

Abstract
Bone marrow (BM)–derived hematopoietic cells, which are major components of tumor stroma, determine the tumor microenvironment and regulate tumor phenotypes. Cyclooxygenase (COX)−2 and endogenous prostaglandins are important determinants for tumor growth and tumor-associated angiogenesis; however, their contributions to stromal formation and angiogenesis remain unclear. In this study, we observed that Lewis lung carcinoma cells implanted in wild-type mice formed a tumor mass with extensive stromal formation that was markedly suppressed by COX-2 inhibition, which reduced the recruitment of BM cells. Notably, COX-2 inhibition attenuated CXCL12/CXCR4 expression as well as expression of several other chemokines. Indeed, in a Matrigel model, prostaglandin (PG) E2 enhanced stromal formation and CXCL12/CXCR4 expression. In addition, a COX-2 inhibitor suppressed stromal formation and reduced expression of CXCL12/CXCR4 and a fibroblast marker (S100A4) in a micropore chamber model. Moreover, stromal formation after tumor implantation was suppressed in EP3−/− mice and EP4−/− mice, in which stromal expression of CXCL12/CXCR4 and S100A4 was reduced. The EP3 or EP4 knockout suppressed S100A4+ fibroblasts, CXCL12+, and/or CXCR4+ stromal cells as well. Immunofluorescent analyses revealed that CXCL12+CXCR4+S100A4+ fibroblasts mainly comprised stromal cells and most of these were recruited from the BM. Additionally, either EP3- or EP4-specific agonists stimulated CXCL12 expression by fibroblasts in vitro. The present results address the novel activities of COX-2/PGE2-EP3/EP4 signaling that modulate tumor biology and show that CXCL12/CXCR4 axis may play a crucial role in tumor stromal formation and angiogenesis under the control of prostaglandins.
Recent advances in tumor biology have identified the stroma as an important regulator of carcinogenesis and a potentially valuable therapeutic target. Although interactions between the epithelium and stroma have long been considered to be important in tumor progression, the efficacy of targeting stromal components as a therapeutic strategy has not been established, because the specific regulators of such interactions remain unclear. In addition to endothelial cells, macrophages and fibroblasts1 are the major stromal components of the tumor microenvironment, and they play key roles in the enhancement of angiogenesis. It has recently been established that bone marrow (BM)–derived hematopoietic cells are the major components of the stroma of tumors, and that they determine the tumor microenvironment2–8; however, the specific factors that enhance the functions of BM-derived precursor cells, and the mechanism of recruitment of these cells during tumor angiogenesis, are not fully understood. Tumor-associated angiogenesis in the tumor stroma is a prerequisite for invasive growth of a tumor larger than 2 to 3 mm in diameter, and then metastasis occurs. Tumor-associated angiogenesis is caused by a shift in the local balance of proangiogenic and antiangiogenic factors toward the proangiogenic state.9–11 This angiogenic switch in the tumor stroma may be important in the control of cancer progression and may be regulated by the recruitment of BM-derived cells.
Chemokines and their receptors play critical roles in leukocyte trafficking during inflammatory processes; however, a growing body of data suggests that a number of chemokines and their receptors also play diverse roles in cancer growth, cancer metastasis, cancer angiogenesis, or modulation of the cancer microenvironment.12–15 Preclinical tumor models indicate that some chemokine receptor antagonists can block cancer growth either directly or by altering the cancer stroma.16–21 However, there is a need to extend our understanding of the signaling pathways by which chemokines and chemokine receptors facilitate cancer processes. Further, the interactions between the inflammatory mediators and chemokine systems are poorly understood.
Cyclooxygenase (COX)−2 is one of two forms of COX and is expressed at sites of inflammation and malignancies, suggesting that COX-2 inhibition may be useful in the treatment or prevention of inflammatory diseases and various cancers.22 In previous studies, we determined that inhibition of the COX-2/VEGF-dependent pathway suppresses tumor-associated angiogenesis and tumor growth in mice.23,24 COX-2–lacking mice were found to be resistant to the development of colorectal neoplasia.25 The same may be true in humans, because epidemiological studies have shown that nonsteroidal anti-inflammatory drugs, the prototypic inhibitors of COX, reduce the risk of several types of cancer.26 The relevant endogenous prostaglandins (PGs) may be PGE2, which is generated from arachidonic acid and other polyunsaturated fatty acids, in a reaction catalyzed by COX-2.27 Four G protein–coupled receptors, which respond to the PGE2 are designated subtypes EP1, EP2, EP3, and EP4.28 Stromal tissues in COX-2 inhibitor–treated mice and EP3 receptor knockout mice are very thin and appeared to be pale in color.23,24 COX-2 inhibition and EP3 receptor knockout markedly reduce the formation of stromal tissues around the tumors besides the attenuation of angiogenesis, suggesting that COX-2–derived PGE2 has a crucial role in tumor stroma formation and also in the enhancement of tumor-associated angiogenesis.
In the present study, we tested whether COX-2–derived PGE2 upregulates the expression of chemokines and their receptors, and further clarified whether the upregulated chemokine systems enhance stromal formation and PG-dependent tumor angiogenesis. We showed that the chemokine systems and PGE2 may be useful therapeutic targets for cancers that modulate tumor microenvironment.
Materials and Methods
Animals and Cells
Eight-week-old male C57BL/6 wild-type mice, obtained from CLEA Japan Inc. (Tokyo, Japan), were used. In some experiments, EP receptor knockout (−/−) mice (eight- to ten-week-old males) lacking EP1, EP2, EP3, and EP4, were used (for EP4−/− mice, EP4WT mice were used for comparison) as described previously.23,29 These mice were housed in a limited access animal facility. The animal room temperature and relative humidity were set to 25 ± 1°C and 60% ± 5%, respectively. Artificial lighting provided a 24-hour cycle of 12 hours light/12 hours dark (8 am to 8 pm). All experiments were performed in accordance with the guidelines for animal experiments of the Kitasato University School of Medicine.
Lewis lung carcinoma (LLC) cells were originally isolated from C57BL/6 mice, and we used CRL-1642 (ATCC, Manassas, VA), which are high-tumorigenic and low-metastatic type mouse LLC cells. No metastasis has been shown for experimental 28 days when these LLC cells are subcutaneously transplanted. Fibroblasts (L929) were obtained from Cell Bank, RIKEN Bioresource Center (Ibaraki, Japan). They were cultured in DMEM supplemented with 10% (v/v) FBS and 100 units/500 ml penicillin, all of which were obtained from Gibco BRL, Life Technologies, Inc. (Rockville, MD), at 37°C in 5% humidified CO2. After harvest, LLC cells were washed three times with PBS solution (PBS; 8 g/L NaCl, 0.2 g/L KCl, 2.9 g/L Na2HPO412H2O, and 0.2 g/L KH2PO4, pH 7.2) and used in the respective experiments.
Mouse Dorsal Matrigel Implantation Assay
Under light ether anesthesia, mice were subcutaneously injected in the back with 0.25 ml of phenol-red-free Matrigel (Japan BD Biosciences, Tokyo, Japan), diluted 1:1 in saline. The day of implantation was defined as day 0. In the PGE2 administration experiment, PGE2 (10 nmol/site/day; Cayman Chemical, Ann Arbor, MI) or the same volume of vehicle (0.2% ethanol) was injected in the center of the Matrigel on the day four to six. In the stromal cell–derived factor (SDF)-1α administration experiment, recombinant murine SDF-1α (500 ng/mouse/day; PeproTech GmbH, Hamburg, Germany) or the same volume of saline was subcutaneously injected in the center of the Matrigel on day one to six. In both experiments, mice were euthanized with an excess dose of ether after seven days, and the Matrigel implants were harvested with the surrounding stromal tissue and skin. After trimming, the wet weights of the stroma and Matrigel were measured and prepared for the other assays.
Mouse Micropore Chamber Assay
LLC cells were washed as described above and resuspended in PBS solution at a density of 1 × 108 cells/ml. Thereafter, 1 × 107 LLC cells in 0.1 ml of solution were placed in a round chamber, which consisted of a ring covered with cellulose ester filters (pore size, 0.45 μm; Millipore, Bedford, MA) on both sides. Then, these chambers were subcutaneously implanted into the dorsum of mice.24,30 The day of implantation was defined as day 0. In the experiment in which COX-2 inhibitor was administered, mice were fed diets containing 1000 ppm COX-2 inhibitor (Celecoxib; Pfizer Inc., New York, NY) starting from day 0 until the end of the experiment. The dose of Celecoxib was approximately 100 mg/kg/day, as estimated from the diets they consumed. The control mice were fed diets without agents. In the CXCR4 neutralizing experiment, CXCR4 neutralizing rat monoclonal IgG antibody, named 2B11 (Japan BD Biosciences, Tokyo, Japan), dissolved in PBS solution at a concentration of 70 μg/ml, was subcutaneously injected into four sites around the chamber, at a dose of 3.5 μg/site (total 14 μg/mouse/day) from day one to six. Similarly, the control mouse group was injected at four sites with normal rat IgG (Santa Cruz Biotechnology Inc., Santa Cruz, CA) dissolved in PBS solution, at the same dose. In both experiments, mice were euthanized on day seven, and the stromal tissues that had formed on the micropore chamber were immediately used in assays.
Subcutaneous Tumor Cell Implantation Models
LLC cells were washed as described above and resuspended in PBS at a density of 1 × 107 cells/ml, and 0.1 ml of the resulting suspension was subcutaneously injected into the right flank of mice. The day of inoculation was defined as day 0. In the experiment of COX-2 inhibition, mice of the Celecoxib group were fed diets containing 1000 ppm Celecoxib, starting from day 0 until the end of the experiment. The control mice were fed diets without agents. On days 7, 14, and 21, mice were euthanized with an excess dose of ether, then tumor including stromal tissue was carefully excised. In the experiment of each EP receptor knockout mice, the mice were similarly sacrificed on day 14. The wet weights (mg) and size (mm3) of excised tissue were measured immediately after harvesting.
Bone Marrow Chimeric Model of Green Fluorescent Protein
Bone marrow transplantation (BMT) was performed as reported previously.29,31 Briefly, BM cells were obtained by flushing the cavities of freshly dissected femurs, tibias, and pelves of donor male green fluorescent protein (GFP) transgenic mice (a gift from Dr. M. Okabe, Genome Information Research Center, Osaka University, Osaka, Japan) with PBS. The flushed BM cells were dispersed by pipetting and resuspended in PBS at a density of 1 × 107 cells/ml. Wild-type mice were lethally irradiated with 9.0 Gy using an MBR-1505R X-ray irradiator (Hitachi Medico Co., Tokyo, Japan) with a filter (copper, 0.5 mm; aluminum, 2 mm), monitoring the cumulative radiation dose. The BM mononuclear cells of GFP mice (2 × 106 cells in 200 μl of PBS) were transplanted into irradiated wild-type mice via the tail vein. After 12 weeks, peripheral blood was collected and analyzed by fluorescence-activated cell sorting. The mice in which more than 90% of the peripheral leukocytes were GFP-positive were used in the tumor implantation experiment.
On day 14, the GFP-BMT-chimeric mice were euthanized with an excess dose of ether, and tumor and stromal tissue were carefully excised. The wet weight (mg) and size (mm3) of excised tissue were measured immediately after harvesting. A 50-mg aliquot of excised tissue was homogenized in 1 ml of PBS. The fluorescence of the supernatants (3000g for 30 minutes, at 4°C) was measured in 96-well plates using a 488 nm excitation wavelength.
Effect of PGE2 and EP-Specific Agonists on Fibroblasts
L929 fibroblasts (3 × 105 cells per well) precultured for six hours in six-well plates were incubated for 24 hours with or without PGE2 and the respective agonists; EP1 agonist (ONO-DI-004), EP2 agonist (ONO-AE1-259-01), EP3 agonist (ONO-AE-248), and EP4 agonist (ONO-AE1-329; Ono Pharmaceutical Co., Osaka, Japan) as described previously.29,32 After harvest, RNA extraction and real-time RT-PCR analysis were performed as described below.
Immunohistochemistry
For histological analysis, excised tissues were immediately fixed with 4% paraformaldehyde in 0.1 mol/L sodium phosphate buffer (pH 7.4) for 24 hours, dehydrated with a graded series of ethanol solutions, and embedded in paraffin. Sections (4 μm thick) were prepared from the paraffin-embedded tissue and mounted on glass slides which had been treated with VECTABOND (Vector Laboratories Inc., Burlingame, CA) according to the manufacturer's instructions. After deparaffinization with xylene and hydration with a degraded series of ethanol solution, the sections were subjected to either H&E staining or immunostaining. For immunostaining, antigen unmasking was performed with Protease-K (DakoCytomation, Glostrup, Denmark) for three minutes, endogenous peroxidase activity was blocked by incubation in 3% hydrogen peroxide/phosphate buffer for five minutes, and nonspecific antibody binding was blocked by incubation with 1% diluted normal horse serum for 30 minutes. Sections were then incubated at 4°C overnight with the following antibodies: rat anti-mouse CD31 monoclonal IgG2a antibody (Abcam plc, CB, UK), to identify endothelial cells of the microvessels; mouse anti-mouse SDF-1 monoclonal IgG1 antibody (P-159X: Santa Cruz Biotechnology, Inc., Santa Cruz, CA), to detect SDF-1α (CXCL12); rat anti-mouse CXCR4 monoclonal IgG2b antibody (2B11: Japan BD Biosciences, Tokyo, Japan), to detect CXCR4; rabbit anti-mouse S100A4 polyclonal IgG antibody (Abcam plc, CB, UK), to identify fibroblasts in the stromal tissue; rat anti-mouse F4/80 monoclonal IgG2a antibody (BM8: Santa Cruz Biotechnology Inc., Santa Cruz, CA) or goat anti-mouse F4/80 polyclonal IgG antibody (A-19: Santa Cruz Biotechnology Inc., Santa Cruz, CA) to identify monocytes; goat anti-mouse CD3ε polyclonal IgG antibody to identify lymphocytes (M-20: Santa Cruz Biotechnology Inc., Santa Cruz, CA), and rabbit anti-mouse α-smooth muscle actin (α-SMA) monoclonal IgG antibody (E184: Abcam plc, CB, UK) or mouse anti-mouse α-SMA monoclonal IgG2a antibody (1A4: Sigma-Aldrich Inc., St. Louis, MO) to identify myofibroblasts. Immune complexes were detected with a Vectastain Universal Elite ABC Kit (Vector Laboratories Inc., Burlingame, CA). Immune complexes of primary antibodies raised in mice were detected with the mouse-on-mouse (M.O.M.) kit (Histofine Mouse Stain Kit, Nichirei Biosciences Inc., Tokyo, Japan), according to the manufacturer's instructions. These immune complexes were revealed using the 3, 3′-diaminobenzidine (DAB) substrate, with nickel ammonium sulfate (Vector Laboratories Inc., Burlingame, CA) as chromogen. Sections were counterstained with hematoxylin.
Immunofluorescence
For double immunofluorescence, samples were fixed with 10% neutral buffered paraformaldehyde at 4°C for 1 hour. After cryoprotection with 30% sucrose/0.1 mol/L phosphate buffer (pH 7.2), cryostat sections about 10 to 20 μm in thickness were cut and nonspecific staining was blocked by the incubation with 1% bovine serum albumin (BSA)/PBS for 1 hour. The sections were incubated with a mixture of primary antibodies in 1% BSA/PBS at room temperature for 1 hour or overnight. Primary antibodies were the same as the above immunohistochemistry. After being washed three times in PBS, the sections were incubated with a mixture of secondary antibodies for 1 hour at room temperature. Negative control staining was conducted by replacing the primary antibodies with control IgG of each immune animal. Secondary antibodies were Alexa Fluor 488, 568, or 647- conjugated donkey anti-mouse, rabbit, rat, or goat IgGs (Molecular Probes, Inc., Eugene, OR). They were then observed using a confocal scanning laser microscope (LSM710: Carl Zeiss MicroImaging GmbH., Oberkochen, Germany). Serial optical sections (collected at 1-μm intervals) in the z axis were collected and overlaid for the final images shown using ZEN-2008 software (Carl Zeiss MicroImaging GmbH., Oberkochen, Germany) installed on LSM710.
After double labeling, four high-power optical fields (400-fold magnification) were randomly selected, and the number of positive cells was counted for every single channel (green, red) as well as the merged channels (yellow) for each animal. At least five animals were analyzed for each marker analysis. All samples were blinded before this morphometric analysis.
Microvessel Density and Microvessel Area
Microvessel density (MVD) and microvessel area (MVA) in the stroma were assessed as a parameter of tumor-associated angiogenesis, according to the established methods described previously.24,33 Briefly, areas of highest neovascularization were found by scanning the stroma around the tumor at low power (40-fold and 100-fold magnification). After the most vascularized area was identified, the individual microvessels were then counted on a 400-fold field. CD31 immunoreactive endothelial cells were clearly and easily differentiated from the stromal cells and other connective tissue elements. MVD was expressed in terms of the number of vessels per unit area (mm2). MVA was also calculated with the image analysis software National Institutes of Health Image (NIH Research Service Branch) and was expressed as the area of vessels per observed area (mm2/mm2). MVD and MVA were calculated in four different fields per section, and four sections were obtained from each animal.
Stromal Thickness
Stromal thickness was assessed on a 100-fold field of an H&E-stained section, calculated in four fields per section, and four sections/individual, and averaged.
Quantitative Real-Time RT-PCR Analysis
Each sample of excised tissue was immediately immersed in RNAlater RNA stabilization Reagent (QIAGEN Japan, Tokyo, Japan), and the samples were homogenized for 60 seconds at 6000 rpm using a MagNA Lyser (Roche diagnostics Inc., Mannheim, Germany). Harvested cells were washed three times with PBS solution and homogenized using QIA Shredder (QIAGEN Japan, Tokyo, Japan). Total RNA was extracted from the homogenized tissues and cells using the RNeasy Mini Kit (QIAGEN Japan, Tokyo, Japan), and single-stranded cDNA was generated from 1 μg of total RNA via reverse transcription using ReverTra Ace (TOYOBO Co., Ltd., Osaka, Japan), according to the manufacturer's instructions. Quantitative PCR amplification was performed using SYBR® Premix Ex Taq (Takara Bio Inc., Shiga, Japan). The gene-specific primers used were described in Table 1. PCR was performed in 20 μl reactions according to the method of the manufacturer. The reaction mixture was then subjected to 35 cycles of amplification in a DNA thermal cycler. Each cycle consisted of a heat-denaturation step at 95°C for 1 minute, an annealing of primers step at 55°C for 1 minute, and an extension step at 72°C for 1 minute. After completion of 35 PCR cycles, the samples were incubated at 72°C for 5 minutes. Data were normalized relative to the expression level of GAPDH for each sample. To confirm that PCR products were derived from target mRNA, the products were cloned using TOPO TA Cloning Kit for Sequencing (Invitrogen, San Diego, CA), and the nucleotide sequences were analyzed. All samples were tested in triplicate.
Table 1.
Primers for Real-Time Quantitative PCR
| Gene | Forward primer sequence | Reverse primer sequence | Annealing temperature |
|---|---|---|---|
| GAPDH | 5′-TTGTGATGGGTGTGAACCAC-3′ | 5′-CATGAGCCCTTCCACAATGCC-3′ | 55 |
| CCL2 | 5′-CGGAACCAAATGAGATCAGAA-3′ | 5′-TTGTGGAAAAGGTAGTGGATG-3′ | 55 |
| CCL3 | 5′-TGACTAAGAGAAACCGGCAGA-3′ | 5′-TCAGGCATTCAGTTCCAGGTC-3′ | 55 |
| CCL4 | 5′-TCCTGCTGTTTCTCTTACACC-3′ | 5′-TGTCTGCCTCTTTTGGTCAGG-3′ | 55 |
| CCL5 | 5′-CTGCTGCTTTGCCTACCTCTC-3′ | 5′-GTGACAAACACGACTGCAAGA-3′ | 55 |
| CCL6 | 5′-TCTTTATCCTTGTGGCTGTCC-3′ | 5′-GCAGCAGTCTGAAGAAGTGTC-3′ | 55 |
| CCL7 | 5′-TGCTTTCAGCATCCAAGTGTG-3′ | 5′-ACCGACTACTGGTGATCCTTC-3′ | 55 |
| CCL8 | 5′-CCAGATAAGGCTCCAGTCACC-3′ | 5′-TTCTCTCGTAGCTTTTCAGCA-3′ | 55 |
| CCL9 | 5′-ACAACTGCTCTTGGAATCTGG-3′ | 5′-AGTCTTGAAAGCCCATGTGAA-3′ | 55 |
| CCL12 | 5′-CATAGCTACCACCATCAGTCC-3′ | 5′-TGCTTGTGATTCTCCTGTAGC-3′ | 55 |
| CXCL1 | 5′-AAACCGAAGTCATAGCCACAC-3′ | 5′-GGGGACACCTTTTAGCATCTT-3′ | 55 |
| CXCL2 | 5′-ATCCAGAGCTTGAGTGTGACG-3′ | 5′-GCCTTGCCTTTGTTCAGTATC-3′ | 55 |
| CXCL3 | 5′-CCCTACCAAGGGTTGATTTTG-3′ | 5′-GGCAAACTTCTTGACCATCCT-3′ | 55 |
| CXCL5 | 5′-GCCCTACGGTGGAAGTCATAG-3′ | 5′-TGCATTCCGCTTAGCTTTCTT-3′ | 55 |
| CXCL7 | 5′-CTGCCCACTTCATAACCTCCA-3′ | 5′-GTCCATGCCATCAGATTTTCC-3′ | 55 |
| CXCL9 | 5′-GTTCTTTTCCTCTTGGGCATC-3′ | 5′-GAGCATCGTGCATTCCTTATC-3′ | 55 |
| CXCL10 | 5′-CGTCATTTTCTGCCTCATCCT-3′ | 5′-CCTATGGCCCTCATTCTCACT-3′ | 55 |
| CXCL11 | 5′-TGCTCAAGGCTTCCTTATGTT-3′ | 5′-CTTTCTCGATCTCTGCCATTT-3′ | 55 |
| CXCL12 | 5′-GCATCAGTGACGGTAAACCAG-3′ | 5′-GCACAGTTTGGAGTGTTGAGG-3′ | 55 |
| CCR1 | 5′-GGCCCTAGCCATCTTAGCTTC-3′ | 5′-TGAAACCTCTTCCACTGCTTC-3′ | 55 |
| CCR2 | 5′-TTACCTCAGTTCATCCACGGC-3′ | 5′-CAAGGCTCACCATCATCGTAG-3′ | 55 |
| CCR3 | 5′-GCAGCATTGCCTGAATTTATC-3′ | 5′-TTTCCAGCTGTCTTCTTCACC-3′ | 55 |
| CCR5 | 5′-GTTATCTCTCAGTGTTCTTCC-3′ | 5′-ATCGGGTATAGACTGAGCTTG-3′ | 55 |
| CCR6 | 5′-TTCACCCCTTTGCTGTTTATG-3′ | 5′-CTGTGCCTCTTGGAGTTCTGG-3′ | 55 |
| CCR7 | 5′-TACATCGGCGAGAATACCACG-3′ | 5′-ATACATGAGAGGCAGGAACCA-3′ | 55 |
| CCR8 | 5′-CGACCTGCTCTTTGTCCTCTC-3′ | 5′-CCAGAGACCACCTTACACATC-3′ | 55 |
| CXCR2 | 5′-AACAATACATCCCGTTTGAGG-3′ | 5′-AGTGTGAACCCGTAGCAGAAC-3′ | 55 |
| CXCR3 | 5′-TGTATGGGGTCTCTGTCTGCT-3′ | 5′-ACCTGTGGGAAGTTGTACTGG-3′ | 55 |
| CXCR4 | 5′-CTCTGAAGAAGTGGGGTCTGG-3′ | 5′-AAGTAGATGGTGGGCAGGAAG-3′ | 55 |
| CXCR7 | 5′-TTTGCTGTCCCCTTCACTATC-3′ | 5′-CCAACATACCAGGAAGACCAC-3′ | 55 |
| CX3CR1 | 5′-GTGACATGAAGAGGGACCTGA-3′ | 5′-GGCGTAGATAAAGGGGTTGAG-3′ | 55 |
| GFP | 5′-ACTACAACAGCCACAACGTCT-3′ | 5′-GGTGGTCTGCTGGTAGTGGTC-3′ | 55 |
| VEGF-A | 5′-ACGACAGAAGGAGAGCAGAAG-3′ | 5′-ATGTCCACCAGGGTCTCAATC-3′ | 55 |
| F4/80 | 5′-TATCTTTTCCTCGCCTGCTTC-3′ | 5′-CACCACCTTCAGGTTTCTCAC-3′ | 55 |
| S100A4 | 5′-TGGGGAAAAGGACAGATGAAG-3′ | 5′-ATGCAGGACAGGAAGACACAG-3′ | 55 |
| α-SMA | 5′-GAAGAGCTACGAACTGCCTGA-3′ | 5′-TGAAAGATGGCTGGAAGAGAG-3′ | 55 |
| CD3ɛ | 5′-ACTGGAGCAAGAATAGGAAGG-3′ | 5′-ATAGTCTGGGTTGGGAACAGG-3′ | 55 |
SMA indicates smooth muscle actin.
Measurement of Stromal CXCL12 (SDF-1α) Protein Level
Subcutaneous stromal tissues surrounding tumors were isolated from the surface of tumors, and tissues were homogenized in ninefold volume (w/v) of PBS with 0.1% (v/v) Triton X, and centrifuged for 30 minutes at 3500g at 4°C. The supernatant was recovered, and CXCL12 (SDF-1α) protein levels were determined using a mouse SDF-1α Quantikines ELISA Kit (R&D Systems Inc., Minneapolis, MN). Data are expressed as ng/ml of lysate. All samples in the CXCL12 assay were tested in duplicate.
Statistical Analysis
Statistical computations were performed using a SAS software package (SAS Institute, Cary, NC), StatView version 5.0. Data were expressed as means ± SEM. Comparisons among multiple groups were performed by factorial analysis of variance, followed by Scheffé test. Comparisons between two groups were performed with Student t test. A result was considered statistically significant when the P value was <5% (P < 0.05).
Results
COX-2–Dependent Stromal Formation and Angiogenesis in the Tumor Implantation Model, and Changes in the Expression of Chemokines and Chemokine Receptors in the Tumor Stroma
We first examined the roles of inducible COX-2 in angiogenesis and tumor stromal formation and clarified which chemokines and their receptors were involved in stromal formation. We adopted the tumor implantation model with the use of high-tumorgenic LLC cells. Tumor growth in mice was observed during three-week experimental periods. Celecoxib administration (100 mg/kg/day) suppressed tumor growth significantly throughout the experimental periods (Figure 1A). As shown in Figure 1B, Celecoxib-treated mice exhibited less evidence of angiogenesis, which was visualized as marked red color, than vehicle-treated mice. Consistent with the reduced redness of the tumors, MVD and MVA were significantly reduced in Celecoxib-treated mice in comparison with those in vehicle-treated mice (Figure 1, C and D). Celecoxib treatment significantly reduced the formation of stromal tissues (Figure 1E). To investigate the involvement of chemokine systems in stromal formation, we next tested the expression of a number of chemokines and their receptors in tumor stromal tissues, using real-time RT-PCR analysis (Table 2). Substantial expression of CCL2, CCL3, CCL4, CCL5, CCL6, CCL7, CCL8, CCL9, and CCL12 was detected in the stromal tissues throughout the experiment. Celecoxib treatment did not alter the mRNA levels of CCL2, CCL3, CCL4, CCL5, CCL6, CCL7, CCL8, and CCL9, whereas it did cause the levels of CCL12 mRNA to decrease in the early periods but to increase on day 14. Further, the mRNA expression of chemokine receptors, CCR1, CCR2, CCR3, CCR5, CCR6, CCR7, CCR8, and CX3CR1, did not significantly change with Celecoxib treatment. By contrast, CXCL12 (SDF-1) mRNA levels were reduced by 73% in Celecoxib-treated mice relative to vehicle-treated mice on day 14 (Figure 1F). In line with mRNA expression, CXCL12 protein expression was suppressed by 42% (Figure 1H). Even on day 7, CXCL12 expression tended to be lower in the Celecoxib-treated mice than in the vehicle-control mice (vehicle versus celecoxib, 0.0180 ± 0.0039 and 0.0107 ± 0.0024, P = 0.12), and also at day 21 (0.0095 ± 0.0026 and 0.0055 ± 0.0009, P = 0.12). In addition, the expression of CXCR4, a receptor for CXCL12, was reduced on day 14 (Figure 1G), in Celecoxib-treated mice, suggesting that the CXCL12/CXCR4 axis may be at least in part involved in regulating tumor stromal formation via COX-2 derived PGs. CXCR7, recently deorphanised as an alternative receptor that binds CXCL12,34 did not significantly alter in its mRNA expression by Celecoxib-treatment (Table 2). In addition, we also determined the stromal expression of two important CXC chemokine receptors, CXCR2 and CXCR3, because those chemokines also play a role in angiogenesis and angiostasis, respectively.15,35 Gene expression of angiogenic CXCR2 and its ligands including CXCL1, CXCL2, CXCL3, CXCL5, and CXCL7 did not alter by COX-2 inhibition throughout the experimental period. Gene expression of CXCR3 did not change, though its ligands including CXCL9, CXCL10, and CXCL11 were significantly increased on day 14 (see Supplemental Table S1 at http://ajp.amjpathol.org).
Figure 1.
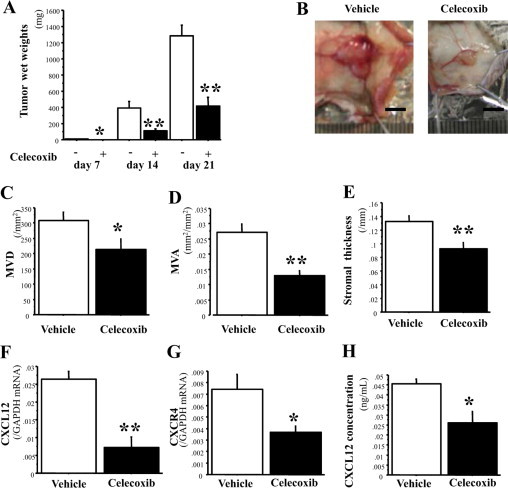
COX-2–dependent stromal formation and angiogenesis in the tumor implantation model, and expression of chemokines and chemokine receptors in tumor stroma. A: Time course of tumor growth in the absence (−; unfilled) and presence (+; filled) of Celecoxib. The day of inoculation was defined as day 0. Celecoxib significantly attenuated tumor growth on days 7, 14, and 21. n = 5, 10, and 7 per group on days 7, 14, and 21, respectively. B: Typical appearance of tumors excised on day 14. Scale bars = 5.0 mm. C and D: Angiogenesis (MVD and MVA) on day 14. E: Stromal formation (stromal thickness) on day 14. F and G: The mRNA expression of CXCL12 and CXCR4 in tumor stroma on day 14. H: CXCL12 protein expression in tumor stroma on day 14. n = 10 per group in C–H. The results represent means ± SEM. *P < 0.05; **P < 0.01 by Student t test (compared with vehicle-treated mice on the same day).
Table 2.
mRNA Expression of Chemokines and Chemokine Receptors in the Stroma of Murine Tumor Model
| mRNA | Celecoxib | Day 7 | P* | Day 14 | P* | Day 21 | P* |
|---|---|---|---|---|---|---|---|
| CCL2 | − | 0.2850 ± 0.1070 | NS | 0.1810 ± 0.0434 | NS | 0.1680 ± 0.0429 | NS |
| + | 0.1370 ± 0.0375 | 0.2180 ± 0.0553 | 0.2940 ± 0.0258 | ||||
| CCL3 | − | 0.0126 ± 0.0076 | NS | 0.0220 ± 0.0120 | NS | 0.0380 ± 0.0181 | NS |
| + | 0.0131 ± 0.0052 | 0.0138 ± 0.0009 | 0.0211 ± 0.0090 | ||||
| CCL4 | − | 0.0035 ± 0.0003 | NS | 0.0044 ± 0.0013 | NS | 0.0063 ± 0.0012 | NS |
| + | 0.0041 ± 0.0012 | 0.0078 ± 0.0028 | 0.0080 ± 0.0021 | ||||
| CCL5 | − | 0.0178 ± 0.0055 | NS | 0.0075 ± 0.0016 | NS | 0.0103 ± 0.0038 | NS |
| + | 0.0064 ± 0.0016 | 0.0070 ± 0.0019 | 0.0269 ± 0.0183 | ||||
| CCL6 | − | 1.090 ± 0.3670 | NS | 0.7200 ± 0.5670 | NS | 0.2430 ± 0.0729 | NS |
| + | 0.3590 ± 0.0714 | 0.3300 ± 0.2270 | 0.2470 ± 0.0959 | ||||
| CCL7 | − | 0.0196 ± 0.0060 | NS | 0.0268 ± 0.0078 | NS | 0.0319 ± 0.0125 | NS |
| + | 0.0184 ± 0.0061 | 0.0283 ± 0.0106 | 0.0257 ± 0.0093 | ||||
| CCL8 | − | 0.9800 ± 0.4190 | NS | 0.3420 ± 0.2400 | NS | 0.1800 ± 0.0780 | NS |
| + | 0.1970 ± 0.0601 | 0.2540 ± 0.1610 | 0.1790 ± 0.1220 | ||||
| CCL9 | − | 0.2540 ± 0.0725 | NS | 0.1820 ± 0.1270 | NS | 0.0867 ± 0.0306 | NS |
| + | 0.0928 ± 0.0099 | 0.0902 ± 0.0453 | 0.0976 ± 0.0232 | ||||
| CCL12 | − | 0.0339 ± 0.0046 | 0.03 | 0.0281 ± 0.0035 | NS (0.07) | 0.0251 ± 0.0030 | NS |
| + | 0.0201 ± 0.0016 | 0.0432 ± 0.0054 | 0.0228 ± 0.0017 | ||||
| CCR1 | − | 0.0216 ± 0.0036 | NS | 0.0200 ± 0.0022 | NS | 0.0158 ± 0.0010 | NS |
| + | 0.0128 ± 0.0021 | 0.0158 ± 0.0013 | 0.0133 ± 0.0007 | ||||
| CCR2 | − | 0.0125 ± 0.0005 | NS | 0.0114 ± 0.0023 | NS | 0.0071 ± 0.0008 | NS |
| + | 0.0098 ± 0.0014 | 0.0141 ± 0.0034 | 0.0047 ± 0.0007 | ||||
| CCR3 | − | 0.0008 ± 0.0003 | NS | 0.0004 ± 0.0001 | NS | 0.0006 ± 0.0002 | NS |
| + | 0.0010 ± 0.0004 | 0.0010 ± 0.0006 | 0.0005 ± 0.0005 | ||||
| CCR5 | − | 0.0358 ± 0.0169 | NS | 0.0215 ± 0.0032 | NS | 0.0082 ± 0.0008 | NS |
| + | 0.0108 ± 0.0031 | 0.0290 ± 0.0028 | 0.0085 ± 0.0018 | ||||
| CCR6 | − | 0.0002 ± 0.0001 | NS | 0.0003 ± 0.0001 | NS | 0.0001 ± 0.0000 | NS |
| + | 0.0002 ± 0.0000 | 0.0003 ± 0.0001 | 0.0001 ± 0.0000 | ||||
| CCR7 | − | 0.0015 ± 0.0002 | NS | 0.0014 ± 0.0004 | NS | 0.0032 ± 0.0025 | NS |
| + | 0.0011 ± 0.0006 | 0.0036 ± 0.0007 | 0.0007 ± 0.0002 | ||||
| CCR8 | − | 0.0001 ± 0.0001 | NS | 0.0002 ± 0.0001 | NS | 0.0001 ± 0.0000 | NS |
| + | 0.0001 ± 0.0000 | 0.0002 ± 0.0001 | 0.0001 ± 0.0000 | ||||
| CXCR7 | − | 0.0038 ± 0.0004 | NS (0.07) | 0.0104 ± 0.0061 | NS | 0.0027 ± 0.0004 | NS |
| + | 0.0081 ± 0.0019 | 0.0080 ± 0.0035 | 0.0021 ± 0.0009 | ||||
| CX3CR1 | − | 0.0005 ± 0.0002 | NS | 0.0010 ± 0.0004 | NS | 0.0009 ± 0.0003 | NS |
| + | 0.0002 ± 0.0001 | 0.0030 ± 0.0017 | 0.0004 ± 0.0001 |
NS indicates not significant. Data are means ± SEM.
Compared with vehicle-treated mice on the same day by Student t test.
COX-2-Dependent Recruitment of BM Cells to the Tumor Stroma
To evaluate the role of COX-2–derived PGs in the recruitment of BM cells to tumor stroma, we produced BM chimeric mice with GFP-positive BM. LLC cells were implanted subcutaneously after intravenous injections of BM cells isolated from GFP-transgenic mice and reconstitution of the BM. Stromal tissues were isolated on day 14, and GFP fluorescence intensity was determined. Celecoxib treatment significantly reduced the stromal fluorescence intensity (see Supplemental Figure S1A at http://ajp.amjpathol.org) and the mRNA levels of GFP significantly (see Supplemental Figure S1B at http://ajp.amjpathol.org). Furthermore, we established that the expression of CXCL12 and CXCR4 was reduced on COX-2 inhibition (see Supplemental Figure S1, C and D at http://ajp.amjpathol.org). These results suggest that COX-2-derived PGs regulate the recruitment of BM cells to the tumor stroma.
Effect of Topical Application of PGE2 on Stromal Formation in a Matrigel Implantation Model
To determine the role of PGE2 in stromal formation, we used the Matrigel implantation model. This model is characterized by the formation of the granulation tissues around the gel implants36 and is designed to mimic tumor-associated angiogenesis. Application of PGE2 enhanced angiogenesis, as gauged by measurements of MVD (see Supplemental Figure S2A at http://ajp.amjpathol.org) and MVA (see Supplemental Figure S2B at http://ajp.amjpathol.org), and stromal thickness (see Supplemental Figure S2C at http://ajp.amjpathol.org). PGE2 also increased the expression of CXCL12 and CXCR4 (see Supplemental Figure S2, D and E at http://ajp.amjpathol.org). These results imply that COX-2–derived PGE2 may enhance stromal formation and angiogenesis.
COX-2–Dependent Enhancement of Stromal Formation and Angiogenesis around Micropore Chambers, and the Involvement of CXCL12/CXCR4 Signaling
To evaluate stromal reactions without infiltration of tumor cells, we used micropore chambers filled with LLC cells, which allowed the secretion of soluble factors from LLC cells, and the separation of stromal tissues from LLC cells. We first tested the effects of COX-2 inhibition on tumor-associated stromal formation. The administration of Celecoxib strongly reduced granulation tissue formation, mimicking the tumor stromal reactions, and was accompanied by suppressed angiogenesis as determined by MVD and MVA around the LLC-containing chambers (Figures 2, A, B, and C). As shown in Figure 2, D and E, Celecoxib suppressed the formation of granulation tissues on day 14. COX-2 inhibition reduced the mRNA expression of CXCL12 (Figure 2F) and CXCR4 (Figure 2G) in the granulation tissues. An ELISA assay for CXCL12 revealed that immunoreactive CXCL12 levels in the granulation tissues were reduced under COX-2 inhibition (Figure 2H). The levels of VEGF-A were also reduced when COX-2 was inhibited (Figure 2I). Immunohistochemical examinations revealed that cells positive for CXCL12 or CXCR4 may have been mononuclear cells resembling macrophages or fibroblasts, and that the populations of CXCL12- and CXCR4-positive cells had been markedly reduced in granulation tissues by COX-2 inhibition (see Supplemental Figure S3, A–D at http://ajp.amjpathol.org). These results suggest that COX-2 facilitates not only tumor-associated angiogenesis, but also stromal formation, and that the CXCL12/CXCR4 axis may be involved in these effects.
Figure 2.
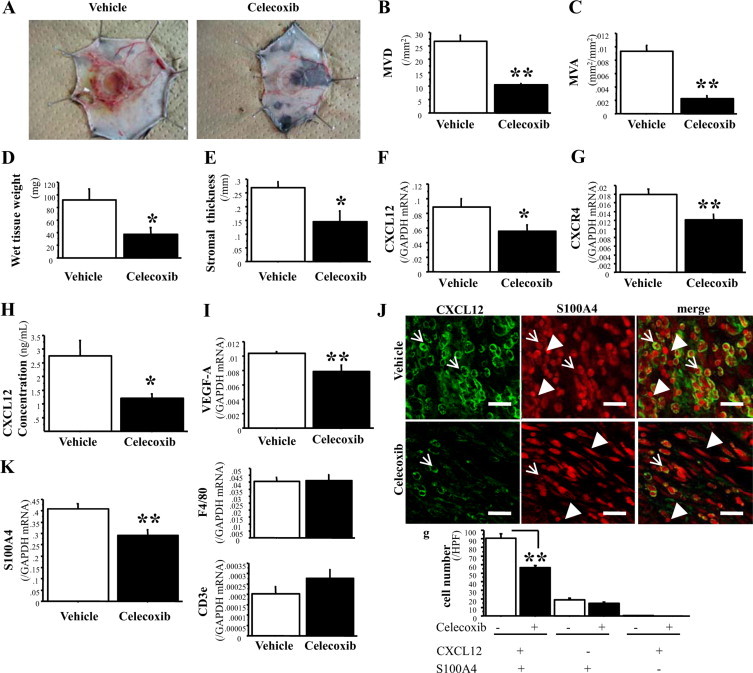
COX-2–dependent enhancement of stromal formation and angiogenesis around micropore chambers, and the involvement of CXCL12/CXCR4 signaling. A: Typical appearance of granulation tissues formed around micropore chambers. Diameter of chamber ring, 13 mm. B and C: Angiogenesis formed around micropore chambers, as measured by MVD and MVA. D and E: Stromal wet weight and stromal thickness. F, G and I: mRNA expression of CXCL12, CXCR4, and VEGF-A, respectively. H: CXCL12 protein level in granulation tissues formed around micropore chambers. J: Immunofluorescence analysis of CXCL12 (left) and S100A4 (middle) expression in tumor stoma from vehicle-treated mice (top row) and Celecoxib-treated mice (bottom row). Almost all (99.7%) of CXCL12+ cells are positive for S100A4; arrows; merged signal, yellow (right). Arrowheads indicate single labeled cells with S100A4. Scale bars = 20 μm. Quantification of each labeled cells is shown. Treatment with Celecoxib decreased the number of CXCL12+S100A4+ cells. K: mRNA expression of S100A4, F4/80, and CD3ε in granulation tissues. The results represent means ± SEM from 10 mice per group. *P < 0.05; **P < 0.01 by Student t test (compared with vehicle-treated mice).
Next, we performed immunohistochemistry and immunofluorescence of major components (fibroblasts, monocytes, and lymphocytes) in stromal cells to identify the cell type that has pivotal role in tumor stromal formation. Especially, fibroblasts are thought to be the main effecter cells in the stromal compartment for cancer cell progression and metastasis.37,38 α-SMA has been used as a marker of cancer-associated myofibroblasts, which are more competent in enhancing tumor progression39; however, the uncertainty in defining the myofibroblasts has been suggested.40 On the other hand, S100A4 expression is increased in activated fibroblasts and myofibroblasts and has been used as a highly specific marker of those cells,41–43 and moreover, S100A4 per se contributes to tumor progression.44 In fact, S100A4 expression was observed in more fibroblasts of normal tissues than α-SMA (see Supplemental Figure S4A at http://ajp.amjpathol.org). Analyzed by double labeling of S100A4 and α-SMA, most fibroblasts were S100A4+α-SMA+ (79.6 ± 3.1%) in the micropore chamber model, whereas only 0.3 ± 0.2% fibroblasts were single positive for α-SMA, as depicted in Supplemental Figure S5 (see Supplemental Figure S5 at http://ajp.amjpathol.org). These fibroblasts lineage rate was not altered, though total numbers of fibroblasts were decreased by COX-2 inhibition. These results suggest that S100A4-positive cells represented tumor stromal fibroblasts in this study. Celecoxib treatment significantly decreased the expression of the S100A4 gene in the granulation tissues, whereas the expression of F4/80 and CD3ε (markers for monocytes and lymphocytes, respectively) was not affected by COX-2 inhibition (Figure 2K). In fact, the numbers of fibroblasts in stromal tissues that stained with the S100A4 antibody were declined on COX-2 inhibition, as observed by immunohistochemical analysis (see Supplemental Figure S3, E and F at http://ajp.amjpathol.org). Because immunohistological stadies suggested that cells positive for CXCL12 or CXCR4 may have been mononuclear cells resembling fibroblast, we performed double immunofluorescence of CXCL12 and S100A4. Interestingly, double labeling assay of CXCL12 and S100A4 revealed that almost all (99.7%) CXCL12-positive cells were S100A4-positive fibroblasts, and the numbers of CXCL12+S100A4+ stromal cells were significantly decreased by COX-2 inhibition (Figure 2J). These results suggest that CXCL12/CXCR4 signaling recruits fibroblasts in a COX-2–dependent manner and thereby has a role in tumor-associated stromal formation.
Most Fibroblasts in Tumor Stroma Were Recruited from BM and Predominantly Produced CXCL12
To identify the cells that express CXCL12 and CXCR4, we also used tumor implantation model (n = 5). Tumor and stromal tissue were excised 14 days after the inoculation of LLC cells. Double labeling of CXCL12 and each cell marker (S100A4, F4/80, and CD3ε) showed that fibroblasts predominantly expressed CXCL12 and CXCR4 in the stroma (94.4 ± 1.5% of CXCL12-positive cells and 87.8 ± 2.5% of CXCR4-positive cells) in accordance with the above micropore chamber model (Figure 3, A–R).
Figure 3.
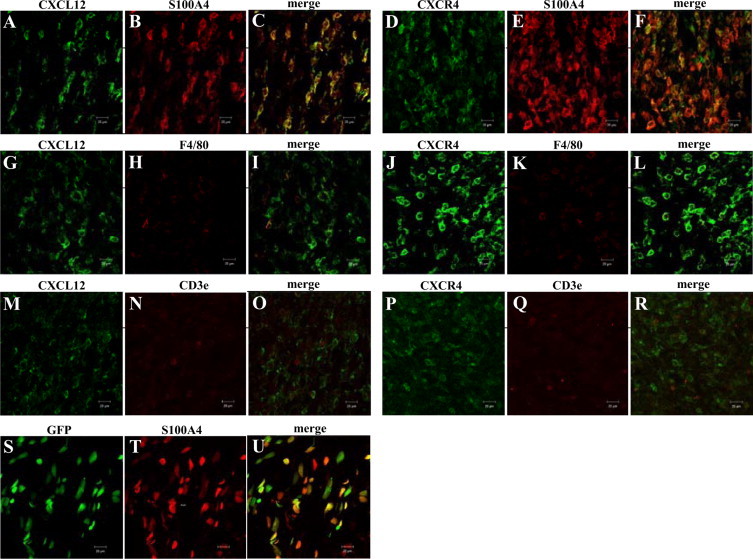
A–R: Double labeling analysis of CXCL12 or CXCR4 with S100A4, F4/80, or CD3ε in tumor stoma 14 days after tumor implantation. Among the CXCL12-positive cells in the tumor stroma, 94.4% are S100A4-positive (A–C), 6.4% are F4/80-positive (G–I), and 0% are CD3ε-positive (M–O). Among CXCR4-positive cells, 87.8% are S100A4-positive (D–F), 5.8% are F4/80-positive (J–L), and 0% are CD3ε-positive (P–R). Most CXCL12+ or CXCR4+ stromal cells were S100A4-positive fibroblasts. S–U: Double labeling assay of GFP and S100A4 in tumor stoma of tumor implantation model in GFP-BM chimeric mice. 92.3% of GFP+ cells were positive for S100A4. And GFP+S100A4+ cells occupied 89.6% of S100A4-positive stromal fibroblasts. Scale bars = 20 μm.
Similarly, we next evaluated tumor implantation model in GFP-BM chimeric mice to discriminate stromal fibroblasts whether they are recruited from BM or not (n = 4). Interestingly, GFP+S100A4+ cells reached 89.6 ± 1.0% of S100A4-positive stromal fibroblasts, suggesting that most tumor stromal fibroblasts were recruited from BM (Figure 3, S–U). These results suggest that most fibroblasts expressing CXCR4 are recruited from BM to tumor stroma and produce CXCL12, leading to further recruitment of stromal fibroblasts.
SDF-1α (CXCL12) Enhances Stromal Formation in a Matrigel Implantation Model
To confirm that CXCL12 has a role in enhancement of stromal formation and angiogenesis, we exposed Matrigel implants to topical injections of CXCL12 (SDF-1α). CXCL12 injections enhanced granulation tissue formation mimicking the stromal formation and angiogenesis (Figure 4, A, B, C, and D) and upregulated the expression of VEGF-A (Figure 4E).
Figure 4.
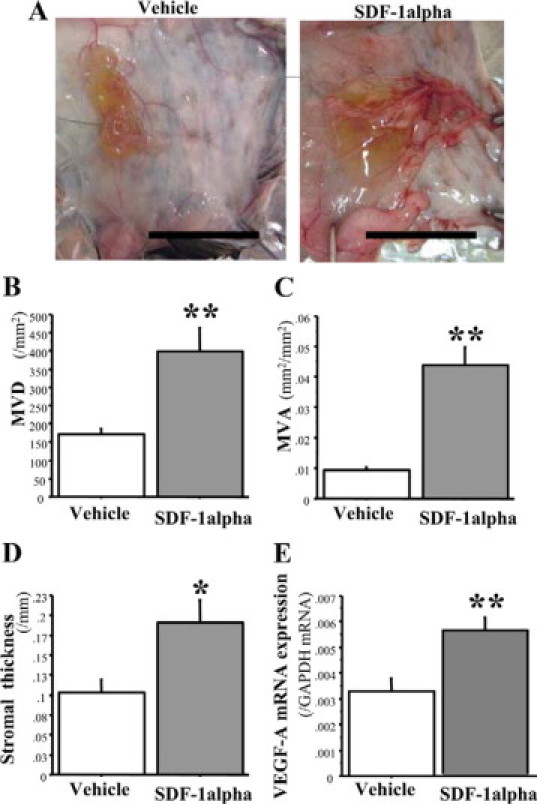
CXCL12 (SDF-1α) enhances granulation tissue formation that mimics the stromal reaction and angiogenesis in a Matrigel implantation model. A: Typical appearance of stromal formation and angiogenesis formed around a Matrigel. Scale bars = 10 mm. B and C: Angiogenesis (MVD and MVA) was significantly promoted by CXCL12 application. D: Stromal thickness was markedly increased by CXCL12 application. E: mRNA expression of VEGF-A in stroma formed around a Matrigel. The results represent means ± SEM from 11 mice per group. *P < 0.05; **P < 0.01 by Student t test (compared with vehicle-treated mice).
CXCR4 Neutralizing Antibody Reduced Angiogenesis and Granulation Tissue Formation around Micropore Chambers
We further tested the effect of topical injections of the neutralizing antibody against CXCR4 (2B11) around a micropore chamber containing LLC cells. The CXCR4 antibody markedly suppressed stromal formation and angiogenesis (Figure 5, A, B, C, D, and E). The expression of VEGF-A mRNA in the stroma was also suppressed by 2B11 (Figure 5F). The stromal expression of S100A4 gene was significantly down-regulated by 2B11 compared with control IgG, whereas expressions of the F4/80 and CD3ε genes were not influenced (Figure 5, G, H, and I). These results suggest that CXCR4 signaling has a crucial role in tumor-associated stromal formation and angiogenesis in this model.
Figure 5.
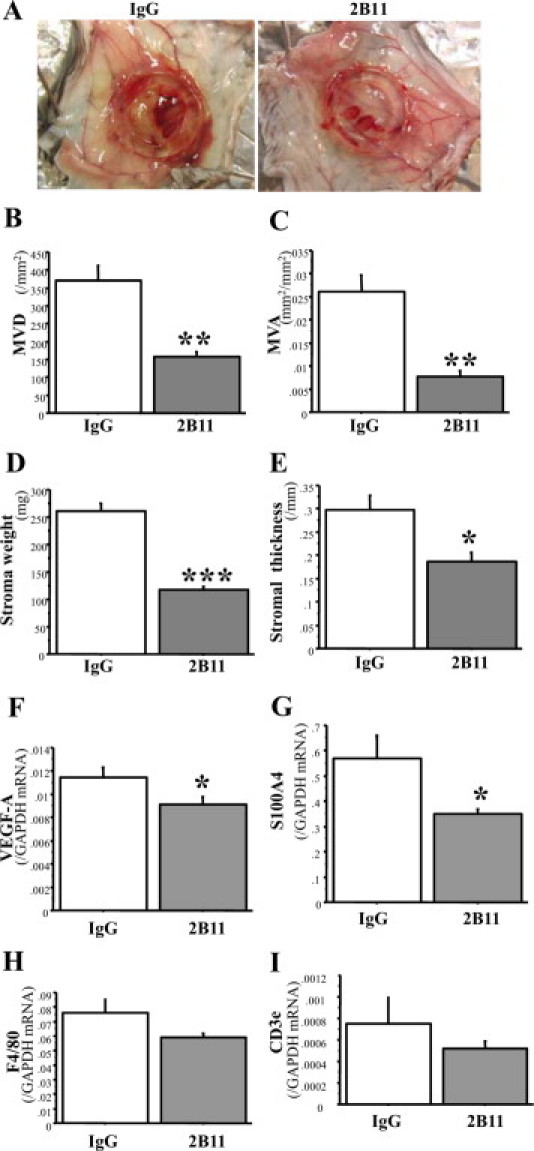
CXCR4-neutralizing antibody (2B11) reduces angiogenesis and granulation tissue formation around micropore chambers. A: Typical appearance of stromal formation and angiogenesis formed around micropore chambers. Diameter of chamber ring, 13 mm. B and C: Angiogenesis (MVD and MVA) in the granulation tissues around micropore chambers. D and E: Stromal formation (stromal wet weight and stromal thickness). F: mRNA expression of VEGF-A in the granulation tissues. G, H and I: mRNA expression of S100A4 (G), F4/80 (H), and CD3ε (I) in granulation tissues. n = 11 in IgG-treated group and n = 15 in 2B11-treated group. The results represent means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 by Student t test (compared with control IgG-treated mice).
Identification of the EP Receptor Signaling that Facilitates Stromal Formation in the Tumor Implantation Model
We previously showed that COX-2/PGE2 promotes tumor angiogenesis and tumor growth.23,24 In the present study, we identified the EP receptors (EP1, EP2, EP3, and EP4) responsible for stromal formation in tumor implantation model. Fourteen days after tumor inoculation, tumor stromal formation was significantly suppressed in EP3−/− mice and EP4−/− mice, whereas disruption of either EP1 or EP2 did not affect tumor stromal formation (Figure 6, A and B). The expressions of CXCL12 and CXCR4 genes (Figure 6, C and D) and of CXCL12 protein (Figure 6E) in stromal tissues were suppressed in EP3−/− mice and in EP4−/− mice as confirmed immunohistochemically in Supplemental Figure S6, A–F at http://ajp.amjpathol.org. The expression of S100A4 gene was significantly suppressed in EP3−/− and EP4−/− mice, but not in EP1−/− and EP2−/− mice (Figure 6F). The populations of S100A4-positive cells were sparse in tumor stroma in EP3−/− mice and EP4−/− mice, compared with wild-type mice (see Supplemental Figure S6, G–I at http://ajp.amjpathol.org). These results suggest that endogenous COX-2–derived PGE2 may enhance tumor stromal formation by increasing the recruitment of fibroblasts via the CXCL12/CXCR4 axis, which is modulated by EP3/EP4 signaling.
Figure 6.
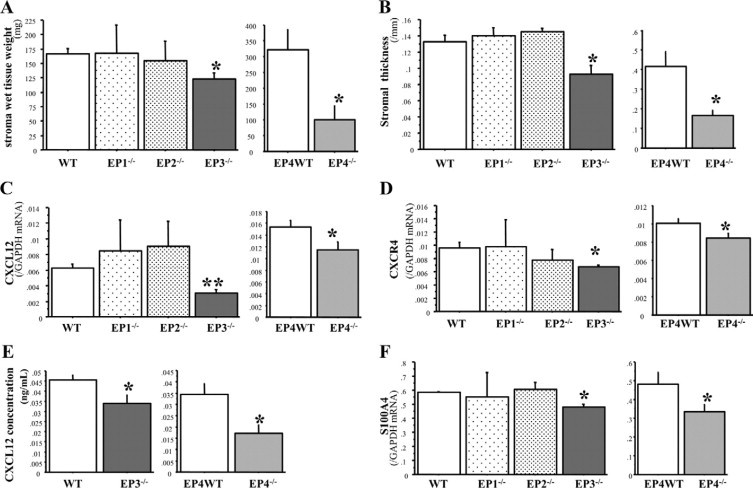
Identification of EP receptor signaling pathways that facilitate of stromal formation in the tumor implantation model. A and B: Stromal wet tissue weight and stromal thickness in each EP receptor knockout mice. C and D: mRNA expression of CXCL12 and CXCR4 in stromal tissue in each EP receptor knockout mice. E: CXCL12 protein level in stromal tissue of wild-type, EP3−/−, and EP4−/− mice. F: mRNA expression of S100A4 in stromal tissue in EP receptor knockout mice. n = 10, 4, 5, 8, 8, and 7 in wild-type (WT), EP1−/−, EP2−/−, EP3−/−, EP4WT, and EP4−/−, respectively. The results represent means ± SEM. *P < 0.05; **P < 0.01 by Student t test (compared with wild-type mice in EP1, 2, and 3−/− mice, EP4WT mice were used for comparison as to EP4−/− mice).
EP3 or EP4 Specific Agonist Stimulates CXCL12 Expression by Fibroblasts
We evaluated the effect of PGE2 and each EP specific agonists on CXCL12 expression by fibroblasts. PGE2 at 1 to 100 nmol/L markedly stimulated CXCL12 gene expression by fibroblasts (Figure 7). Similarly, either EP3 or EP4 specific stimulation significantly increased CXCL12 expression as the extent of PGE2 effects, whereas neither EP1 nor EP2 stimulation did. These results suggest that CXCL12 induction elicited by endogenous PGE2 is mediated by both EP3 and EP4 receptors on fibroblasts.
Figure 7.
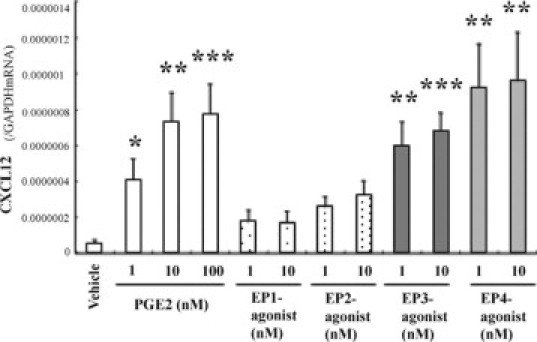
Effects of EP1, EP2, EP3, and EP4 specific agonists on CXCL12 gene expression by L929 fibroblasts. L929 fibroblasts (3 × 105 cells per well) cultured in six-well plates were incubated for 24 hours with the respective agonists, then CXCL12 gene expression was measured. Data were expressed as the mean ± SEM of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001 by Student t test (compared with vehicle-treated cells).
Discussion
In the present study, we have shown that COX-2–derived PGE2 upregulates the expression of chemokines and their receptors, CXCL12-CXCR4. Furthermore, we established that the upregulation of this chemokine system enhanced stromal formation, in which fibroblast recruitment was predominantly regulated by COX-2–derived PGE2, and facilitated tumor-associated angiogenesis. These effects were mediated by EP3/EP4 signaling in the host tissues. Thus, we propose that the chemokine systems, CXCL12-CXCR4, together with COX-2 and the EP receptors, may be used as effective therapeutic targets to treat cancer.
Growth of carcinomas, the most common types of human cancer, is known to be regulated by the tumor microenvironment, which consists of extracellular matrix and a variety of cells, including inflammatory cells, endothelial cells, and fibroblasts, which can generate or release various cytokines and growth factors.45 In previous studies, we showed that COX-2/PGE2 signaling is crucial for tumor angiogenesis, and that this signaling was transduced through the EP3 receptor on the host stromal cells rather than on the tumor cells.23 Moreover, the stromal cells expressing EP3 receptors, which were recruited from BM, have a significant role in enhancing angiogenesis and tumor growth.31 One of the most striking phenotypes of tumors treated with COX-2 inhibitor, and also of tumors in EP3 receptor knockout mice, is a reduced formation of tumor stromal tissues around tumors. These results suggest that COX-2 and host signaling of EP receptors may regulate the stromal tissue formation that constitutes the tumor microenvironment, and that the microenvironment could affect tumor-associated angiogenesis and tumor growth in a PG-dependent manner. Indeed, the tumor implantation model in GFP-BM chimeric mice revealed that recruitment of BM cells around the tumors was dependent on COX-2 activity (see Supplemental Figure S1 at http://ajp.amjpathol.org). These results allow us to test the involvement of chemokines and their receptors in tumor stromal formation, which determines the tumor microenvironment.
Chemokines were initially characterized as being chemoattractants for leukocytes that infiltrate into inflamed tissues46,47; however, recent works have demonstrated novel roles for chemokines and their receptors in regulating various aspects of the transformed phenotype, such as tumor growth, angiogenesis, invasion, and metastasis. Here, we tested the involvement of chemokines and chemokine receptors in tumor stromal formation in relation to tumor–stroma interactions which regulate tumor growth and tumor-associated angiogenesis. We characterized the expression of several chemokines and their receptors in tumor stromal tissues after inoculation of LLC cells (Table 2). Substantial expression of CCL2, CCL3, CCL4, CCL5, CCL6, CCL7, CCL8, CCL9, and CCL12 was detected in the stromal tissues, but COX-2 inhibition did not alter the mRNA levels of these chemokines. The CCR1, CCR2, CCR3, CCR5, CCR6, CCR7, CCR8, and CX3CR1 receptors were detected in tumor stromal tissues, and a COX-2 inhibitor did not significantly alter their expression pattern. In the present study, we found that the expression of CXCL12 and its receptor CXCR4 was reduced on COX-2 inhibition, suggesting that the CXCL12/CXCR4 axis may be involved in the regulation of tumor stromal formation by COX-2–derived PGs. Additionally, the gene expression of CXCR7, an alternative receptor for CXCL12, tended to increase only at early time point (Table 2). Recent data have suggested that CXCR7 expression on cancer cells may promote tumor progression and metastasis.34,48,49 On the other hand, CXCR7 may impair CXCR4 in its ability to activate G proteins after CXCL12 binding and modulate CXCL12-mediated chemotaxis by heterodimerization with CXCR4.50 Thus, a role of CXCR7 in the recruitment of stromal cells also needs to be assessed in future studies. Besides, CXC chemokines containing an ELR motif (glutamic acid-leucine-arginine) are thought to promote angiogenesis, whereas CXC chemokines lacking this sequence are considered to be angiostatic (with the exception of CXCL12).15,35 In the present study, the gene expressions of angiostatic CXCR3 chemokine family were increased by COX-2 inhibition, whereas those of angiogenic CXCR2 chemokine family were not affected. These results may imply suppressive COX-2 activity against angiostatic CXCR3 chemokine family in tumor stroma, although further studies are needed.
In the present study, we used a micropore chamber filled with LLC cells to further investigate how BM cells are recruited to form the stroma. With this model, one can separate the stromal tissues from the tumor cells and avoid contamination of tumor cells in the tested sample of stromal tissues. We clarified that endogeneous COX-2–derived PGs regulated tumor stromal formation via the CXCL12/CXCR4 axis. Recruitment of fibroblasts was regulated by COX-2–derived PG in this chamber model. Fibroblasts are the major components of stromal tissues in various cancers.51–53 Although fibroblasts are known to produce growth factors that influence cancer cell progression, they had been thought to have a passive role in cancer progression, responding to signals from cancer cells. However, it has recently been reported that tumor stromal fibroblasts, named cancer-associated fibroblasts, are crucial for cancer progression,51,54,55 and, moreover, may be potential inducers of certain carcinomas.56 Epidemiological analysis revealed that elevated levels of stromal fibroblasts are correlated with a poor prognosis for human carcinoma patients.57 Therefore, modifications in the stromal fibroblasts may regulate the overall development of cancer.54 Cancer-associated fibroblasts have been reported to originate partly from BM-derived progenitor cells.58–60 α-SMA has been used as a marker of cancer-associated myofibroblasts, which are major component of cancer-associated fibroblasts,39 although the uncertainty in defining the myofibroblasts exists.40 We used S100A4 as a marker of tumor stromal fibroblasts in this study. S100A4 expression is increased in activated fibroblasts and myofibroblasts and has been used as a highly specific marker of those cells,41–43 and moreover, S100A4 per se causes tumor progression.44 Intriguingly, most fibroblasts positive for α-SMA were also S100A4-positive, and the rate of fibroblast lineages (α-SMA+/S100A4+) was not altered by COX-2 inhibition notwithstanding total fibroblasts were decreased. These results suggest that S100A4 may be a good marker for tumor-associated activated fibroblast in tumor stroma. In the present study, COX-2/PGE2 was found to regulate the recruitment of stromal cells from BM, which was mainly composed of fibroblasts that expressed CXCR4 and producing CXCL12, resulting in tumor growth.
The principal COX-2 product in tumors is PGE2, which acts through four G protein–coupled receptors designated EP1, EP2, EP3, and EP4.61,62 In the present study, EP3 and EP4 receptor signaling was found to be crucial for tumor stromal formation and tumor growth. We previously considered the AP-1–dependent upregulation of VEGF to be important in tumor-associated angiogenesis mediated by the EP3 receptor, because some splicing variants of EP3 that link to the activation of adenylate cyclases may result in tumor-associated angiogenesis23; however, the EP3 receptor also activates the Raf and MAPK pathway.63 EP4 receptor signaling is known to increase cAMP levels in some types of cells.28 As shown in this study, EP3 and EP4 receptors have similar roles in the enhancement of stromal formation. Thus, the activation of adenylate cyclase through binding to G proteins stimulates cAMP production and, in turn, activation of cAMP-dependent protein kinase A may be involved in tumor stromal formation.64 EP4 receptor also activates Akt and ERK-1/2.65 Further, EP4 agonist–induced cancer cell migration is known to be regulated by ERK,66,67 suggesting that tumor stromal formation may also be regulated by ERK activation. CXCR4 expression was reported to be upregulated at least in part by a cAMP-responsive element in human T lymphocytes.68 ERK activation was reported to induce CXCL12 and CXCR4 in neurons.69 Thus, ERK may enhance EP3-/EP4-dependent stromal formation.
Stimulation of VEGF and bFGF was reported to increase the expression of CXCR4 on endothelial cells. Further, PGE2 has been shown to mediate the effects of bFGF and VEGF by upregulating CXCR4 expression in human microvascular endothelial cells.70 These findings indicate that PGE2 is a mediator of VEGF- and bFGF-induced CXCR4-dependent neovessel assembly in vivo and show that the angiogenic effects of PGE2 require CXCR4 expression.70 In the present study, the recruitment of fibroblasts expressing CXCL12 and CXCR4 to the tumor stroma was found to have a significant role in enhancing angiogenesis and tumor growth. CXCL12 may act on the fibroblasts in an autocrine or paracrine fashion in tumor stroma. PGE2-EP3/EP4 signaling in tumor stroma may promote tumor stromal formation and tumor growth mainly through the host CXCL12/CXCR4 axis by recruiting stromal fibroblasts.
In conclusion, PGE2-EP3/EP4 signaling appears to be crucial for tumor-associated stromal formation and tumor growth. EP3 and EP4 signaling on the stromal fibroblasts was correlated with the induction of a potent chemotactic and proangiogenic cytokine, CXCL12, in stromal fibroblasts. The subsequent upregulation of CXCL12/CXCR4 signaling facilitated tumor stromal formation by accelerating the recruitment of fibroblasts, which resulted in tumor growth. Highly selective EP3, EP4, and CXCR4 antagonists may therefore serve as novel therapeutic tools to treat cancer. These results may elucidate how nonsteroidal anti-inflammatory drugs prevent tumor development.
Acknowledgements
We thank Ms. Michiko Ogino and Ms. Kyoko Yoshikawa for their technical assistance, and Bioedit for correcting grammatical errors.
Footnotes
Supported by a grant from the Research Program of the Graduate School of Medical Sciences, Kitasato University.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
mRNA expression of angiogenic/angiostatic CXC chemokines and those receptors in the stroma of murine tumor model
References
- 1.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cher ML, Towler DA, Rafii S, Rowley D, Donahue HJ, Keller E, Herlyn M, Cho EA, Chung LW. Cancer interaction with the bone microenvironment: a workshop of the National Institutes of Health Tumor Microenvironment Study Section. Am J Pathol. 2006;168:1405–1412. doi: 10.2353/ajpath.2006.050874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, Chadburn A, Heissig B, Marks W, Witte L, Wu Y, Hicklin D, Zhu Z, Hackett NR, Crystal RG, Moore MA, Hajjar KA, Manova K, Benezra R, Rafii S. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 5.Seandel M, Butler J, Lyden D, Rafii S. A catalytic role for proangiogenic marrow-derived cells in tumor neovascularization. Cancer Cell. 2008;13:181–183. doi: 10.1016/j.ccr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, Blanchard D, Bais C, Peale FV, van Bruggen N, Ho C, Ross J, Tan M, Carano RA, Meng YG, Ferrara N. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450:825–831. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- 7.Shojaei F, Zhong C, Wu X, Yu L, Ferrara N. Role of myeloid cells in tumor angiogenesis and growth. Trends Cell Biol. 2008;18:372–378. doi: 10.1016/j.tcb.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Tejada ML, Yu L, Dong J, Jung K, Meng G, Peale FV, Frantz GD, Hall L, Liang X, Gerber HP, Ferrara N. Tumor-driven paracrine platelet-derived growth factor receptor alpha signaling is a key determinant of stromal cell recruitment in a model of human lung carcinoma. Clin Cancer Res. 2006;12:2676–2688. doi: 10.1158/1078-0432.CCR-05-1770. [DOI] [PubMed] [Google Scholar]
- 9.Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 10.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 11.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–18. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 12.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 13.Mantovani A, Allavena P, Sozzani S, Vecchi A, Locati M, Sica A. Chemokines in the recruitment and shaping of the leukocyte infiltrate of tumors. Semin Cancer Biol. 2004;14:155–160. doi: 10.1016/j.semcancer.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 15.Strieter RM, Burdick MD, Mestas J, Gomperts B, Keane MP, Belperio JA. Cancer CXC chemokine networks and tumour angiogenesis. Eur J Cancer. 2006;42:768–778. doi: 10.1016/j.ejca.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Ao M, Franco OE, Park D, Raman D, Williams K, Hayward SW. Cross-talk between paracrine-acting cytokine and chemokine pathways promotes malignancy in benign human prostatic epithelium. Cancer Res. 2007;67:4244–4253. doi: 10.1158/0008-5472.CAN-06-3946. [DOI] [PubMed] [Google Scholar]
- 17.Darash-Yahana M, Pikarsky E, Abramovitch R, Zeira E, Pal B, Karplus R, Beider K, Avniel S, Kasem S, Galun E, Peled A. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004;18:1240–1242. doi: 10.1096/fj.03-0935fje. [DOI] [PubMed] [Google Scholar]
- 18.Ishida T, Ueda R. CCR4 as a novel molecular target for immunotherapy of cancer. Cancer Sci. 2006;97:1139–1146. doi: 10.1111/j.1349-7006.2006.00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Issa A, Le TX, Shoushtari AN, Shields JD, Swartz MA. Vascular endothelial growth factor-C and C-C chemokine receptor 7 in tumor cell-lymphatic cross-talk promote invasive phenotype. Cancer Res. 2009;69:349–357. doi: 10.1158/0008-5472.CAN-08-1875. [DOI] [PubMed] [Google Scholar]
- 20.Menu E, De Leenheer E, De Raeve H, Coulton L, Imanishi T, Miyashita K, Van Valckenborgh E, Van Riet I, Van Camp B, Horuk R, Croucher P, Vanderkerken K. Role of CCR1 and CCR5 in homing and growth of multiple myeloma and in the development of osteolytic lesions: a study in the 5TMM model. Clin Exp Metastasis. 2006;23:291–300. doi: 10.1007/s10585-006-9038-6. [DOI] [PubMed] [Google Scholar]
- 21.Robinson SC, Scott KA, Wilson JL, Thompson RG, Proudfoot AE, Balkwill FR. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res. 2003;63:8360–8365. [PubMed] [Google Scholar]
- 22.Dannenberg AJ, Subbaramaiah K. Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell. 2003;4:431–436. doi: 10.1016/s1535-6108(03)00310-6. [DOI] [PubMed] [Google Scholar]
- 23.Amano H, Hayashi I, Endo H, Kitasato H, Yamashina S, Maruyama T, Kobayashi M, Satoh K, Narita M, Sugimoto Y, Murata T, Yoshimura H, Narumiya S, Majima M. Host prostaglandin E(2)-EP3 signaling regulates tumor-associated angiogenesis and tumor growth. J Exp Med. 2003;197:221–232. doi: 10.1084/jem.20021408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshida S, Amano H, Hayashi I, Kitasato H, Kamata M, Inukai M, Yoshimura H, Majima M. COX-2/VEGF-dependent facilitation of tumor-associated angiogenesis and tumor growth in vivo. Lab Invest. 2003;83:1385–1394. doi: 10.1097/01.lab.0000090159.53224.b9. [DOI] [PubMed] [Google Scholar]
- 25.Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 26.Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 27.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 28.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 29.Kamoshita E, Ikeda Y, Fujita M, Amano H, Oikawa A, Suzuki T, Ogawa Y, Yamashina S, Azuma S, Narumiya S, Unno N, Majima M. Recruitment of a prostaglandin E receptor subtype. EP3-expressing bone marrow cells is crucial in wound-induced angiogenesis. Am J Pathol. 2006;169:1458–1472. doi: 10.2353/ajpath.2006.051358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayashi I, Amano H, Yoshida S, Kamata K, Kamata M, Inukai M, Fujita T, Kumagai Y, Furudate S, Majima M. Suppressed angiogenesis in kininogen-deficiencies. Lab Invest. 2002;82:871–880. doi: 10.1097/01.lab.0000018885.36823.d6. [DOI] [PubMed] [Google Scholar]
- 31.Ogawa Y, Suzuki T, Oikawa A, Hosono K, Kubo H, Amano H, Kitasato H, Hayashi I, Kato T, Sugimoto Y, Narumiya S, Watanabe M, Majima M. Bone marrow-derived EP3-expressing stromal cells enhance tumor-associated angiogenesis and tumor growth. Biochem Biophys Res Commun. 2009;382:720–725. doi: 10.1016/j.bbrc.2009.03.094. [DOI] [PubMed] [Google Scholar]
- 32.Suzawa T, Miyaura C, Inada M, Maruyama T, Sugimoto Y, Ushikubi F, Ichikawa A, Narumiya S, Suda T. The role of prostaglandin E receptor subtypes (EP1. EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the respective EPs. Endocrinology. 2000;141:1554–1559. doi: 10.1210/endo.141.4.7405. [DOI] [PubMed] [Google Scholar]
- 33.Fujita M, Hayashi I, Yamashina S, Itoman M, Majima M. Blockade of angiotensin AT1a receptor signaling reduces tumor growth, angiogenesis, and metastasis. Biochem Biophys Res Commun. 2002;294:441–447. doi: 10.1016/S0006-291X(02)00496-5. [DOI] [PubMed] [Google Scholar]
- 34.Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold ME, Sunshine MJ, Littman DR, Kuo CJ, Wei K, McMaster BE, Wright K, Howard MC, Schall TJ. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203:2201–2213. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vandercappellen J, Van Damme J, Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008;267:226–244. doi: 10.1016/j.canlet.2008.04.050. [DOI] [PubMed] [Google Scholar]
- 36.Kleinman HK, McGarvey ML, Liotta LA, Robey PG, Tryggvason K, Martin GR. Isolation and characterization of type IV procollagen, laminin, and heparan sulfate proteoglycan from the EHS sarcoma. Biochemistry. 1982;21:6188–6193. doi: 10.1021/bi00267a025. [DOI] [PubMed] [Google Scholar]
- 37.Lloyd BH, Platt-Higgins A, Rudland PS, Barraclough R. Human S100A4 (p9Ka) induces the metastatic phenotype upon benign tumour cells. Oncogene. 1998;17:465–473. doi: 10.1038/sj.onc.1201948. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt-Hansen B, Klingelhofer J, Grum-Schwensen B, Christensen A, Andresen S, Kruse C, Hansen T, Ambartsumian N, Lukanidin E, Grigorian M. Functional significance of metastasis-inducing S100A4(Mts1) in tumor-stroma interplay. J Biol Chem. 2004;279:24498–24504. doi: 10.1074/jbc.M400441200. [DOI] [PubMed] [Google Scholar]
- 39.Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–1601. doi: 10.4161/cc.5.15.3112. Epub 2006 Aug 1591. [DOI] [PubMed] [Google Scholar]
- 40.Eyden B, Banerjee SS, Shenjere P, Fisher C. The myofibroblast and its tumours. J Clin Pathol. 2009;62:236–249. doi: 10.1136/jcp.2008.061630. [DOI] [PubMed] [Google Scholar]
- 41.Ambartsumian NS, Grigorian MS, Larsen IF, Karlstrom O, Sidenius N, Rygaard J, Georgiev G, Lukanidin E. Metastasis of mammary carcinomas in GRS/A hybrid mice transgenic for the mts1 gene. Oncogene. 1996;13:1621–1630. [PubMed] [Google Scholar]
- 42.Ryan DG, Taliana L, Sun L, Wei ZG, Masur SK, Lavker RM. Involvement of S100A4 in stromal fibroblasts of the regenerating cornea. Invest Ophthalmol Vis Sci. 2003;44:4255–4262. doi: 10.1167/iovs.03-0578. [DOI] [PubMed] [Google Scholar]
- 43.Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grum-Schwensen B, Klingelhofer J, Berg CH, El-Naaman C, Grigorian M, Lukanidin E, Ambartsumian N. Suppression of tumor development and metastasis formation in mice lacking the S100A4(mts1) gene. Cancer Res. 2005;65:3772–3780. doi: 10.1158/0008-5472.CAN-04-4510. [DOI] [PubMed] [Google Scholar]
- 45.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–337. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsushima K, Morishita K, Yoshimura T, Lavu S, Kobayashi Y, Lew W, Appella E, Kung HF, Leonard EJ, Oppenheim JJ. Molecular cloning of a human monocyte-derived neutrophil chemotactic factor (MDNCF) and the induction of MDNCF mRNA by interleukin 1 and tumor necrosis factor. J Exp Med. 1988;167:1883–1893. doi: 10.1084/jem.167.6.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshimura T, Matsushima K, Tanaka S, Robinson EA, Appella E, Oppenheim JJ, Leonard EJ. Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines. Proc Natl Acad Sci USA. 1987;84:9233–9237. doi: 10.1073/pnas.84.24.9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iwakiri S, Mino N, Takahashi T, Sonobe M, Nagai S, Okubo K, Wada H, Date H, Miyahara R. Higher expression of chemokine receptor CXCR7 is linked to early and metastatic recurrence in pathological stage I nonsmall cell lung cancer. Cancer. 2009;115:2580–2593. doi: 10.1002/cncr.24281. [DOI] [PubMed] [Google Scholar]
- 49.Miao Z, Luker KE, Summers BC, Berahovich R, Bhojani MS, Rehemtulla A, Kleer CG, Essner JJ, Nasevicius A, Luker GD, Howard MC, Schall TJ. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci US. 2007;104:15735–15740. doi: 10.1073/pnas.0610444104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood. 2009;113:6085–6093. doi: 10.1182/blood-2008-12-196618. [DOI] [PubMed] [Google Scholar]
- 51.Beacham DA, Cukierman E. Stromagenesis: the changing face of fibroblastic microenvironments during tumor progression. Semin Cancer Biol. 2005;15:329–341. doi: 10.1016/j.semcancer.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 52.Sappino AP, Schurch W, Gabbiani G. Differentiation repertoire of fibroblastic cells: expression of cytoskeletal proteins as marker of phenotypic modulations. Lab Invest. 1990;63:144–161. [PubMed] [Google Scholar]
- 53.Sappino AP, Skalli O, Jackson B, Schurch W, Gabbiani G. Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues. Int J Cancer. 1988;41:707–712. doi: 10.1002/ijc.2910410512. [DOI] [PubMed] [Google Scholar]
- 54.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 55.Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 56.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 57.Surowiak P, Suchocki S, Gyorffy B, Gansukh T, Wojnar A, Maciejczyk A, Pudelko M, Zabel M. Stromal myofibroblasts in breast cancer: relations between their occurrence, tumor grade and expression of some tumour markers. Folia Histochem Cytobiol. 2006;44:111–116. [PubMed] [Google Scholar]
- 58.Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, Alison MR, Wright NA. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004;64:8492–8495. doi: 10.1158/0008-5472.CAN-04-1708. [DOI] [PubMed] [Google Scholar]
- 59.Direkze NC, Jeffery R, Hodivala-Dilke K, Hunt T, Playford RJ, Elia G, Poulsom R, Wright NA, Alison MR. Bone marrow-derived stromal cells express lineage-related messenger RNA species. Cancer Res. 2006;66:1265–1269. doi: 10.1158/0008-5472.CAN-05-3202. [DOI] [PubMed] [Google Scholar]
- 60.Ishii G, Sangai T, Oda T, Aoyagi Y, Hasebe T, Kanomata N, Endoh Y, Okumura C, Okuhara Y, Magae J, Emura M, Ochiya T, Ochiai A. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem Biophys Res Commun. 2003;309:232–240. doi: 10.1016/s0006-291x(03)01544-4. [DOI] [PubMed] [Google Scholar]
- 61.Fulton AM, Ma X, Kundu N. Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer Res. 2006;66:9794–9797. doi: 10.1158/0008-5472.CAN-06-2067. [DOI] [PubMed] [Google Scholar]
- 62.Majima M, Amano H, Hayashi I. Prostanoid receptor signaling relevant to tumor growth and angiogenesis. Trends Pharmacol Sci. 2003;24:524–529. doi: 10.1016/j.tips.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 63.Namba T, Sugimoto Y, Negishi M, Irie A, Ushikubi F, Kakizuka A, Ito S, Ichikawa A, Narumiya S. Alternative splicing of C-terminal tail of prostaglandin E receptor subtype EP3 determines G-protein specificity. Nature. 1993;365:166–170. doi: 10.1038/365166a0. [DOI] [PubMed] [Google Scholar]
- 64.Chell S, Kadi A, Williams AC, Paraskeva C. Mediators of PGE2 synthesis and signalling downstream of COX-2 represent potential targets for the prevention/treatment of colorectal cancer. Biochim Biophys Acta. 2006;1766:104–119. doi: 10.1016/j.bbcan.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 65.Fujino H, Regan JW. Prostanoid receptors and phosphatidylinositol 3-kinase: a pathway to cancer? Trends Pharmacol Sci. 2003;24:335–340. doi: 10.1016/S0165-6147(03)00162-7. [DOI] [PubMed] [Google Scholar]
- 66.Pozzi A, Yan X, Macias-Perez I, Wei S, Hata AN, Breyer RM, Morrow JD, Capdevila JH. Colon carcinoma cell growth is associated with prostaglandin E2/EP4 receptor-evoked ERK activation. J Biol Chem. 2004;279:29797–29804. doi: 10.1074/jbc.M313989200. [DOI] [PubMed] [Google Scholar]
- 67.Rao R, Redha R, Macias-Perez I, Su Y, Hao C, Zent R, Breyer MD, Pozzi A. Prostaglandin E2-EP4 receptor promotes endothelial cell migration via ERK activation and angiogenesis in vivo. J Biol Chem. 2007;282:16959–16968. doi: 10.1074/jbc.M701214200. [DOI] [PubMed] [Google Scholar]
- 68.Cristillo AD, Highbarger HC, Dewar RL, Dimitrov DS, Golding H, Bierer BE. Up-regulation of HIV coreceptor CXCR4 expression in human T lymphocytes is mediated in part by a cAMP-responsive element. FASEB J. 2002;16:354–364. doi: 10.1096/fj.01-0744com. [DOI] [PubMed] [Google Scholar]
- 69.Luo Y, Lathia J, Mughal M, Mattson MP. SDF1alpha/CXCR4 signaling, via ERKs and the transcription factor Egr1, induces expression of a 67-kDa form of glutamic acid decarboxylase in embryonic hippocampal neurons. J Biol Chem. 2008;283:24789–24800. doi: 10.1074/jbc.M800649200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Salcedo R, Zhang X, Young HA, Michael N, Wasserman K, Ma WH, Martins-Green M, Murphy WJ, Oppenheim JJ. Angiogenic effects of prostaglandin E2 are mediated by up-regulation of CXCR4 on human microvascular endothelial cells. Blood. 2003;102:1966–1977. doi: 10.1182/blood-2002-11-3400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
mRNA expression of angiogenic/angiostatic CXC chemokines and those receptors in the stroma of murine tumor model