

Abstract
Prostaglandin E2, which is known to contribute to cancer progression, is inactivated by the catabolic enzyme, 15-hydroxyprostaglandin dehydrogenase (PGDH), which has tumor-suppressor activity in lung, colon, breast, and gastric cancers. Therefore, we evaluated the expression of PGDH in human bladder cancer tissue specimens and cell lines. Immunoperoxidase staining of bladder cancer tissues demonstrated that (1) PGDH is highly expressed by normal urothelial cells but (2) reduced in many low stage (Ta/Tis) bladder cancers, and (3) PGDH is completely lost in most invasive bladder cancers. Of eight cancer cell lines tested, only two relatively well-differentiated bladder cancer cell lines, RT4 and UM-UC9, expressed PGDH. Moreover, inhibition of PGDH expression in well-differentiated RT4 cells using small inhibitory RNA or short hairpin RNA resulted in a more aggressive phenotype with increased motility and anchorage-independent growth. Additionally, PGDH knockdown affected prostaglandin E2 signaling as measured by cAMP generation. These data indicate that loss of PGDH expression contributes to a more malignant bladder cancer phenotype and may be necessary for bladder cancer development and/or progression.
Inflammation has long been recognized as linked to cancer development and progression.1–3 Prostaglandins are part of the inflammatory milieu. One type of prostaglandin, prostaglandin E2 (PGE2), has been shown to contribute significantly to carcinogenesis.4,5 Many authors have examined the synthesis of PGE2 by the inducible enzyme, cyclooxygenase 2 (COX2). Indeed, COX2 is up-regulated in many cancers,6 including bladder cancer.7 However, specific therapeutic inhibition of COX2 can result in cardiovascular toxicities.8 An alternative approach to regulating tumor PGE2 is to increase PGE2 breakdown.
PGE2 catabolism is mediated by 15-hydroxyprostaglandin dehydrogenase (PGDH).9 PGDH expression has been shown to regulate PGE2 levels in lung cancer.10 PGDH expression is reduced in colon, breast, gastric and lung cancers, and restoration can inhibit tumorigenesis in xenografts, thus PGDH is recognized as a tumor suppressor in those cancers.11–15 Therefore, PGDH is important for regulating cancer development and progression.
Studies on PGDH expression in the urinary bladder have been limited. Celis et al16 showed that PGDH expression is reduced in bladder cancers by using a proteomics approach. More recently, a study of mouse bladder cancer chemical carcinogenesis showed that COX2 expression was increased while PGDH levels reduced as bladder cancers developed.17 Work from our laboratory showed that small inhibitory RNA (siRNA) inhibition of PGDH expression in the well-differentiated RT4 human bladder cancer cell line disrupts E-cadherin complex assembly.18 Thus, a more detailed study of PGDH expression in bladder cancer is warranted. In this report, we document the loss of PGDH expression early in bladder cancer progression and evaluate the biological effects of inhibiting PGDH expression on PGE2 signaling, catabolism, cell motility, and anchorage-independent growth.
Materials and Methods
Cell Culture
Bladder cancer cells were developed in our laboratory or obtained from the American Type Culture Collection (Manassas, VA) and were cultured in Dulbecco's modified Eagle's medium containing 10% of fetal calf serum and antibiotics, as previously described.19
Immunostaining
Tissues were obtained from surgical resections and/or cystectomies after informed consent. Some of the staining was performed on a tissue microarray prepared by the tissue procurement core of the University of Michigan Cancer Center. Antibodies were prepared by a commercial source (Affinity BioReagents, Golden, CO) by immunizing of rabbits with a KLH-conjugated 21 amino acid peptide for the C-terminal sequence of human PGDH as previously described.18 Immunostaining was performed as previously described.18 Antibody staining was evaluated by a pathologist (L.P.K.). The results are reported as an intensity score, combining the strength of antibody staining (1 = negative, 2 = weak, 3 = moderate, and 4 = strong) multiplied by an estimate of the percentage of tumor staining. Thus the results are expressed in a range from 0 to 400.
Western Blotting
Cell lysates were prepared by homogenized cells directly in the wells with cell lysis buffer (150 mmol/L of NaCl, 10 mmol/L of Tris-HCl, pH 7.2, 0.5% of Na-deoxycholate, 5 mmol/L of EDTA, and 0.1% of NP40) supplemented with protease inhibitors (Sigma-Aldrich, St Louis, MO) and cleared by centrifugation at 10,000 rpm at 4°C for 5 minutes. Equal amounts of protein were electrophorectically separated on 8% to 16% of SDS-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membrane. The membrane was blocked with 5% skim milk in Tris-buffered saline with 0.1% of Tween-20 (TBST) at 4°C overnight with constant shaking. The next day, the membrane was incubated with anti-PGDH antibody at 1:5000 dilution in 5% milk/TBST at room temperature for 2 hours. This PGDH antibody was the same one used for immunostaining. After three washes with TBST, the membrane was incubated with horseradish peroxidase-conjugated anti-rabbit antibody (Cell Signaling, Danvers, MA; 1:5000 in 5% milk/TBST) at room temperature for 1 hour, followed by three washes using TBST. The membrane was then developed by using Immobulon Western chemiluminescent substrate (Millipore, Billerica, MA) and was exposed to X-ray film.
PGDH siRNA
Diced siRNA pool was synthesized by using the BLOCK-iT complete Dicer RNAi kit (Invitrogen, Carlsbad, CA) as previously described.18 PGDH siRNA 9115 and 9027 were purchased from Ambion (Austin, TX). RT4 cells were transfected with PGDH and/or control siRNA by using lipofectamine 2000 (Invitrogen) in 6-well plates. SiRNA (4 μg in 250-μl media) was mixed with 20 μl of lipofectamine 2000, incubated at room temperature for 20 minutes, and added into each well. Fresh media was added 24 hours after each transfection.
PGDH Short Hairpin RNA Lentivirus
On the basis of the results of transient PGDH knock-down experiment in RT4 cells using the Ambion PGDH specific-siRNA, the sequences of 9027 and 9115 were used for PGDH short hairpin RNA (shRNA) construction at the University of Michigan Cancer Center Vector Core. The oligos containing sequences of 9027 and/or 9115 siRNA were ordered from Invitrogen. These oligos also contain the sequences required for efficient transcription, for loop structure formation, as well as BglII and HindIII site overhang on each end for cloning. Their sequences are as follows: for 9027, the forward oligo is 5′-GATCCCCGGTGAAGGCGGCATCATTATTCAAGAGATAATGATGCCGCCTTCACCTTTTTGGAAA-3′ and the reverse oligo is 3′-GGGCCACTTCCGCCGTAGTAATAAGTTCTCTATTACTACGGCGGAAGTGGAAAAACCTTTTCGA-5′; for 9115, the forward oligo is 5′-GATCCCCGGGAATTCATTTTCAAGACTTCAAGAGAGTCTTGAAAATGAATTCCCTTTTTGGAAA-3′ and the reverse oligo is 5′-GGGCCCTTAAGTAAAAGTTCTGAAGTTCTCTCAGAACTTTTACTTAAGGGAAAAACCTTTTCGA-3′ (sequences representing 9027 and/or 9115 siRNA are bolded and italicized). The oligos were resuspended in 1X annealing buffer from Invitrogen to final concentration of 3 μg/ml. Forward and reverse oligos (10 μg each) were placed in a boiling water bath for 3 minutes and allowed to cool down at room temperature for 2 hours to anneal. The resulting double-stranded oligos were first cloned onto pSUPER5 vector via BglII and HindIII sites. The resulting plasmids were then digested with XbaI and XhoI to obtain an XbaI-XhoI fragment containing the siRNA sequences, which were then subcloned onto pFG12 vector to yield pFG12-9027 and/or pFG12-9115.
To produce the lentiviruses, HEK 293T cells were transfected with pFG12, pFG12-9027, and/or pFG12-9115 separately along with pRCE, pHCMV, and pRSV Rev. Cells were incubated at 37°C/5%CO2 for 2 days. The media containing the virus was collected and filtered through a 0.45-μm syringe filter separately, to which final concentration of 28 μmol/L of chloroquinone was added. The media was mixed well and added onto the RT4 cells that have been seeded a day before, and the media was incubated at 37°C/5%CO2 for 4 hours. Chloroquinone containing media was then replaced with fresh media. RT4 cells were incubated at 37°C/5% CO2 for 2 to 3 days. Green fluorescent protein expressing cells, an indication of lentiviral infection, were sorted and collected by flow cytometry. These cells were called empty vector (EV), 9027, and/or 9115, respectively. The knock-down effect of shRNA was verified by both PGDH RT-PCR and Western blotting.
Prostaglandin E2 Metabolism Assay
A commercially available enzyme immunoassay was used to evaluate prostaglandin E2 metabolism following the manufacturer's instructions (Cayman Chemical, Ann Arbor, MI) as previously described.18
cAMP Generation
cAMP levels after prostaglandin E2 stimulation were evaluated by using a commercially available kit (Cayman Chemical) following the manufacturer's instructions.
Wound Healing Migration Assay
To evaluate cell migration, a monolayer scrape “wound healing” model was used.20 Cells were plated into a 24-well plate 1 day after the second transfection with siRNA. The cells were serum-starved overnight. A wound was generated in the monolayer in a 24-well plate by using a 1-ml pipette tip. After scraping, fresh serum-containing media was added and the cultures were incubated at 37°C/5% CO2 and monitored for cell motility into the wound. Pictures were taken at different time points to document the rate of wound healing, and migration was quantitated by image analysis (NIH Image J).
Anchorage-Independent Growth Assay
Soft agar assays were performed as previously described.21 Briefly, dishes were coated with a 1:1 mixture of appropriate medium for the cell line being studied and 1% of Bactoagar (Difco, BD Biosciences, Franklin Lakes, NJ). Cells were plated at 1 × 105 cells/well in a 1:1 mixture of appropriate medium and 0.3% of Bactoagar. Cells were fed three times over 1 week and stained with 500 μg/ml of ρ-iodonitrotetrazolium violet (Sigma-Aldrich) overnight. Colonies of different sizes were counted by using an Accucount 10000 (BioLogics, Gainesville, VA) and were photographed.
Statistical Analysis
Statistical analysis was performed by using computer software programs (SAS, SAS Institute, Carey, NC; SigmaStat 3.5, Systat Software, Inc, Port Richmond, CA). Differences were considered significant at the P < 0.05 level and were indicated with asterisks in figures. Error bars on figures represent ±1 SD. All assays were performed at least in duplicate and were replicated at least twice.
Results
Expression of PGDH is Lost Early in Bladder Cancer Progression
Immunohistochemical staining was performed on a series of normal human bladder and ureter samples as well as bladder cancer tissues, as shown in Figure 1, A–E, and summarized in Table 1. As previously reported,18 PGDH is strongly expressed in normal urothelium in a manner consistent with increased differentiation; that is, strongest expression is in the most differentiated urothelial cells at the bladder lumen, and reduced expression is in less differentiated cells near the lamina propria. Reduced and patchy PGDH expression was noted in noninvasive bladder cancers (stages Ta and Tis/carcinoma in situ). Most invasive bladder cancers showed little or no staining. These results are statistically significant (P < 0.001 by χ2 test).
Figure 1.
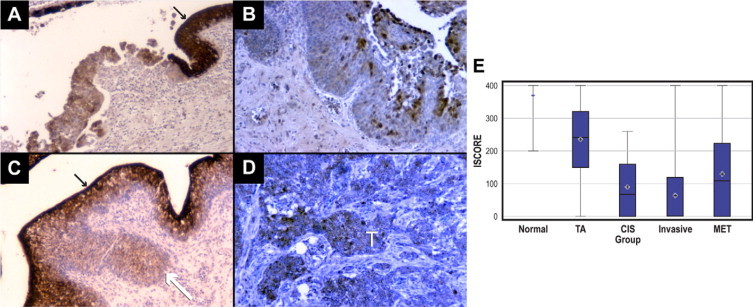
Immunoperoxidase staining of human bladder cancer tissues with PGDH antibodies. A: Bladder carcinoma in situ (CIS/Tis) showing loss of expression in the tumor compared with strong expression in adjacent normal urothelium (black arrow). B: Superficial Ta bladder cancer showing very patchy expression. C: T1 bladder cancer (white arrow) showing loss of expression, with the overlying and surrounding normal urothelium strongly positive (black arrow). D: Invasive bladder cancer showing greatly reduced PGDH staining in tumor (marked with white T). E: Box plot. Results for staining intensity are presented as box plots. The endpoints (whiskers) represent the minimum and maximum observations, while the box stretches from the 25th to the 75th percentiles. The line in the middle of the box is the median and the plus sign is the mean. Strong staining in normal samples is significantly different from all tumor groups (P < 0.01). Superficial Ta staining is greater than carcinoma in situ (CIS) and invasive bladder cancer samples (P < 0.01).
Table 1.
PGDH Immunostaining in Bladder Cancer Samples
| Staining score |
||||
|---|---|---|---|---|
| Tissue | 0–100 | 101–200 | 201–300 | 301–400 |
| Normal | 0 | 1 | 2 | 16 |
| Tis | 9 | 0 | 3 | 0 |
| Superficial (Ta) | 4 | 11 | 9 | 13 |
| Invasive (T1-4) | 53 | 14 | 2 | 4 |
| Metastases | 11 | 4 | 4 | 3 |
The box plot shows six summary statistics for staining of the normal tissues and tissues from the four groups with cancer. The PGDH distributions for the four groups with cancer are very different from the PGDH distribution for the normal group. The mean PGDH level is lower for all four cancer groups than it is for the normal group. The variability of the PGDH level is much higher among the cancer groups than for the normal group. The absence of a box for the normal group indicates that the interquartile range is zero—both the 25th and 75th percentiles are 400. There is no overlap in the boxes for the normal group compared with the cancer group. This result indicates that none of the normal samples exhibited a loss of PGDH as large as that found in the cancer groups. Compared with normal tissue, samples from the all cancer groups were statistically significantly lower (P < 0.01, Mann-Whitney U test). Additionally, the superficial (Ta) cancer tissues had significantly higher staining than the other cancer tissue samples (P < 0.01, Mann-Whitney U test). Finally, the metastatic group was significantly higher than the invasive group (P < 0.01, Mann-Whitney U test). The other groups were not significantly different from each other. These data are consistent with metaanlysis (Oncomine) of bladder cancer gene profiling studies as shown in Figure 2, B–D, showing a significant loss of PGDH mRNA expression from samples representing bladder cancer tumor progression in three separate studies.22–24
Figure 2.
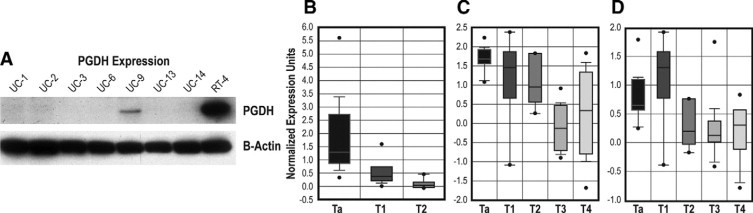
Cell line expression of PGDH and Oncomine results. A: PGDH protein expression in bladder cancer cell lines (Western blot) is shown. Actin staining in the lower panel is to demonstrate equal protein loading in each lane. Only two cell lines, RT4 (well-differentiated) and weakly UM-UC9 (moderately differentiated), show expression of PGDH. Other negative cell lines are poorly differentiated. B: Oncomine metaanalysis of RNA expression result from Dyrskjøt et al.23 PGDH RNA levels were significantly lower in higher stage invasive T1 and T2 tumors compared with superficial Ta bladder cancers (P = 6.9 × 10−13 by t-test). C: Oncomine metaanalysis of RNA expression from Stransky et al.24 PGDH RNA levels were significantly lower in samples from more invasive tumors (P = 5.8 × 10−8). D: Oncomine metaanalysis of RNA expression from Blaveri et al.22 PGDH RNA levels were significant lower in samples from more invasive tumors (P = 2.1 × 10−5).
PGDH Expression in Human Bladder Cancer Cell Lines
A series of human bladder cancer cell lines were tested for PGDH expression by Western blotting, as shown in Figure 2A. Of the eight cell lines tested, only two, RT4 and UM-UC9, show expression of PGDH; RT4 shows strong expression, whereas UM-UC9 is weakly positive. RT4 is recognized to be a very well differentiated cell line,18,19,25 and UM-UC9 shows some differentiated characteristics,19 whereas the other cell lines in the panel are poorly differentiated. These data are consistent with our previous studies indicating the PGDH expression is associated with urothelial differentiation.18
Effects of PGDH Knockdown in RT4 Cells
In our previous studies, we used siRNA to transiently knock down PGDH expression in RT4 cells.18 In that report, we showed that PGDH levels and prostaglandin E2 metabolism were reduced after siRNA transfection. We evaluated the migration of control cells with PGDH expression and siRNA treated cells by using the wound healing model.20 As shown in Figure 3A, cells treated with PGDH siRNA showed increased wound healing, whereas control cells (either parental RT4 or RT4 cells transfected with control sequences) showed statistically significantly less migration into the wound area (P < 0.001, t-test).
Figure 3.
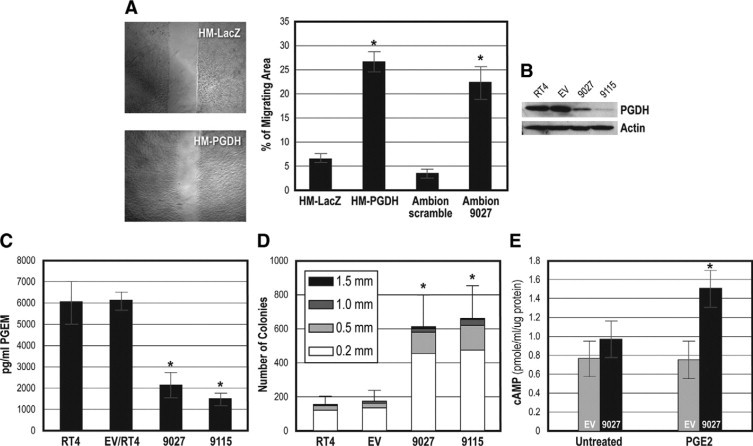
Biological effects of PGDH inhibition. A: Migration in a wound healing model after PGDH knockdown with siRNA treatment. HM-LacZ (siRNA control) cells show sharp “wound” edges, indicating no migration after 5 hours, whereas HM-PGDH knockdown cells have begun to migrate into fill the defect. The graph shows quantification of migration including Ambion control and 9027 PGDH siRNA knockdown cells. Significantly greater migration was found in the PGDH knockdown cells (*P < 0.001 by t-test). B: Lentivirus shRNA knockdown of PGDH expression in RT4 cells, shown by Western blotting. RT4 are the parental cells; EV are RT4 cells infected with virus without insert; 9027 and 9115 represent infection of RT4 with lentivirus containing 9027 or 9115 shRNA sequences. PGDH protein levels are reduced in both knockdown cell types. C: PGDH activity in PGDH shRNA knockdown cells. PGDH enzymatic activity is reduced in the RT4 shRNA lentivirus infected knockdown cells (*P < 0.001 by one-way analysis of variance; reduction in 9027 and 9115 compared with EV, P < 0.014 by multiple comparison using the Holm-Sidak method). D: Anchorage-independent growth in control and PGDH shRNA knockdown cells. Inset indicates the colony size. PDH shRNA lentivirus knockdown cells 9027 and 9115 show significantly greater growth in soft agar (*P < 0.05 by Mann-Whitney U test). E: cAMP responses to PGE2 stimulus in control and shRNA knockdown cells. PGDH shRNA lentivirus knockdown 9027 cells respond to PGE2 stimulus at 10 minutes by generation of cAMP, whereas parental EV cells show no stimulation. Results between EV, 9027 unstimulated and 9027 after PGE2 stimulation were statistically significant (*P < 0.014 by multiple comparison Holm-Sidak method).
To establish long-term cells with PGDH knockdown, we used a shRNA method via lentivirus infection. As shown in Figure 3B, RT4 cells infected with shRNA with the 9027 or 9115 sequences have reduced expression of PGDH by Western blotting, whereas cells infected with virus without insert (EV) or parental uninfected RT4 cells continue to show high PGDH expression. PGDH knockdown with shRNA also significantly reduced PGDH enzymatic function, as shown in Figure 3C, that the 9115 and 9027 lentivirus infected cell lines produce fewer PGE2 metabolites, whereas the control parental and EV-infected RT4 cells maintain metabolic activity. Additionally, loss of PGDH expression enhanced signaling responses to PGE2 as shown by increased generation of cAMP in 9027 lentivirus-infected cells compared with the EV control (Figure 3E). Anchorage-independent growth was evaluated in the lentivirus infected RT4 cells, as shown in Figure 3D. PGDH knockdown 9115 and 9027 RT4 cells show increased growth in soft agar, whereas parental uninfected RT4 or control EV-infected cells show minimal growth.
Discussion
Our findings indicate that loss of PGDH expression contributes to bladder cancer progression. Inhibition of PGDH in the well-differentiated RT4 cell line increased two parameters associated with cancer aggressiveness: increased motility and anchorage independent growth. Our previous study18 showed that siRNA inhibition of PGDH expression resulted in lack of E-cadherin assembly. E-cadherin is also widely recognized as a tumor suppressor,26 so that change also reflects increased malignancy of RT4 after inhibition of PGDH expression. Further, loss of PGDH expression correlated with tumor stage in bladder cancer, and that loss occurs very early in bladder cancer development. These data are similar to the proteomic analysis of Celis et al,16 although our immunohistochemical study reveals a greater loss of PGDH expression in minimally-invasive bladder cancers than was detected by using proteomics. Taken together, these data strongly support a role for PGDH in regulation of bladder cancer progression and possibly in cancer initiation. These data are consistent with other reports showing that PGDH acts as a tumor suppressor in breast, colon, lung, and gastric cancers.11–15 Studies of PGDH expression in bladder cancer are aided by the fact that the urinary bladder is an easily accessible organ, and many tumors in various tumor stages are treated with surgical resection.27 Thus, studies of bladder cancer allow more detailed evaluation of changes during cancer development.
We also observed a re-expression of PGDH in some bladder cancer metastases. Although the number of these samples is small (n = 22), staining revealed a subset with significant PGDH staining. The phenomenon of genes/proteins with reduced expression in primary tumors and re-expression in metastases has been observed in other cancers. For example, although expression of E-cadherin (a tumor suppressor)26 is lost or reduced in primary prostate cancers, E-cadherin is re-expressed in most tumors at metastatic sites.28 Because PGDH expression is epigenetically regulated through methylation and histone deacetylation, as well as by transcriptional repressors such as snail,29 re-expression may reflect either clonal selection or release of epigenetic repression. In any case, PGDH expression does not appear to prevent bladder cancer growth at metastatic sites.
In previous studies, the biological effects of PGDH expression implied an impact on PGE2 signaling, although a direct effect had not previously been demonstrated. In fact, previous work has suggested that the inhibition of tumor growth by PGDH expressing cells was due more to a regional effect of PGE2 reduction, such as reduced angiogenesis, and less on the direct effects on tumor cells.11 Tumor PGDH expression clearly affects the tumor milieu, as shown by reduction of PGE2 levels in lung cancers10 and alteration of antitumoral immune responses.30 In our study, we show that PGDH knockdown in 9027 shRNA lentivirus-infected RT4 cells permitted PGE2 signaling as measured by cAMP generation, whereas signaling was suppressed in the PGDH-expressing parental lines (Figure 3E). Therefore, PGDH also has a direct impact on PGE2 signaling in cancer cells as well as effects on local concentrations (and effects) of PGE2.
The significant loss of PGDH expression early in bladder cancer development also supports the potential use of PGDH expression in bladder cancer diagnosis. As shown in the box plot for the staining intensity (Figure 1E), there is a clear, statistically significant separation between the normal samples and any of the cancer samples. The identification in tissue specimens or cytology of cytokeratin positive, PGDH negative epithelial cells would be a strong indicator for the presence of bladder cancer.
Our data on PGDH expression in bladder cancer, combined with previous work showing that PGDH is a tumor suppressor in lung, breast, and colon cancers, support the concept that restoring PGDH expression is an important therapeutic opportunity. Our finding that PGDH expression is lost earlier in cancer progression that was previously shown also suggests that PGDH restoration may be a valuable target for chemoprevention. Other laboratories are investigating additional agents that may drive or restore PGDH expression in cancer, such as vitamin D.31 Indeed, a recent report has shown that PGDH expression is an important component for a therapeutic response to COX2 inhibitors in a colon cancer clinical trial.32
Bladder cancers present an exceptional opportunity for chemoprevention or therapeutic targeting of PGDH expression.33 The bladder functions as a storage vessel for urine, so that therapeutic agents are often directly delivered to the bladder (intravesical delivery).27,34 Additionally, patients with superficial bladder cancers are known to be at risk for bladder cancer recurrence and are monitored frequently in the first year after treatment.27,34 Thus, the clinical management of early stage bladder cancer presents an unusual opportunity to attempt a chemotherapeutic intervention to reduce recurrence.
In the most recent projections, over 68,000 new bladder cancer cases and over 14,000 deaths due to bladder cancer were estimated for 2008.35 Further, because superficial bladder cancers have a long clinical course, the total bladder cancer prevalence is much higher and is thought to be around 500,000 patients in the United States.36 Additionally, bladder cancer is among the costliest cancers to manage, due not only to treatment costs but also to surveillance.36 The identification of the loss of PGDH in bladder cancer confirms the broader importance of this pathway for PGE2 regulation and as a therapeutic target.
Footnotes
Supported by grants R21-DK-66077 and P50-DK065313 from National Institute of Diabetes and Digestive and Kidney Diseases, NIH (to M.L.); grant RPG-96-044-MGO from the American Cancer Society (to M.L.), Cancer Center Support grant 5 P30 CA46592; and research funds from the Department of Urology, University of Michigan.
References
- 1.Federico A, Morgillo F, Tuccillo C, Ciardiello F, Loguercio C. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer. 2007;121:2381–2386. doi: 10.1002/ijc.23192. [DOI] [PubMed] [Google Scholar]
- 2.Kundu JK, Surh YJ. Inflammation: gearing the journey to cancer. Mutat Res. 2008;659:15–30. doi: 10.1016/j.mrrev.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 4.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55:115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang D, Mann JR, DuBois RN. The role of prostaglandins and other eicosanoids in the gastrointestinal tract. Gastroenterology. 2005;128:1445–1461. doi: 10.1053/j.gastro.2004.09.080. [DOI] [PubMed] [Google Scholar]
- 6.Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, Lipsky PE. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–1073. [PubMed] [Google Scholar]
- 7.Mohammed SI, Knapp DW, Bostwick DG, Foster RS, Khan KN, Masferrer JL, Woerner BM, Snyder PW, Koki AT. Expression of cyclooxygenase-2 (COX-2) in human invasive transitional cell carcinoma (TCC) of the urinary bladder. Cancer Res. 1999;59:5647–5650. [PubMed] [Google Scholar]
- 8.Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, Lines C, Riddell R, Morton D, Lanas A, Konstam MA, Baron JA, Adenomatous Polyp Prevention on Vioxx (APPROVe) Trial Investigators Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092–1102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 9.Tai HH, Cho H, Tong M, Ding Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: structure and biological functions. Curr Pharm Des. 2006;12:955–962. doi: 10.2174/138161206776055958. [DOI] [PubMed] [Google Scholar]
- 10.Hughes D, Otani T, Yang P, Newman RA, Yantiss RK, Altorki NK, Port JL, Yan M, Markowitz SD, Mazumdar M, Tai HH, Subbaramaiah K, Dannenberg AJ. NAD+-dependent 15-hydroxyprostaglandin dehydrogenase regulates levels of bioactive lipids in non-small cell lung cancer. Cancer Prev Res (Phila Pa) 2008;1:241–249. doi: 10.1158/1940-6207.CAPR-08-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang G, Eisenberg R, Yan M, Monti S, Lawrence E, Fu P, Walbroehl J, Löwenberg E, Golub T, Merchan J, Tenen DG, Markowitz SD, Halmos B. 15-Hydroxyprostaglandin dehydrogenase is a target of hepatocyte nuclear factor 3beta and a tumor suppressor in lung cancer. Cancer Res. 2008;68:5040–5048. doi: 10.1158/0008-5472.CAN-07-6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolf I, O'Kelly J, Rubinek T, Tong M, Nguyen A, Lin BT, Tai HH, Karlan BY, Koeffler HP. 15-hydroxyprostaglandin dehydrogenase is a tumor suppressor of human breast cancer. Cancer Res. 2006;66:7818–7823. doi: 10.1158/0008-5472.CAN-05-4368. [DOI] [PubMed] [Google Scholar]
- 13.Ding Y, Tong M, Liu S, Moscow JA, Tai HH. NAD+-linked 15-hydroxyprostaglandin dehydrogenase (15-PGDH) behaves as a tumor suppressor in lung cancer. Carcinogenesis. 2005;26:65–72. doi: 10.1093/carcin/bgh277. [DOI] [PubMed] [Google Scholar]
- 14.Thiel A, Ganesan A, Mrena J, Junnila S, Nykänen A, Hemmes A, Tai HH, Monni O, Kokkola A, Haglund C, Petrova TV, Ristimäki A. 15-hydroxyprostaglandin dehydrogenase is down-regulated in gastric cancer. Clin Cancer Res. 2009;15:4572–4580. doi: 10.1158/1078-0432.CCR-08-2518. [DOI] [PubMed] [Google Scholar]
- 15.Yan M, Rerko RM, Platzer P, Dawson D, Willis J, Tong M, Lawrence E, Lutterbaugh J, Lu S, Willson JK, Luo G, Hensold J, Tai HH, Wilson K, Markowitz SD. 15-Hydroxyprostaglandin dehydrogenase, a COX-2 oncogene antagonist, is a TGF-beta-induced suppressor of human gastrointestinal cancers. Proc Natl Acad Sci USA. 2004;101:17468–17473. doi: 10.1073/pnas.0406142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Celis JE, Ostergaard M, Basse B, Celis A, Lauridsen JB, Ratz GP, Andersen I, Hein B, Wolf H, Orntoft TF, Rasmussen HH. Loss of adipocyte-type fatty acid binding protein and other protein biomarkers is associated with progression of human bladder transitional cell carcinomas. Cancer Res. 1996;56:4782–4790. [PubMed] [Google Scholar]
- 17.Taylor JA, 3rd, Ristau B, Bonnemaison M, Voznesensky OS, Hegde P, Kuchel GA, Pilbeam CC. Regulation of the prostaglandin pathway during development of invasive bladder cancer in mice. Prostaglandins Other Lipid Mediat. 2009;88:36–41. doi: 10.1016/j.prostaglandins.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tseng-Rogenski S, Lee IL, Gebhardt D, Fischer SM, Wood C, Park JM, Liebert M. Loss of 15-hydroxyprostaglandin dehydrogenase expression disrupts urothelial differentiation. Urology. 2008;71:346–350. doi: 10.1016/j.urology.2007.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liebert M, Wedemeyer GA, Stein JA, Washington RW, Jr, Flint A, Ren LQ, Grossman HB. Identification by monoclonal antibodies of an antigen shed by human bladder cancer cells. Cancer Res. 1989;49:6720–6726. [PubMed] [Google Scholar]
- 20.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 21.Woods Ignatoski K, Ethier SP. Constitutive activation of pp 125fak in eleven newly isolated breast cancer cell lines. Breast Cancer Res and Treatment. 1999;54:173–182. doi: 10.1023/a:1006135331912. [DOI] [PubMed] [Google Scholar]
- 22.Blaveri E, Simko JP, Korkola JE, Brewer JL, Baehner F, Mehta K, Devries S, Koppie T, Pejavar S, Carroll P, Waldman FM. Bladder cancer outcome and subtype classification by gene expression. Clin Cancer Res. 2005;11:4044–4055. doi: 10.1158/1078-0432.CCR-04-2409. [DOI] [PubMed] [Google Scholar]
- 23.Dyrskjøt L, Thykjaer T, Kruhøffer M, Jensen JL, Marcussen N, Hamilton-Dutoit S, Wolf H, Orntoft TF. Identifying distinct classes of bladder carcinoma using microarrays. Nat Genet. 2003;33:90–96. doi: 10.1038/ng1061. [DOI] [PubMed] [Google Scholar]
- 24.Stransky N, Vallot C, Reyal F, Bernard-Pierrot I, de Medina SG, Segraves R, de Rycke Y, Elvin P, Cassidy A, Spraggon C, Graham A, Southgate J, Asselain B, Allory Y, Abbou CC, Albertson DG, Thiery JP, Chopin DK, Pinkel D, Radvanyi F. Regional copy number-independent deregulation of transcription in cancer. Nat Genet. 2006;38:1386–1396. doi: 10.1038/ng1923. [DOI] [PubMed] [Google Scholar]
- 25.Liebert M, Hubbel A, Chung M, Wedemeyer G, Lomax MI, Hegeman A, Yuan TY, Brozovich M, Wheelock MJ, Grossman HB. Expression of mal is associated with urothelial differentiation in vitro: identification by differential display reverse-transcriptase polymerase chain reaction. Differentiation. 1997;61:177–185. doi: 10.1046/j.1432-0436.1997.6130177.x. [DOI] [PubMed] [Google Scholar]
- 26.Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene. 2008;27:6920–6929. doi: 10.1038/onc.2008.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barocas DA, Clark PE. Bladder cancer. Curr Opin Oncol. 2008;20:307–314. doi: 10.1097/CCO.0b013e3282f8b03e. [DOI] [PubMed] [Google Scholar]
- 28.De Marzo AM, Knudsen B, Chan-Tack K, Epstein JI. E-cadherin expression as a marker of tumor aggressiveness in routinely processed radical prostatectomy specimens. Urology. 1999;53:707–713. doi: 10.1016/s0090-4295(98)00577-9. [DOI] [PubMed] [Google Scholar]
- 29.Backlund MG, Mann JR, Holla VR, Shi Q, Daikoku T, Dey SK, DuBois RN. Repression of 15-hydroxyprostaglandin dehydrogenase involves histone deacetylase 2 and snail in colorectal cancer. Cancer Res. 2008;68:9331–9337. doi: 10.1158/0008-5472.CAN-08-2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eruslanov E, Kaliberov S, Daurkin I, Kaliberova L, Buchsbaum D, Vieweg J, Kusmartsev S. Altered expression of 15-hydroxyprostaglandin dehydrogenase in tumor-infiltrated CD11b myeloid cells: a mechanism for immune evasion in cancer. J Immunol. 2009;182:7548–7557. doi: 10.4049/jimmunol.0802358. [DOI] [PubMed] [Google Scholar]
- 31.Moreno J, Krishnan AV, Peehl DM, Feldman D. Mechanisms of vitamin D-mediated growth inhibition in prostate cancer cells: inhibition of the prostaglandin pathway. Anticancer Res. 2006;26:2525–2530. [PubMed] [Google Scholar]
- 32.Yan M, Myung SJ, Fink SP, Lawrence E, Lutterbaugh J, Yang P, Zhou X, Liu D, Rerko RM, Willis J, Dawson D, Tai HH, Barnholtz-Sloan JS, Newman RA, Bertagnolli MM, Markowitz SD. 15-Hydroxyprostaglandin dehydrogenase inactivation as a mechanism of resistance to celecoxib chemoprevention of colon tumors. Proc Natl Acad Sci USA. 2009;106:9409–9413. doi: 10.1073/pnas.0902367106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Golijanin DJ, Kakiashvili D, Madeb RR, Messing EM, Lerner SP. Chemoprevention of bladder cancer. World J Urol. 2006;24:445–472. doi: 10.1007/s00345-006-0123-x. [DOI] [PubMed] [Google Scholar]
- 34.Dalbagni G. The management of superficial bladder cancer. Nat Clin Pract Urol. 2007;4:254–260. doi: 10.1038/ncpuro0784. [DOI] [PubMed] [Google Scholar]
- 35.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 36.Botteman MF, Pashos CL, Redaelli A, Laskin B, Hauser R. The health economics of bladder cancer: a comprehensive review of the published literature. Pharmacoeconomics. 2003;21:1315–1330. doi: 10.1007/BF03262330. [DOI] [PubMed] [Google Scholar]