 by the PAR1-specific peptide AP1 (TFFLR-NH2; 100 μmol/L) subsequent incubation with KLK4 did not induce any Ca2+ elevation (Figure 5D). As expected, desensitization of PAR2 by the PAR2-specific peptide 2-furoyl-LIGRLO-NH2 did not affect subsequent KLK4-induce Ca2+ elevation (data not shown). As control, addition of carbachol to the same cells in Figure 5 (C and D) induced Ca2+ elevation, showing the absence of intracellular calcium depletion in the cells (data not shown). These results strongly suggest that KLK4 activates preferentially PAR1 and not PAR2 in HT29 cells.
Figure 5.
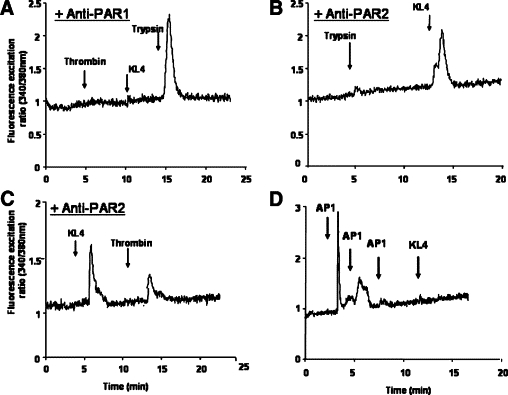
KLK4 initiates changes in intracellular Ca2+ mobilization via PAR1 but not PAR2 in HT29 cells. HT29 cells were preincubated for 2 hours with or without antibodies directed against PAR1 (A) or PAR2 (B and C) activation site and then were loaded with Fura-2/AM for 60 minutes. A: Cells preincubated with anti-PAR1 were first challenged with thrombin (0.01 μmol/L) followed by a second challenge with KLK4 (1 μmol/L) and a third challenge with trypsin (0.01 μmol/L). B: Cells preincubated with anti-PAR2 were challenged first with trypsin (0.01 μmol/L) followed by a second challenge with KLK4 (1 μmol/L). Note that cells are still responsive to KLK4 in the presence of PAR2 blocking antibodies. C: Cells preincubated with anti-PAR2 were challenged with KLK4 (1 μmol/L) and then with thrombin (0.01 μmol/L). D: HT29 cells were challenged first three times with the activating peptide AP1 (TFFLR-NH2; 100 μmol/L) and then with KLK4 (1 μmol/L). Note that AP1 (TFFLR-NH2) abrogated KLK4 response. All of the compounds were given at the arrows. These results are representative of three others.
KLK4 Enhanced ERK1/2 Activation in Human Colon Cancer Cells
Because we have previously shown that PAR1 and PAR2 activation plays a pivotal role in extracellular-regulated kinase (ERK1/2)-induced activity in colon cancer,12,14,15 we next investigated the effect of KLK4 on ERK1/2 phosphorylation. Addition of KLK4 (1 μmol/L) to quiescent HT29 cells for various times induced a rapid and significant phosphorylation of p42/p44, reaching a maximum within 5 to 10 minutes (Figure 6A). KLK4 induces ERK1/2 phosphorylation in the range of concentrations between 0.01 μmol/L and 0.5 μmol/L. Significant ERK1/2 phosphorylation was obtained already with 0.01 μmol/L of KLK4, which is comparable with the signal seen with 0.01 μmol/L of thrombin at 5 minutes (Figure 6B). These experiments suggest that KLK4 signals via PAR1 to activate the MAP kinase pathway in colon cancer cells and possibly playing an important role in colon tumorigenesis.
Figure 6.
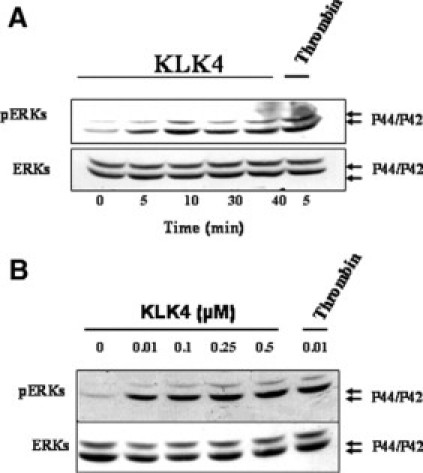
KLK4 activates p42/p44 MAPK in HT29 cells. A: Immunoblot with phospho-specific p42/p44 MAPK antibodies on quiescent HT29 cell lysates treated with or without KLK4 (0.5 μmol/L) or thrombin (0.01 μmol/L) for the indicated time periods (shown in minutes). B: Dose-dependent activation of p42/p44 MAP-kinase phosphorylation by KLK4. Quiescent HT29 cells were stimulated with the indicated concentrations of KLK4 or with thrombin (0.01 μmol/L) for 10 minutes. To confirm equal protein loading, the membranes were stripped and incubated with p42/p44 antibodies. Results are representative of two independent experiments.
Discussion
We report here that KLK4, a member of the tumor-associated KLK family, is ectopically expressed in human colon cancer cells. To our knowledge, this is the first evidence of KLK4 expression in colon cancer. In contrast, KLK4 is barely detected in epithelial cells of normal human colon. In the colon cancer–derived cell line HT29, KLK4 appears to promote efficient calcium mobilization through the ectopically expressed receptor PAR1 and subsequent ERK1/2-MAP kinase activation. Therefore, KLK4 could be considered as a potential endogenous PAR activator in colon cancer owing to the aberrant expression of its receptor PAR1.
Our observation of the increased expression of KLK4 in colon cancer tumors relative to normal colonic epithelia parallels with our previous results showing aberrant expression of PAR1 in colonic adenocarcinomas compared with the normal mucosa.12 In fact, KLK4 is coexpressed in tumors that have been shown to express PAR1 (data not shown). The low level of KLK4 in normal colonic tissues is in agreement with two previous reports. First, Shaw and coauthors reported an absence of KLK4 in normal colonic tissue extracts by ELISA.32 Second, Kontos et al reported recently that KLK4 mRNA is upregulated in colon cancer compared with normal colon.37 However, an in silico analysis of kallikrein-related peptidase gene expression in colon cancer did not report the presence of detectable KLK4; this discrepancy with our findings may be attributable to differences in sensitivity between the two detection methods.38 The mechanism whereby the KLK4 gene is switched on in colon cancer is unknown, but this ectopic expression could possibly be under the same gene regulation as in other cancers. Indeed, KLK4 has been reported to be highly expressed in breast carcinoma,25 prostate adenocarcinoma,39 and ovarian adenocarcinoma.40 Specific CpG dinucleotides in the proximal promoter have been shown to regulate gene expression of some human KLKs, including KLK4 via DNA methylation.41
Additional studies are required to determine whether endogenously released KLK4 contributes to colon cancer development. KLK4 is synthesized and secreted as a zymogen.23,42 This fact raises the question of how endogenously released KLK4 might contribute to colon tumorigenesis. Although physiological activators of KLK4 have not been reported, in vitro biochemical studies suggest that some KLKs may participate in an enzymatic cascade to activate each other.43 Whether these KLKs also process pro-KLK4 has yet to be demonstrated.
Our data from microscopic analysis clearly showed a loss of both PAR1 and PAR2 from the cell surface of HT29 cells after KLK4 incubation. Further analysis of calcium mobilization in HT29 cells that express both PAR1 and PAR211,12 have yielded important insights into the ability of KLK4 to induce signaling via PAR1 but not PAR2. In enzymatics experiments we performed to test the capacity of this enzyme to cleave peptidic substrates mimicking PAR1 and PAR2, we found that KLK4 cleaves both receptor-derived PAR1 and PAR2 peptides in vitro (data not shown). This is consistent with previously reported observations that cleavage of peptides spanning the activation site of PARs by trypsin or thrombin may not accurately reflect the ability of enzymes to activate and/or disarm their respective receptors on intact cells44 and further that individual KLKs can function as both an activator and a disarmer of individual PARs.19
Several lines of evidence indicate that KLK4 signals in HT29 cells only through PAR1 but not PAR2: (i) PAR1 blocking antibodies abrogated completely the effect of KLK4 on calcium transients; (ii) desensitization of PAR1 with AP1 (TFFLR-NH2), which specifically targets PAR1,45 abrogated all KLK4–induced calcium transients; (iii) blocking cleavage of PAR2 with a monoclonal antibody antagonist failed to attenuate the KLK4-induced Ca2+ flux. The observation that PAR1 blocking antibodies abrogated completely the effect of KLK4 on calcium transients whereas the response to trypsin remained intact suggests that KLK4 cleavage of PAR2, in a cellular context, is probably upstream of the activation site.
Our results are consistent with two recent reports showing that KLK4 can signal in prostate cancer cell lines by a PAR-dependent mechanism.21,22 In contrast, in these studies, the authors reported the involvement of both PAR1 and PAR2 in prostate cancer KLK4-induced signaling. These discrepancies could be attributable to differences in protease preparations, levels of receptor expression, or cell type. Indeed, both studies were conducted mostly on murine lung fibroblasts (LMF or KOLF) from PAR1-null mice46,47 that overexpress human PAR1 or PAR221,22 or on cells overexpressing PAR2-GFP.21 Further studies on other cancer cells that naturally express PARs are necessary to clarify these issues. Another possible explanation could be that posttranslational modifications, such as receptor glycosylation, might also affect KLK4 signaling via PAR2. Such phenomena have been reported for other serine proteases such as tryptase-mediated activation of PAR2.48 Differential glycosylation could potentially modify enzyme kinetic parameters or restrict KLK4 from gaining access to the cleavage site of the receptor.
The specificity of KLK4 toward PAR1 could be facilitated by direct interaction with the receptor, which might localize the protease and allosterically modulate its activity toward PAR1. Interestingly, KLK4 shows a negatively charged surface patch, which may represent an exosite for prime-side substrate recognition.26 Such an interaction between the anion-binding exosite of thrombin and an acidic region at the C terminus of PAR1 has been shown to increase thrombin affinity toward the receptor.49
Further research is needed to establish how KLK4 acts specifically on PAR1 to generate a functional ligand and induce signaling. KLK4-induced PAR1 activation resulted in rapid ERK1/2 phosphorylation in HT29 cells. Similarly, we previously showed that PAR1 activation leads to activation of the MAP kinase pathway in colon cancer cells.12,15 Interestingly, we observed that KLK4-induced ERK1/2 phosphorylation was initiated with a lower KLK4 concentration than that needed for induction of calcium mobilization. It is possible that at this concentration, KLK4 activates the MAP-kinase pathway without calcium signaling. Indeed, a recent report by Ramachandran et al50 has shown that PARs can signal through multiple pathways that are differentially triggered by distinct protease-revealed tethered ligands depending on the site cleavage. It is of note that at low concentrations KLK4 could process membrane proteins other than PARs, such as the urokinase-receptor,29 and, thus, potentially, signal via alternative pathways. On the other hand, PAR-dependent ERK1/2 activation by KLK4 might also be initiated via a calcium independent pathway leading to activation of receptor tyrosine kinases signaling.15 The mechanism by which KLK4 induces PAR1 signaling in colon cancer is not known, but it is possible that activation of PAR1 by KLK4 couples with different effectors and elicits distinct cellular responses than that induced by thrombin. Such mechanism has been recently reported for activated protein C by Russo et al who revealed that compartmentalization of PAR1 in caveolae is critical for protease-selective signaling.51,52
Whether KLK4 signaling through the aberrantly expressed PAR1 in colon cancer can directly influence tumor development in vivo clearly deserves further investigations. However, concomitant upregulation of KLK4 and PAR1 in colonic tumors would suggest that KLK4-mediated PAR1 activation could play an important role in colon tumorigenesis.
Acknowledgements
We are very grateful to Dr. Francine Walker (Pathology Department of Bichat Hospital, Paris) for providing the specimens for these studies. We thank Dr. Thérèse Lehy for the helpful discussions, and Markus Magdolen, Sandra Baur, Samira Benadda, and Jeff Tracy for technical assistance.
Footnotes
Supported by the Association pour la Recherche sur le Cancer (Contract: N°3937) and Institut National de la Santé et de la Recherche Médicale (INSERM), the Kommission für Klinische Forschung (KKF) of the TU Munich, and by a travel grant from the Bayerisch-Französisches Hochschulzentrum (BFHZ/CCUFB).
References
- 1.Chung DC. The genetic basis of colorectal cancer: insights into critical pathways of tumorigenesis. Gastroenterology. 2000;119:854–865. doi: 10.1053/gast.2000.16507. [DOI] [PubMed] [Google Scholar]
- 2.Wong J, Hawkins N, Ward R. Colorectal cancer: a model for epigenetic tumorigenesis. Gut. 2007;56:140–148. doi: 10.1136/gut.2005.088799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mook OR, Frederiks WM, Van Noorden CJ. The role of gelatinases in colorectal cancer progression and metastasis. Biochim Biophys Acta. 2004;1705:69–89. doi: 10.1016/j.bbcan.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7:800–808. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]
- 5.Dery O, Corvera CU, Steinhoff M, Bunnett NW. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am J Physiol. 1998;274:C1429–C1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- 6.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–282. [PubMed] [Google Scholar]
- 7.Ramsay AJ, Reid JC, Adams MN, Samaratunga H, Dong Y, Clements JA, Hooper JD. Prostatic trypsin-like kallikrein-related peptidases (KLKs) and other prostate-expressed tryptic proteinases as regulators of signalling via proteinase-activated receptors (PARs) Biol Chem. 2008;389:653–668. doi: 10.1515/BC.2008.078. [DOI] [PubMed] [Google Scholar]
- 8.Hollenberg MD, Oikonomopoulou K, Hansen KK, Saifeddine M, Ramachandran R, Diamandis EP. Kallikreins and proteinase-mediated signaling: proteinase-activated receptors (PARs) and the pathophysiology of inflammatory diseases and cancer. Biol Chem. 2008;389:643–651. doi: 10.1515/BC.2008.077. [DOI] [PubMed] [Google Scholar]
- 9.Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol Rev. 2004;84:579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- 10.Hollenberg MD, Compton SJ. International Union of Pharmacology. XXVIII. Proteinase-Activated Receptors. Pharmacol Rev. 2002;54:203–217. doi: 10.1124/pr.54.2.203. [DOI] [PubMed] [Google Scholar]
- 11.Darmoul D, Marie JC, Devaud H, Gratio V, Laburthe M. Initiation of human colon cancer cell proliferation by trypsin acting at protease-activated receptor-2. Br J Cancer. 2001;85:772–779. doi: 10.1054/bjoc.2001.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darmoul D, Gratio V, Devaud H, Lehy T, Laburthe M. Aberrant expression and activation of the thrombin receptor protease-activated receptor-1 induces cell proliferation and motility in human colon cancer cells. Am J Pathol. 2003;162:1503–1513. doi: 10.1016/S0002-9440(10)64283-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gratio V, Walker F, Lehy T, Laburthe M, Darmoul D. Aberrant expression of proteinase-activated receptor 4 promotes colon cancer cell proliferation through a persistent signaling that involves Src and ErbB-2 kinase. Int J Cancer. 2009;124:1517–1525. doi: 10.1002/ijc.24070. [DOI] [PubMed] [Google Scholar]
- 14.Darmoul D, Gratio V, Devaud H, Laburthe M. Protease-activated receptor 2 in colon cancer: trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J Biol Chem. 2004;279:20927–20934. doi: 10.1074/jbc.M401430200. [DOI] [PubMed] [Google Scholar]
- 15.Darmoul D, Gratio V, Devaud H, Peiretti F, Laburthe M. Activation of proteinase-activated receptor 1 promotes human colon cancer cell proliferation through epidermal growth factor receptor transactivation. Mol Cancer Res. 2004;2:514–522. [PubMed] [Google Scholar]
- 16.Ramachandran R, Hollenberg MD. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br J Pharmacol. 2008;153:S263–S282. doi: 10.1038/sj.bjp.0707507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knecht W, Cottrell GS, Amadesi S, Mohlin J, Skaregarde A, Gedda K, Peterson A, Chapman K, Hollenberg MD, Vergnolle N, Bunnett NW. Trypsin IV or mesotrypsin and p23 cleave protease-activated receptors 1 and 2 to induce inflammation and hyperalgesia. J Biol Chem. 2007;282:26089–26100. doi: 10.1074/jbc.M703840200. [DOI] [PubMed] [Google Scholar]
- 18.Cottrell GS, Amadesi S, Grady EF, Bunnett NW. Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J Biol Chem. 2004;279:13532–13539. doi: 10.1074/jbc.M312090200. [DOI] [PubMed] [Google Scholar]
- 19.Oikonomopoulou K, Hansen KK, Saifeddine M, Vergnolle N, Tea I, Diamandis EP, Hollenberg MD. Proteinase-mediated cell signalling: targeting proteinase-activated receptors (PARs) by kallikreins and more. Biol Chem. 2006;387:677–685. doi: 10.1515/BC.2006.086. [DOI] [PubMed] [Google Scholar]
- 20.Stefansson K, Brattsand M, Roosterman D, Kempkes C, Bocheva G, Steinhoff M, Egelrud T. Activation of proteinase-activated receptor-2 by human kallikrein-related peptidases. J Invest Dermatol. 2008;128:18–25. doi: 10.1038/sj.jid.5700965. [DOI] [PubMed] [Google Scholar]
- 21.Ramsay AJ, Dong Y, Hunt ML, Linn M, Samaratunga H, Clements JA, Hooper JD. Kallikrein-related peptidase 4 (KLK4) initiates intracellular signaling via protease-activated receptors (PARs). KLK4 and PAR-2 are co-expressed during prostate cancer progression. J Biol Chem. 2008;283:12293–12304. doi: 10.1074/jbc.M709493200. [DOI] [PubMed] [Google Scholar]
- 22.Mize GJ, Wang W, Takayama TK. Prostate-specific kallikreins-2 and -4 enhance the proliferation of DU-145 prostate cancer cells through protease-activated receptors-1 and -2. Mol Cancer Res. 2008;6:1043–1051. doi: 10.1158/1541-7786.MCR-08-0096. [DOI] [PubMed] [Google Scholar]
- 23.Borgono CA, Michael IP, Diamandis EP. Human tissue kallikreins: physiologic roles and applications in cancer. Mol Cancer Res. 2004;2:257–280. [PubMed] [Google Scholar]
- 24.Obiezu CV, Shan SJ, Soosaipillai A, Luo LY, Grass L, Sotiropoulou G, Petraki CD, Papanastasiou PA, Levesque MA, Diamandis EP. Human kallikrein 4: quantitative study in tissues and evidence for its secretion into biological fluids. Clin Chem. 2005;51:1432–1442. doi: 10.1373/clinchem.2005.049692. [DOI] [PubMed] [Google Scholar]
- 25.Borgono CA, Diamandis EP. The emerging roles of human tissue kallikreins in cancer. Nat Rev Cancer. 2004;4:876–890. doi: 10.1038/nrc1474. [DOI] [PubMed] [Google Scholar]
- 26.Debela M, Beaufort N, Magdolen V, Schechter NM, Craik CS, Schmitt M, Bode W, Goettig P. Structures and specificity of the human kallikrein-related peptidases KLK 4, 5, 6, and 7. Biol Chem. 2008;389:623–632. doi: 10.1515/BC.2008.075. [DOI] [PubMed] [Google Scholar]
- 27.Debela M, Magdolen V, Schechter N, Valachova M, Lottspeich F, Craik CS, Choe Y, Bode W, Goettig P. Specificity profiling of seven human tissue kallikreins reveals individual subsite preferences. J Biol Chem. 2006;281:25678–25688. doi: 10.1074/jbc.M602372200. [DOI] [PubMed] [Google Scholar]
- 28.Brass LF, Vassallo RR, Jr, Belmonte E, Ahuja M, Cichowski K, Hoxie JA. Structure and function of the human platelet thrombin receptor. Studies using monoclonal antibodies directed against a defined domain within the receptor N terminus. J Biol Chem. 1992;267:13795–13798. [PubMed] [Google Scholar]
- 29.Beaufort N, Debela M, Creutzburg S, Kellermann J, Bode W, Schmitt M, Pidard D, Magdolen V. Interplay of human tissue kallikrein 4 (hK4) with the plasminogen activation system: hK4 regulates the structure and functions of the urokinase-type plasminogen activator receptor (uPAR) Biol Chem. 2006;387:217–222. doi: 10.1515/BC.2006.029. [DOI] [PubMed] [Google Scholar]
- 30.McKiernan E, O'Brien K, Grebenchtchikov N, Geurts-Moespot A, Sieuwerts AM, Martens JW, Magdolen V, Evoy D, McDermott E, Crown J, Sweep FC, Duffy MJ. Protein kinase Cdelta expression in breast cancer as measured by real-time PCR, western blotting and ELISA. Br J Cancer. 2008;99:1644–1650. doi: 10.1038/sj.bjc.6604728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaw JL, Diamandis EP. Distribution of 15 human kallikreins in tissues and biological fluids. Clin Chem. 2007;53:1423–1432. doi: 10.1373/clinchem.2007.088104. [DOI] [PubMed] [Google Scholar]
- 33.French bioethics law, no. 2004-800. August 6, 2004, Art. L. 1232-1, Art. L. 1235-2, Art. L. 1245-2
- 34.Trejo J. Protease-activated receptors: new concepts in regulation of G protein-coupled receptor signaling and trafficking. J Pharmacol Exp Ther. 2003;307:437–442. doi: 10.1124/jpet.103.052100. [DOI] [PubMed] [Google Scholar]
- 35.Hollenberg MD, Saifeddine M, al-Ani B, Kawabata A. Proteinase-activated receptors: structural requirements for activity, receptor cross-reactivity, and receptor selectivity of receptor-activating peptides. Can J Physiol Pharmacol. 1997;75:832–841. [PubMed] [Google Scholar]
- 36.Nitschke R, Leipziger J, Greger R. Agonist-induced intracellular Ca2+ transients in HT29 cells. Pflugers Arch. 1993;423:519–526. doi: 10.1007/BF00374950. [DOI] [PubMed] [Google Scholar]
- 37.Kontos C, Papadopoulos I, Scorilas A. In: Quantitative expression analysis and prognostic significance of KLK4 mRNA expression in colon cancer. Tissue Kallikrein and Kallikrein-Related Peptidase: Biochemistry, Structure, Function, and (Patho)physiology. Schmitt MFH, Sommerhoff C, Magdolen V, editors. Hieronymus Druck und Verlag; Munich: 2009. p. 86. [Google Scholar]
- 38.Yousef GM, Borgono CA, Popalis C, Yacoub GM, Polymeris ME, Soosaipillai A, Diamandis EP. In-silico analysis of kallikrein gene expression in pancreatic and colon cancers. Anticancer Res. 2004;24:43–51. [PubMed] [Google Scholar]
- 39.Obiezu CV, Soosaipillai A, Jung K, Stephan C, Scorilas A, Howarth DH, Diamandis EP. Detection of human kallikrein 4 in healthy and cancerous prostatic tissues by immunofluorometry and immunohistochemistry. Clin Chem. 2002;48:1232–1240. [PubMed] [Google Scholar]
- 40.Obiezu CV, Scorilas A, Katsaros D, Massobrio M, Yousef GM, Fracchioli S, Rigault de la Longrais IA, Arisio R, Diamandis EP. Higher human kallikrein gene 4 (KLK4) expression indicates poor prognosis of ovarian cancer patients. Clin Cancer Res. 2001;7:2380–2386. [PubMed] [Google Scholar]
- 41.Pampalakis G, Diamandis EP, Sotiropoulou G. The epigenetic basis for the aberrant expression of kallikreins in human cancers. Biol Chem. 2006;387:795–799. doi: 10.1515/BC.2006.100. [DOI] [PubMed] [Google Scholar]
- 42.Takayama TK, McMullen BA, Nelson PS, Matsumura M, Fujikawa K. Characterization of hK4 (prostase), a prostate-specific serine protease: activation of the precursor of prostate specific antigen (pro-PSA) and single-chain urokinase-type plasminogen activator and degradation of prostatic acid phosphatase. Biochemistry. 2001;40:15341–15348. doi: 10.1021/bi015775e. [DOI] [PubMed] [Google Scholar]
- 43.Clements JA, Willemsen NM, Myers SA, Dong Y. The tissue kallikrein family of serine proteases: functional roles in human disease and potential as clinical biomarkers. Crit Rev Clin Lab Sci. 2004;41:265–312. doi: 10.1080/10408360490471931. [DOI] [PubMed] [Google Scholar]
- 44.Al-Ani B, Hollenberg MD. Selective tryptic cleavage at the tethered ligand site of the amino terminal domain of proteinase-activated receptor-2 in intact cells. J Pharmacol Exp Ther. 2003;304:1120–1128. doi: 10.1124/jpet.102.043844. [DOI] [PubMed] [Google Scholar]
- 45.Kawabata A, Saifeddine M, Al-Ani B, Leblond L, Hollenberg MD. Evaluation of proteinase-activated receptor-1 (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: activation of PAR2 by PAR1-targeted ligands. J Pharmacol Exp Ther. 1999;288:358–370. [PubMed] [Google Scholar]
- 46.Andrade-Gordon P, Maryanoff BE, Derian CK, Zhang HC, Addo MF, Darrow AL, Eckardt AJ, Hoekstra WJ, McComsey DF, Oksenberg D, Reynolds EE, Santulli RJ, Scarborough RM, Smith CE, White KB. Design, synthesis, and biological characterization of a peptide-mimetic antagonist for a tethered-ligand receptor. Proc Natl Acad Sci USA. 1999;96:12257–12262. doi: 10.1073/pnas.96.22.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trejo J, Connolly AJ, Coughlin SR. The cloned thrombin receptor is necessary and sufficient for activation of mitogen-activated protein kinase and mitogenesis in mouse lung fibroblasts. Loss of responses in fibroblasts from receptor knockout mice. J Biol Chem. 1996;271:21536–21541. doi: 10.1074/jbc.271.35.21536. [DOI] [PubMed] [Google Scholar]
- 48.Compton SJ, Renaux B, Wijesuriya SJ, Hollenberg MD. Glycosylation and the activation of proteinase-activated receptor 2 (PAR(2)) by human mast cell tryptase. Br J Pharmacol. 2001;134:705–718. doi: 10.1038/sj.bjp.0704303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vu TK, Wheaton VI, Hung DT, Charo I, Coughlin SR. Domains specifying thrombin-receptor interaction. Nature. 1991;353:674–677. doi: 10.1038/353674a0. [DOI] [PubMed] [Google Scholar]
- 50.Ramachandran R, Mihara K, Mathur M, Rochdi M, Bouvier M, Defea K, Hollenberg MD. Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol Pharmacol. 2009;76:791–801. doi: 10.1124/mol.109.055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Russo A, Soh UJ, Trejo J. Proteases display biased agonism at protease-activated receptors: location matters! Mol Interv. 2009;9:87–96. doi: 10.1124/mi.9.2.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Russo A, Soh UJ, Paing MM, Arora P, Trejo J. Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc Natl Acad Sci USA. 2009;106:6393–6397. doi: 10.1073/pnas.0810687106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Uncited reference
- 31.Debela M, Magdolen V, Grimminger V, Sommerhoff C, Messerschmidt A, Huber R, Friedrich R, Bode W, Goettig P. Crystal structures of human tissue kallikrein 4: activity modulation by a specific zinc binding site. J Mol Biol. 2006;362:1094–1107. doi: 10.1016/j.jmb.2006.08.003. [DOI] [PubMed] [Google Scholar]