

Abstract
The metalloprotease ADAMTS9 participates in melanoblast development and is a tumor suppressor in esophageal and nasopharyngeal cancer. ADAMTS9 null mice die before gastrulation, but, ADAMTS9+/− mice were initially thought to be normal. However, when congenic with the C57Bl/6 strain, 80% of ADAMTS9+/− mice developed spontaneous corneal neovascularization. β-Galactosidase staining enabled by a lacZ cassette targeted to the ADAMTS9 locus showed that capillary endothelial cells (ECs) in embryonic and adult tissues and in capillaries growing into heterotopic tumors expressed ADAMTS9. Heterotopic B.16-F10 melanomas elicited greater vascular induction in ADAMTS9+/− mice than in wild-type littermates, suggesting a potential inhibitory role in tumor angiogenesis. Treatment of cultured human microvascular ECs with ADAMTS9 small-interfering RNA resulted in enhanced filopodial extension, decreased cell adhesion, increased cell migration, and enhanced formation of tube-like structures on Matrigel. Conversely, overexpression of catalytically active, but not inactive, ADAMTS9 in ECs led to fewer tube-like structures, demonstrating that the proteolytic activity of ADAMTS9 was essential. However, unlike the related metalloprotease ADAMTS1, which exerts anti-angiogenic effects by cleavage of thrombospondins and sequestration of vascular endothelial growth factor165, ADAMTS9 neither cleaved thrombospondins 1 and 2, nor bound vascular endothelial growth factor165. Taken together, these data identify ADAMTS9 as a novel, constitutive, endogenous angiogenesis inhibitor that operates cell-autonomously in ECs via molecular mechanisms that are distinct from those used by ADAMTS1.
Following de novo formation of the vasculature from undifferentiated mesodermal precursors during embryogenesis (vasculogenesis), endothelial cells (ECs) proliferate and migrate to generate new capillaries from pre-existing blood vessels, a process termed angiogenesis.1,2 Angiogenesis is critical for embryonic development, subsequent organ growth, and physiological processes such as menstruation and wound healing.3 It is a prominent feature of age-related macular degeneration and diabetic retinopathy, and tumor growth is angiogenesis-dependent.1,2 Owing to this considerable relevance for human pathology, the fundamental mechanisms that regulate angiogenesis have been the subject of intense investigation.
Proteases are considered to be key participants in angiogenesis because of their traditional roles in remodeling of the vascular basement membrane during EC sprouting and migration.4,5 However, proteases also influence EC behavior by shedding cell-surface receptors and growth factors, exposing cryptic adhesion sites, or releasing bioactive fragments.3,6 Thus, a role for proteases as negative regulators of angiogenesis is also accepted.3,7–9 The ADAMTS protease family contains 19 secreted mammalian metalloproteases that localize to the cell-surface and/or extracellular matrix.10,11 Several ADAMTS metalloproteases have specialized physiological roles that were revealed by analysis of human genetic disorders, or naturally occurring and engineered animal mutations,12–14 but the functions of other members of this family, such as ADAMTS9, are poorly understood. ADAMTS proteases share a complex modular structure, comprising a metalloprotease domain coupled to a large ancillary domain containing thrombospondin type-1 repeats (TSRs), which are the hallmark of this family.10 The TSRs of ADAMTS proteases are similar to those of the anti-angiogenic molecules thrombospondin-1 (TSP-1) and TSP-2.15–17 Indeed, ADAMTS1 was previously shown to inhibit angiogenesis via both proteolytic and non-proteolytic mechanisms, ie, by cleavage of TSP-1 and TSP-2, releasing anti-angiogenic fragments, as well as by sequestration of the pro-angiogenic growth factor vascular endothelial growth factor (VEGF)165 by the TSR-containing ancillary domain.9,18 Adamts1 mRNA is widely expressed during embryogenesis and in adult tissues, but its expression in the vasculature appears to be restricted to smooth muscle cells, and is not reported in capillary endothelium.19,20 Although Adamts1 null mice have significant perinatal lethality,21,22 and Adamts1−/− ovaries have fewer and larger ovarian blood vessels than normal, generalized defects in vascular development have not been reported.23 However, ADAMTS1 produced by keratinocytes and skin fibroblasts regulates the migration of EC during wound healing.24
ADAMTS9 is the most highly conserved member of the ADAMTS family, being similar to Caenorhabditis elegans Gon-1, which is required for nematode morphogenesis.25 ADAMTS9 not only has an identical active site sequence as ADAMTS1, but contains 15 potentially anti-angiogenic TSRs.26 ADAMTS9 was previously identified as a tumor suppressor gene in esophageal and nasopharyngeal cancer.27,28 Recent work demonstrated that ADAMTS9 worked cooperatively with another Gon-1-related protease, ADAMTS20, in the colonization of skin by neural crest-derived melanoblasts.29 Using in situ hybridization during murine development, we previously found that ADAMTS9 was expressed in capillaries.30 However, early embryonic lethality of ADAMTS9 null mice appeared to preclude analysis of its role in vascular development, as well as angiogenesis in the tumor context.29 We demonstrate here that when congenic with the C57Bl/6 strain, ADAMTS9+/− mice develop spontaneous corneal neovascularization within a few weeks of weaning and that heterotopic tumors in these mice attract more vasculature than wild-type mice. It is shown that ADAMTS9 acts via a cell-autonomous mechanism in microvascular endothelium and that it represents a nonredundant anti-angiogenic activity, since it does not share the mechanisms used by ADAMTS1, and differs from it in other fundamental respects.
Materials and Methods
Transgenic Animals, Tumor Implantation, and Tissue Analysis
Transgenic mice with targeted inactivation of ADAMTS9 by insertion of an IRES-lacZ cassette in exon 12 (encoding TSR 1) were obtained under license from Deltagen (San Carlos, CA). The targeted allele was bred for 10 generations into inbred C57Bl/6 mice to achieve a congenic strain, and minimize the influence of genetic variation on the phenotype. For analysis of ADAMTS9 expression during wound healing, full-thickness, circular, 10-mm diameter excisional wounds were made in dorsal skin of an 8-week-old male mouse. The wound bed with surrounding skin was excised 5 days later, fixed in 4% paraformaldehyde, stained for β-galactosidase (β-gal) as described below, and embedded in paraffin. For analysis of ADAMTS9 expression during tumor growth, isogenic tumor cells (B.16-F10 melanoma and Lewis lung carcinoma) were injected subcutaneously over the flank of 8-week-old male mice. Tumors were excised with surrounding tissue after 1 week, stained for β-gal as described below, and embedded in paraffin. For comparative analysis of tumor-induced vasculature in ADAMTS9+/− and wild-type littermates, 2 × 106 B.16-F10 melanoma cells were injected (day 0) on each flank of 8-week-old mice (n = 8 for ADAMTS9+/− mice, n = 10 for wild-type controls) and tumor-induced angiogenesis was assayed on day 9 essentially as previously described.31–33 These experiments were done in a double-blind design in which the mice were assigned a code and the author performing the tumor study (D.J.L.) was not provided with the genotype. Following completion of quantitative analysis, the code was broken and the data were analyzed using a Student's t-test. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic.
Histology, β-Gal Staining, and Immunohistochemistry
For routine histological analysis, mouse eyes and other tissues were fixed in 4% paraformaldehyde, followed by paraffin embedding and staining of 5- to 10-μm-thick sections with H&E. β-gal histochemistry was undertaken at different gestational ages and in adult (>3-month-old) tissues, excisional wound beds, and tumors as previously described.34 Immunohistochemistry of β-gal-stained paraffin sections was done using anti-PECAM-1 (rat monoclonal antibody MEC13.3, Pharmingen), anti-endomucin (rat monoclonal V.7C7)35 provided by Dr. Dietmar Vestweber, rat monoclonal anti-podoplanin antibody 8.1.1 (Developmental Studies Hybridoma Bank),36 rabbit polyclonal anti-NG2 (Millipore Corp., Billerica, MA) and anti-smooth muscle α-actin (SMA) monoclonal antibody (Sigma-Aldrich) in a conventional indirect immunoalkaline phosphatase method. Before incubation with the anti-endomucin primary antibody, sections were treated by antigen retrieval. Briefly, slides were immersed in citrate-EDTA buffer (10 mmol/L citric acid, 2 mmol/L EDTA, 0.05% v/v Tween-20, pH 6.2) and microwaved four times for 1.5 minutes at 50% power in a domestic microwave oven. The buffer was allowed to stand for 30 seconds between each microwave cycle. After four cycles of microwaving, slides were allowed to cool for 20 to 30 minutes in citrate-EDTA buffer.
Cell Culture, Transfection of Expression Plasmids and Small-Interfering RNA, and Reverse Transcription-PCR
Immortalized human brain microvascular ECs (HBMECs)37 were routinely cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum. Clonetics microvascular ECs from human dermis (HDMECs) and heart (HHMECs) were cultured according to the supplier's instructions (Cambrex Corporation, East Rutherford, NJ). Human umbilical vein ECs (HUVECs) were cultured in M199 medium (Invitrogen, Carlsbad, CA) containing 15% fetal bovine serum, 10 units/ml heparin, and fibroblast growth factor-2 (PeproTech, Rocky Hill, NJ, 3 ng/ml). Human alveolar basal epithelial carcinoma cell line A549 was grown as recommended by the supplier (ATCC, Manassas, VA).
Mammalian expression plasmids for expression of full-length ADAMTS9 or catalytically inactive mutants (Glu435Gln or Glu435Ala), for C-terminally truncated ADAMTS9 extending up to TSR 8 (designated ADAMTS9 N-L2), or its mutants containing substitution of Ala for Arg74/209/287 (Arg74/209/287Ala) or Ala for Glu435 (Glu435Ala, catalytically inactive) have been previously described.26,38,39 The ADAMTS1 expression plasmid was kindly provided by Dr. Luisa Iruela-Arispe. All ADAMTS9 plasmids encode a C-terminal myc tag or FLAG tag. Since ADAMTS9 N-L2 is more robustly expressed by transfected cells than full-length ADAMTS9 (which loses the C-terminal tag by proteolysis, rendering its detection difficult), and cleaves versican, indicating that proteolytic activity is preserved,39 it was used instead of full-length ADAMTS9 in several experiments. Because furin cleavage following Arg287 reduces proteolytic activity,39 the Arg74/209/287Ala mutant, which abrogates furin processing of ADAMTS9, was used to obtain enhanced activity relative to the wild-type construct. Mouse TSP-1 and TSP-2 expression plasmids were provided by Dr. Olga Stenina and Dr. Paul Bornstein, respectively. COS-1 or HEK293F cells were transfected with expression plasmids using FuGENE6 (Roche Applied Sciences, Indianapolis, IN). HBMECs and HUVECs (plated on vitronectin) were transfected using Targefect-HUVEC according to the supplier's instructions (Targeting Systems, El Cajon, CA). Four pre-designed human ADAMTS9 small interfering (si)RNAs and a negative control siRNA were purchased from Applied Biosystems/Ambion, Austin, TX. Fifty nmol/L of siRNA was added to the cells in combination with Lipofectamine 2000 (Invitrogen) for transfection, and incubated for 48 hours. Total RNA was isolated from siRNA-transfected cells using Trizol (Invitrogen), and cDNA was generated using SuperScript III First-Strand kit (Invitrogen). Primer sets were designed for reverse transcription (RT)-PCR as follows; for human ADAMTS9, forward primer 5′-CGGTTTGTAGAAGTCTTG-3′ and reverse primer 5′-CAGGTTCGTTAAGCAAAC-3′ with an expected amplicon of 622 bp; glyceraldehyde-3-phosphate dehydrogenase (Gapdh), forward primer 5′-GATTTGGTCGTATTGGGC-3′ and 5′-CGTGTTGTCATACTTCTC-3′, with an expected amplicon of 401 bp was used as a control. One pre-designed siRNA gave maximal suppression and was used in subsequent experiments.
Adhesion, Migration, and Tube-Forming Assays
For assaying cell adhesion, HBMECs were transfected with control or ADAMTS9 siRNA and detached 48 hours later using a PBS-based cell dissociation buffer (Invitrogen); 1 × 105 cells were plated on fibronectin (Sigma-Aldrich), vitronectin, laminin-1 (Sigma-Aldrich, St. Louis, MO), or collagen IV (Sigma-Aldrich) coated 96-well culture plates in serum-free medium and allowed to adhere for 1 hour. The cells were gently washed with serum-free medium and fixed with 3.7% formaldehyde. Attached cells were stained with 0.5% Crystal violet, washed, lysed in 1% SDS, and color intensity was measured at 570 nm, with background absorbance subtracted at 630 nm. Cell spreading was investigated on uncoated or Matrigel-coated culture slides (BD Biosciences, Franklin Lakes, NJ). The cells (1 × 103) were incubated for 3 hours, and actin filaments were stained with fluorescein isothiocyanate-phalloidin (Sigma-Aldrich). Photographs were taken under an Olympus inverted fluorescence microscope.
For comparison of proliferation of ADAMTS9 siRNA-treated cells with control siRNA-transfected cells, HBMEC (1 × 104) were plated in 24-well culture plates and incubated at 37°C for 48 hours. Cell numbers were obtained with a Coulter counter, as well as by the WST-1 assay (Roche Applied Science). An in vitro cell monolayer wounding assay was performed to evaluate HBMEC and A549 cell migration. The siRNA-transfected cells were cultured to confluence in six-well culture plates coated with Matrigel (100 mg/ml) in 50 mmol/L NaHCO3 and a linear scratch was made using a 200-μL pipette tip. Photographs were recorded every 10 minutes by time-lapse photomicroscopy in a temperature- and gas-controlled environment with a time-lapse inverted microscope, until closing of the scratch was observed.
A tube-formation assay was performed using Matrigel as a substrate, and 150 μl/well of growth factor depleted Matrigel (9 g/L) was added to 24-well culture plates and allowed to gel for 15 minutes at 37°C. HBMECs, HDMECs, or HHMECs transfected by siRNA or expression plasmid were detached by brief treatment with 0.05% trypsin solution, and cells (5 × 104) were added to the culture plates after resuspension in medium containing 5% fetal bovine serum and VEGF165 (PeproTech, 40 mg/L). After 6 hours incubation (siRNA treatment) or 24 hours (overexpression experiments) the cells were fixed with 3.7% formaldehyde and the lengths of the tube-like structures were measured using ImageJ software (NIH).
Analysis of Proteolytic Activity of ADAMTS9 against TSP-1 and TSP-2
ADAMTS9 or ADAMTS1 expression plasmids were cotransfected with TSP-1 or TSP-2 into COS-1 cells or HEK293F cells. Forty-eight hours (18 hours for COS-1 cells) following transfection, cells were incubated in 293 SFM-II medium (Invitrogen) for 24 hours (HEK293F cells) or serum-free Dulbecco's modified Eagle's medium (DMEM) for 72 hours (COS-1 cells), and conditioned medium and cell-lysate was collected and subjected to reducing or non-reducing SDS-polyacrylamide gel electrophoresis. Western blotting was done using anti-FLAG M2 (Sigma-Aldrich) or anti-myc monoclonal antibody for detection of ADAMTS9, anti-ADAMTS1 monoclonal antibody (Antibody 2197, R&D systems, Minneapolis, MN), anti-TSP1 monoclonal antibody (Catalog No. SC-59887, Santa Cruz Biotechnology, Santa Cruz, CA) and anti-mouse TSP-2 monoclonal antibody (Catalog No. 611150, BD Biosciences). For detection of cleavage of TSP-1 at the Glu311-Leu312 peptide bond, previously described guinea-pig antibodies numbered 78 and 79 that recognize the new N-terminus and C-terminus of cleaved TSP1, respectively, and antibody 80, which recognizes the uncleaved spanning peptide across the cleavage site9 (kindly provided by Dr. Luisa Iruela-Arispe) were used for Western blotting.
Binding of VEGF165 to ADAMTS9 and ADAMTS1
ADAMTS9 N-L2 or ADAMTS1 was immunoprecipitated from conditioned media of transfected HEK293F cells using anti-myc or anti-ADAMTS1 respectively. Protein G-sepharose beads were washed three times with PBS and then with binding buffer [HEPES (50 mmol/L), NaCl (150 mmol/L), 0.5% Nonidet P-40, 0.5% bovine serum albumin, CaCl2 (1 mmol/L), MgCl2 (1 mmol/L)]. Then, 200 ng of VEGF165 and 200 ng of heparin (Sigma-Aldrich) were added to the beads in a final volume of 500 μl of binding buffer, and incubated for 2 hours at 4°C with rotation. The bound protein was eluted with reducing SDS-sample buffer. The eluted samples were analyzed by Western blotting with anti-VEGF polyclonal antibody (Cat No. AB1442, Millipore, Billerica, MA). The presence of co-immunoprecipitated ADAMTS9 N-L2 and ADAMTS1 was verified by Western blotting using anti-myc monoclonal antibody 9E10 (Invitrogen, Carlsbad, CA) and anti-ADAMTS1 monoclonal antibody respectively.
VEGF-Receptor Phosphorylation Assay
HUVECs were cultured to confluence and further incubated for 6 hours in serum-free medium. Conditioned medium from COS-1 cells expressing ADAMTS9, ADAMTS9 N-L2, ADAMTS1, or vector alone, were mixed with VEGF165 (25 ng/ml) and incubated for 10 minutes at 37°C. HUVECs were treated with the VEGF-supplemented conditioned media for 3 minutes at 37°C. They were washed with cold PBS containing Na3VO4 (0.2 mmol/L) and lysed in lysis buffer [1% Triton X-100, Tris-HCl (50 mmol/L), pH 7.5, EDTA (5 mmol/L), NaCl (150 mmol/L), sodium orthovanadate, protease inhibitor mixture (Roche Diagnostics)] at 4°C for 15 minutes and centrifuged. The soluble portion of the lysate was transferred to a fresh tube and incubated overnight with rabbit anti-VEGF receptor (R)2 monoclonal antibody (Cat No. 2479, Cell Signaling Technology, Danvers, MA) and protein G-Sepharose (GE Healthcare, Piscataway, NJ) at 4°C. The immunoprecipitates were pelleted at 1000 rpm in a microcentrifuge and were washed four times in lysis buffer. The supernatant was discarded, and the bound protein was eluted with reducing SDS-sample buffer. The eluted samples were analyzed by Western blotting with anti-phosphotyrosine monoclonal antibody (Cell Signaling Technology). Comparable protein loading was assured by Western blotting with anti-VEGFR2 monoclonal antibody. Expression and secretion of ADAMTS9, ADAMTS9 N-L2, and ADAMTS1 in the transfected cells was verified by Western blotting of the conditioned media using rabbit anti-ADAMTS9 polyclonal antibody (antibody 359740), anti-myc monoclonal antibody 9E10 (Invitrogen), and anti-ADAMTS1 monoclonal antibody respectively.
Results
ADAMTS9+/− Mice Develop Spontaneous Corneal Neovascularization
ADAMTS9+/− mice were generated in a hybrid genetic background (129Sv X C57Bl/6) in which no obvious anomalies were noted. The role of ADAMTS9 in melanoblast development is only apparent in mice homozygous for the mutant Adamts20 allele Belted (bt) and heterozygous for the ADAMTS9 targeted allele.29 After six to eight backcrosses into the C57Bl/6 strain, we noticed corneal opacity and neovascularization in some ADAMTS9+/− mice that was not accompanied by a purulent discharge and did not appear to cause discomfort (ie, the mice did not scratch or rub their eyes). The incidence and severity of this phenotype were formally determined after the targeted allele was crossed 10 generations into C57Bl/6. Although the eyes appear externally normal at weaning, 20% of mice have visible abnormalities by 5 weeks of age, and 80% of ADAMTS9+/− mice over 25 weeks of age are affected (Figure 1A). Involvement was unilateral in approximately half the affected mice, with no preferential involvement of the left or right eye, and was bilateral in the other half. In addition to opacity and neovascularization (Figure 1B), adhesion of the lens to the cornea (anterior synechiae) with an irregular pupil was noted. The corneal stroma was disorganized when invading capillaries were present (Figure 1C) when compared with unaffected eyes. Histological analysis did not reveal classical features of Peters’ anomaly, a congenital anomaly of the anterior chamber in which there is deficiency of neural-crest derived lineages, resulting in a shallow anterior chamber, and persistence of a lens-cornea stalk.41,42 However, a subtle developmental anomaly cannot be formally excluded, and is being currently investigated. β-gal positive (β-gal+) cells were noted lining the corneal capillaries (Figure 1C), and were stained with antibodies to PECAM-1 (Figure 1D), identifying them as endothelial cells. No inflammatory cells or smooth muscle actin (SMA)+ cells were found in ADAMTS9+/− corneas. These anomalies are specific to ADAMTS9+/− mice, since they were not seen in wild-type littermates or in other transgenic C57Bl/6 mice in our mouse colony.
Figure 1.
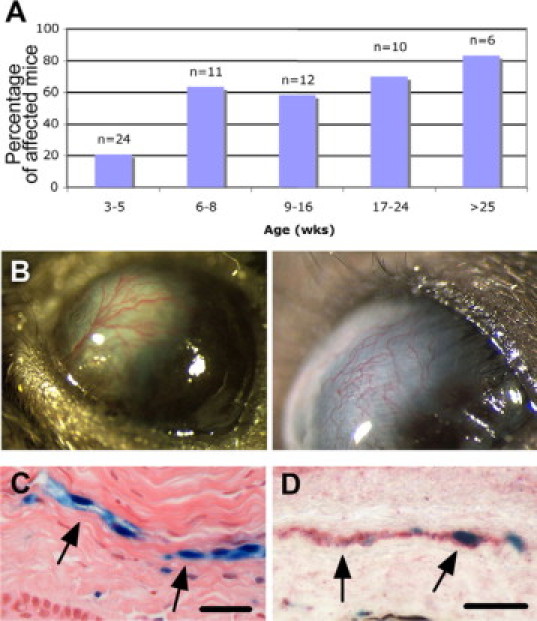
A spontaneous neovascularization in ADAMTS9+/− corneas. A: Age-dependent incidence of corneal neovascularization in ADAMTS9+/− mice. B: Photographs of eyes from mature ADAMTS9+/− mice showing corneal neovascularization. C: H&E-stained section of ADAMTS9+/− cornea showing capillaries with β-gal+ lining cells (arrows, blue) in the corneal stroma. D: Combined β-gal histochemistry (blue) and PECAM-1 immunohistochemistry (red) of an ADAMTS9+/− cornea with vascularization shows that β-gal+ cells (arrows) in corneal stroma are endothelial cells. Scale bars: 11 μm (left) and 6 μm (right).
ADAMTS9 Is Expressed during Angiogenesis in the Mouse Embryo and in Adult Mouse Organs
The presence of β-gal stained ECs in the neovascularized corneas led us to undertake a detailed analysis of ADAMTS9 expression in embryonic and adult vasculature. In previous in situ hybridization analysis using a radioisotope-labeled ADAMTS9 probe, we had noted that capillaries in uterine decidua and in the craniofacial mesenchyme of the mouse embryo expressed ADAMTS9,30 but the scatter of silver-grains did not permit conclusive identification of the expressing cells as ECs. Since β-gal expression faithfully reproduces the expression pattern of Adams9 mRNA, we used it to comprehensively investigate ADAMTS9 expression during vascular development and in adult vasculature. At embryonic day (E) 6.5 and E7.5, β-gal was not seen in the prospective yolk sac (data not shown), indicating that ADAMTS9 was not expressed during vasculogenesis. However, at E8.5, embryonic capillary EC in the trunk mesenchyme and forebrain, defined by endomucin expression, but not EC lining the heart tube or the aorta were β-gal+ (Figure 2, A and B). Although the endocardium or ECs lining blood vessels larger than capillaries, such as the developing aorta, did not express β-gal (Figure 2, A,C, and D), ECs lining the umbilical vein (but not the artery, Figure 2C) were consistently positive. In addition to continued expression in capillaries of the trunk and craniofacial regions during later embryonic development, strong expression was noted in capillaries of the limb, brain, spinal cord and intersomitic vessels (E13.5 and E14.5) (eg, Figure 2, D−F at E13.5).
Figure 2.
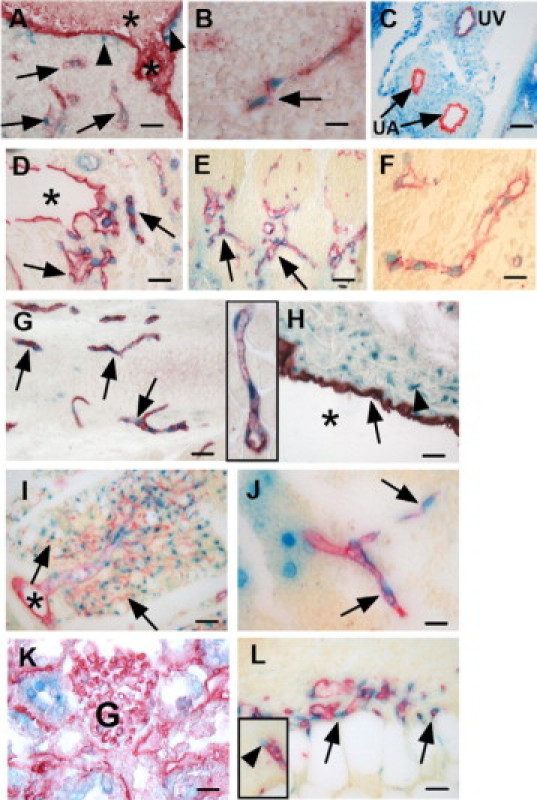
ADAMTS9 is expressed in microvascular endothelial cells (ECs) and aortic mural cells during mouse embryonic angiogenesis and in adult mice. All images show combined β-gal histochemistry (blue) and PECAM-1 (A, B, red) or endomucin (C−F, red) immunohistochemistry from 9.5-day embryos (A, B), 11.5-day umbilical cord (C), 13.5-day mouse embryos (D−F) and adult tissues (G−L). Arrows indicate endomucin+, β-gal+ cells. The asterisks in A, D, H, and I indicate a large blood vessel. A: ECs of mesenchymal capillaries (arrows) posterior to the dorsal aorta (asterisk), but not ECs lining the aorta are β-gal+. A β-gal+ putative mural cell is indicated by an arrowhead. B: β-gal−stained ECs in forebrain. C: ECs of the umbilical vein (UV), but not the arteries (UA) are β-gal+. Note that β-gal is strongly expressed in mural cells of umbilical vessels. D: β-gal+ ECs are seen in capillaries (arrows) arising from a larger blood vessel (asterisk), but not the ECs lining the large vessel (asterisk). E: ECs lining somitic capillaries are β-gal+. F: High power view of a capillary from E13.5 spinal cord illustrates that β-gal + nuclei line the capillary lumen and are surrounded by endomucin+ cytoplasm. G: Adult myocardial capillaries all contain β-gal+ ECs (inset: myocardial capillary at higher magnification). H: Adult aorta. Arrowhead indicates β-gal+ SMC in the tunica media. Note the absence of β-gal staining in ECs (arrow). I: Perirenal fat. Note the absence of β-gal staining in large vessel ECs (asterisk) whereas every capillary ECs express β-gal. J: Thymus. Note that in addition to EC (arrows), some non-EC thymic cells are also β-gal positive. K: In the renal cortex, the glomerular tuft (G) does not express β-gal. L: Section through the bed of a 5-day-old excisional skin wound. EC (arrows) invading the thrombus are β-gal+. Inset shows a β-gal+ stained tip cell invading the thrombus. Scale bars: 37 μm (A−E, G, I) and 11 μm (F, H, J, K, L).
β-Gal staining was present in capillary EC of several adult tissues, such as the myocardium (Figure 2G and inset), adipose tissue (Figure 2I), brain and spinal cord, skeletal muscle, intestine (including intestinal villi), exocrine pancreas, thymus (Figure 2J), and lung. Indeed, in adult mice, microvascular ECs represent the major site of ADAMTS9 expression. ECs lining the aorta (Figure 2H), other adult arteries and veins, and endocardium were consistently negative for β-gal (β-gal-), but mural cells in large vessels (identified as smooth muscle cells by SMA immunohistochemistry (not shown), were β-gal+ (Figure 2H). Capillary ECs of the renal glomerulus (Figure 2K), adrenal gland, parathyroid gland, thyroid gland, and liver, which are lined by fenestrated endothelium, expressed β-gal weakly or not at all. Excisional skin wounds in ADAMTS9+/− mice were analyzed 5 days after wounding, when invasion of the provisional fibrin matrix of the wound bed by capillaries is ongoing. β-gal staining was present in invading EC at the edge, as well as the bed of the wound (Figure 2L). Notably, EC nuclei at the tip of capillary sprouts (Figure 2L, inset), expressed ADAMTS9. Lymphatic ECs, identified by podoplanin immunohistochemistry in adult organs, did not express ADAMTS9 (data not shown).
Because pericytes are closely apposed to capillary endothelial cells, and may be difficult to distinguish morphologically, we immunostained β-gal stained embryo and adult sections with antibody to NG2 or SMA respectively as markers of the embryonic and adult vascular smooth muscle phenotype. No NG2+ or SMA+ cells were seen around the capillaries in the cornea. In embryo and adult tissue sections, the NG2 and SMA staining pattern was clearly distinct from that seen with EC markers (Figures 1 and 2), where red cytoplasmic staining surrounded blue nuclei. Instead, the smooth muscle markers clearly stained cells whose nuclei were not blue, but were adjacent to blue nuclei (see Supplemental Figure 1 at http://ajp.amjpathol.org). This finding was consistently present in all sections stained. Thus, in contrast to mural cells in large arteries, pericytes do not express ADAMTS9.
ADAMTS9 Is Expressed in Microvascular EC Invading Tumors, and Heterotopic Tumors Implanted in ADAMTS9+/− Mice Induce More Vasculature
To determine whether ECs of the microvasculature induced by implanted tumors expressed ADAMTS9, Lewis lung carcinoma cells and B.16-F10 melanoma cells, which are isogenic with ADAMTS9+/− mice, were injected subcutaneously. β-gal staining of the growing tumors showed that host endothelial cells invading both tumor types strongly expressed ADAMTS9 (Figure 3A shows B.16-F10 derived tumors). These ADAMTS9 expressing cells did not stain with SMA antibody or anti-podoplanin (data not shown). To determine whether ADAMTS9+/− mice mounted a similar angiogenic response to B.16-F10 derived tumors as wild-type mice, we compared the tumor-induced vasculature in ADAMTS9+/− mice and in wild-type littermates using a double-blind experimental design. As indicated by the representative images (Figure 3, B and C) and analysis of mean vessel counts in each genotype, there was a statistically significant increase in the vessel count in tumors of ADAMTS9+/− mice (Figure 3D).
Figure 3.
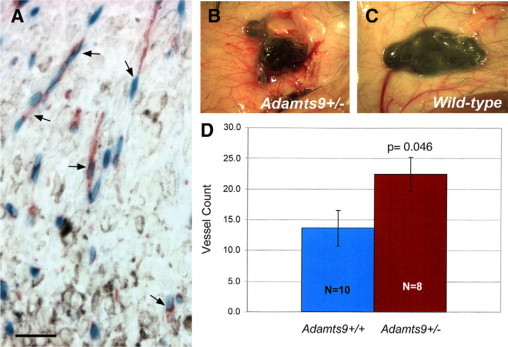
ADAMTS9 is expressed in tumor capillaries and influences the induction of tumoral vessels. A: Section through the periphery of a B.16-F10 melanoma implanted in ADAMTS9+/− mice shows that ADAMTS9 (β-gal staining, blue) is expressed by capillary endothelial cells (arrows) (red, endomucin) invading the melanoma (pigmented cells). Scale bar = 50 μm. B, C: Representative images of a B.16-F10 melanoma in ADAMTS9+/− mice (B) and wild-type (C) mice showing the enhanced vasculature around the tumor in ADAMTS9+/− mice. D: Quantitative analysis of in vivo angiogenic activity induced by B.16-F10 melanomas in ADAMTS9+/− and wild-type mice. Mice were sacrificed 9 days after intradermal injection of tumor cells. Radially oriented blood vessels entering the periphery of the tumors were counted by an observed blinded to the genotype of the mice. The mean vessel count ± the SEM is shown. The P value is shown.
ADAMTS9 mRNA Is Expressed in Cultured Microvascular ECs and HUVECs, and siRNA Knockdown Leads to Enhanced Microvascular EC Spreading, Migration, and Formation of Tube-Like Structures
Taken together, the preceding studies suggested that ADAMTS9 was a constitutive product of microvascular ECs and umbilical vein ECs that suppressed angiogenesis. Accordingly, we asked whether corresponding cell isolates expressed ADAMTS9 mRNA in vitro. ADAMTS9 mRNA was detected in HBMECs, HHMECs, HDMECs, and HUVECs using RT-PCR, and was suppressed substantially by one of four predesigned ADAMTS9 siRNAs that we tested (Figure 4A)(see Supplemental Figure 2 at http://ajp.amjpathol.org). siRNA knockdown in these ECs of different origins resulted in a consistent and statistically significant increase in formation of tube-like structures when compared with the control siRNA-transfected ECs (Figure 4, B and C) (see Supplemental Figure 2 at http://ajp.amjpathol.org). Since capillary ECs demonstrate specific morphological changes, as well as altered migration and cell-adhesion during angiogenesis, we analyzed how these parameters were altered by ADAMTS9 knockdown in HBMEC. Three hours after plating on Matrigel, the ADAMTS9 siRNA-treated HBMEC formed long filopodial extensions, typical of early capillary sprouting, which were not yet evident in the control siRNA treated cells (Figure 5A). Adhesion of HBMECs to vitronectin, laminin, and collagen was reduced following siRNA transfection (Figure 5B). In vitro scratch ‘wounding’ of a HBMEC monolayer demonstrated enhanced migration of ADAMTS9 siRNA-treated cells relative to control siRNA- treated cells (Figure 5C). Interestingly, we also observed enhanced migration of the nonendothelial tumor cell line A549 (see Supplemental Figure 3 at http://ajp.amjpathol.org), suggesting that ADAMTS9 may act on a more general cellular mechanism than an endothelium-specific one. ADAMTS9 siRNA-treated ECs had similar proliferation rates as control siRNA-transfected cells (see Supplemental Figure 4 at http://ajp.amjpathol.org).
Figure 4.
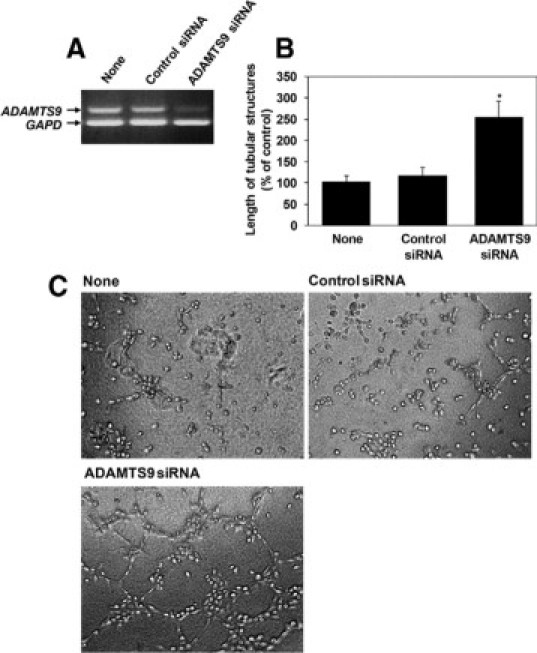
Knockdown of ADAMTS9 in human microvascular EC leads to enhanced tube formation on Matrigel. A: RT-PCR showing expression of ADAMTS9 mRNA in HBMECs and successful knockdown with siRNA. B: Quantitative analysis of the length of tube-like structures measured under the indicated conditions and shown as a percentage of the control. *P < 0.001. C: Representative phase-contrast photomicrographs of tube-like structures formed under the indicated conditions. Corresponding data for microvascular ECs from human heart and dermis and human umbilical vein EC are presented in Supplemental Figure 2 at http://ajp.amjpathol.org.
Figure 5.
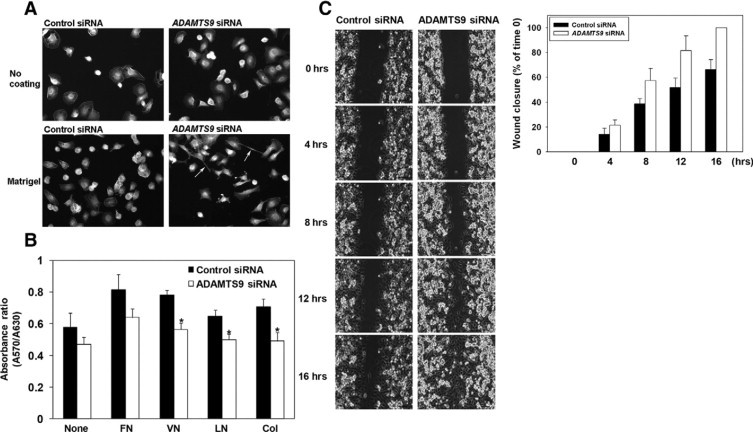
Enhanced spreading, decreased adhesion, and increased migration of HBMECs on ADAMTS9 knockdown. A: Effect of ADAMTS9 siRNA on cell spreading was evaluated in HBMECs permitted to attach to an uncoated or Matrigel-coated substrate. The arrows indicate filopodia, which were not seen in control siRNA-treated cells at this time point. B: Comparison of the effect of ADAMTS9 siRNA and control siRNA on cell attachment to fibronectin (FN), vitronectin (VN), laminin (LN), and collagen IV (Col). *P < 0.001. C: Enhanced migration of microvascular endothelial cells on ADAMTS9 knockdown. A representative example of three independent experiments is illustrated on the left. The panel at right shows quantitative analysis of three independent migration assays.
Overexpression of Catalytically Active ADAMTS9 Suppresses Formation of Tube-Like Structures by Cultured ECs
To determine whether overexpression of ADAMTS9 had opposite effects to siRNA suppression, as well as to determine whether ADAMTS9 proteolytic activity was required for the observed effects, we overexpressed ADAMTS9 and ADAMTS9 Glu435Gln in HBMEC (Figure 6A) and HUVECs (see Supplemental Figure 5 at http://ajp.amjpathol.org). Although HBMECs transfected with the vector control formed tube-like structures with comparable efficiency as untransfected cells, transfection with wild-type ADAMTS9, or ADAMTS1 significantly inhibited tube formation (Figure 6, A and B). ADAMTS9 Glu435Gln-transfected HBMECs demonstrated essentially no inhibition of formation of tube-like structures, suggesting that the noncatalytic domains such as the TSRs contribute little to the observed effect, but indicating that the catalytic activity of ADAMTS9 was necessary. Although some suppression of tube formation was seen in HUVECs, the effect was not as pronounced as in HBMECs, and was not statistically significant (see Supplemental Figure 5 at http://ajp.amjpathol.org).
Figure 6.
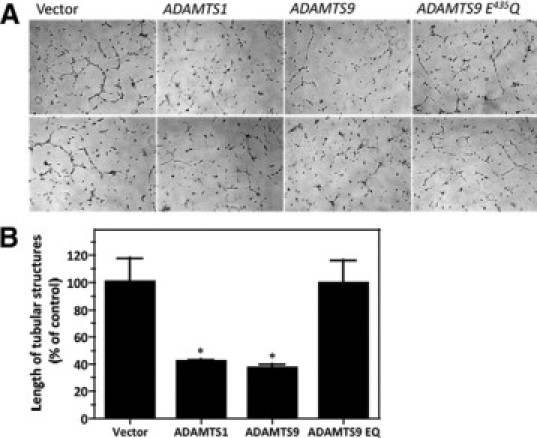
The proteolytic activity of ADAMTS9 is required for its anti-angiogenic effect. A: Two representative fields each show the effect of overexpressing the vector alone, ADAMTS1, ADAMTS9, or ADAMTS9 (E435Q) in HBMECs. B: Quantitative analysis illustrates a statistically significant reduction in tube-like structures following expression of ADAMTS1 and ADAMTS9, but not ADAMTS9 E435Q. *P < 0.05.
ADAMTS9 Does Not Cleave TSP-1 and TSP-2, Sequester VEGF, or Affect VEGF-Receptor Phosphorylation
ADAMTS9-transfected COS-1 cells did not produce fragments of cotransfected TSP-1 when the conditioned medium of cells cotransfected with ADAMTS9 and TSP-1 was analyzed using either reducing or nonreducing conditions (Figure 7A). In parallel experiments, ADAMTS1 transfected cells generated a 110-kDa band (reducing conditions) with a concomitant decrease in the level of intact TSP-1 (Figure 7A). Antibodies that specifically react with neo-epitopes in TSP-1 generated by ADAMTS1 cleavage9 showed no reactive bands in the medium of ADAMTS9 or ADAMTS9 N-L2 transfected cells, in contrast to consistent appearance of cleaved N- and C-terminal TSP1 epitopes present in the medium of ADAMTS1-transfected cells (Figure 7B). Furin-resistant ADAMTS9 N-L2 R74/209/287A, which was previously shown to be more enzymatically active than furin-processed ADAMTS9 zymogen,39 also failed to produce proteolytic fragments of TSP-1 (Figure 7B). A similar cell-based analysis of TSP-2 processing was undertaken and showed that ADAMTS1 consistently generated a 35-kDa TSP-2 fragment in conditioned medium, with concomitant reduction of the levels of intact TSP-2 (Figure 7C). However, fragments of TSP-2 were not observed in ADAMTS9-transfected cells, nor was quantitative reduction of full-length TSP-2 evident (Figure 7C). Thus, under the conditions of these assays, ADAMTS9 does not share TSP-1 or TSP-2 cleavage activity with ADAMTS1.
Figure 7.
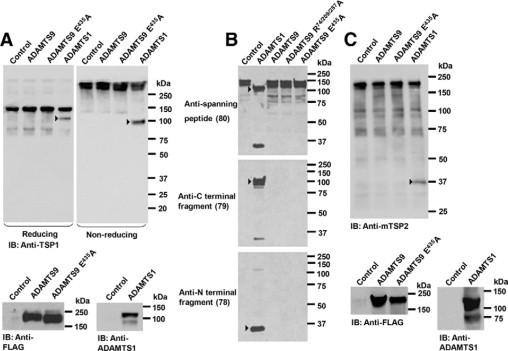
ADAMTS9 does not cleave thrombospondin-1 and -2. A: Analysis of TSP-1 cleavage using Western blotting with an anti-TSP-1 antibody under reducing and nonreducing conditions. The TSP-1 fragment released by ADAMTS1-transfected cells is indicated by the arrowheads. The panels at the bottom show Western blotting with anti-FLAG M2 or anti-ADAMTS1 monoclonal antibody to verify expression of the transfected ADAMTS plasmids. B: Analysis of TSP-1 cleavage using the neoepitope antibodies 78 and 79 and the cleavage-site−spanning antibody 80. The arrowheads indicate the ADAMTS1 cleavage fragments. C: Analysis of TSP-2 cleavage: The arrowhead indicates the ADAMTS1 cleaved fragment. The panels at the bottom show Western blotting with anti-FLAG M2 or anti-ADAMTS1 monoclonal antibody to verify expression of the transfected ADAMTS plasmids.
Immunoprecipitation of the medium of HEK293F cells transfected with either ADAMTS1 or ADAMTS9 confirmed that ADAMTS1 bound VEGF165 but ADAMTS9 N-L2 did not bind VEGF165 despite efficient pull-down using anti-myc antibody (Figure 8A). We could also confirm that ADAMTS1 sequestration of VEGF165 reduced VEGFR2 phosphorylation (Figure 8B). However, neither ADAMTS9 nor ADAMTS9 N-L2 affected VEGFR2 phosphorylation (Figure 8B).
Figure 8.
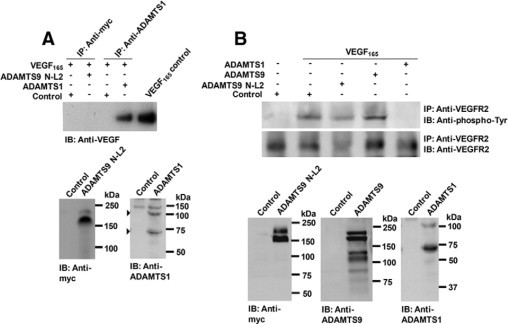
Unlike ADAMTS1, ADAMTS9 does not sequester VEGF165, or affect VEGFR2 signaling. A: Analysis of VEGF165 sequestration by ADAMTS9 and ADAMTS1 was done as described in Materials and Methods. Note that VEGF165 is sequestered by ADAMTS1, but not by ADAMTS9. The panels at the bottom demonstrate that both proteases were present in the respective media. B: ADAMTS9 does not influence VEGFR2 phosphorylation. Note reduction of VEGFR2 phosphorylation in the presence of ADAMTS1, implying sequestration of VEGF165 by ADAMTS1. The lower panels demonstrate expression of the ADAMTS9 and ADAMTS1 plasmids.
Discussion
Taken together, the widespread, constitutive expression of ADAMTS9 observed in EC of several microvascular beds and cultured EC, the enhancement of tube formation and migration by cultured EC on ADAMTS9 knockdown, the reduction of tube formation on overexpression of ADAMTS9, and the occurrence of neovascularization in ADAMTS9+/− corneas, and enhanced tumor vessel counts, is strongly suggestive of a physiological anti-angiogenic role for ADAMTS9. Corneal neovascularization, which appears to occur spontaneously in ADAMTS9+/− mice in the C57Bl/6 strain, is unusual since avascularity of the cornea is normally ensured by intrinsic anti-angiogenic factors.43 The inability of these intrinsic mechanisms to stave off invading ADAMTS9 haploinsufficient EC suggested that ADAMTS9 directly modified the endothelial pro-angiogenic activity. Indeed, the potential anti-angiogenic role of ADAMTS9 suggested by corneal neovascularization and enhanced tumor-induced vasculature is likely to be quite a profound one, since the observed effects are seen despite the presence of an intact ADAMTS9 allele, ie, the present in vivo studies underestimate the significance of ADAMTS9 in the context of angiogenesis.
Detailed analysis of ADAMTS9 expression by β-gal staining supported expression in capillary ECs during embryonic and regenerative angiogenesis (wound healing), and widespread, constitutive expression in several, although not all adult capillary beds. The identification of β-gal+ cells as ECs is based on the observation that β-gal+ nuclei were surrounded by endomucin or PECAM-1-positive cytoplasm, and lined the lumen of capillaries. The lack of cytoplasmic SMA or NG2 staining of capillary-associated β-gal+ nuclei suggested that ADAMTS9 was not expressed by pericytes although our data show that it is expressed by SMC of large vessels, but not their ECs. The significance of this reciprocal expression pattern in microvessels and large vessels is not clear. However, given the anti-angiogenic properties of ADAMTS9, we speculate it may have a role in promoting capillary maturation and stabilization, whereas such a function is probably rendered unnecessary in large arteries.
We asked whether ADAMTS9 was a cell-autonomous regulator of capillary endothelium using ECs of different tissue origins. Having confirmed that ADAMTS9 mRNA was present in a variety of human microvascular ECs and HUVECs by RT-PCR, we suppressed its expression using ADAMTS9 siRNA. ADAMTS9 knockdown led to enhanced migration and formation of tube-like structures by microvascular ECs. However, the effect of ADAMTS9 overexpression on HUVECs was not as dramatic as in microvascular ECs, suggesting that its anti-angiogenic role was possibly less significant in the umbilical vein. Thus, one physiological function of ADAMTS9 in the vasculature may be to exert a suppressive effect on pro-angiogenic mechanisms, which is compatible with the anomalous ingrowth of capillaries into the cornea of ADAMTS9+/− mice and enhanced vascular induction in implanted tumors.
To determine the possible mechanisms by which ADAMTS9 may influence angiogenesis, as well as to address its possible redundancy with ADAMTS1, we investigated several mechanisms by which ADAMTS1 is known to act. ADAMTS9 is considerably larger than ADAMTS1, having 12 TSRs more than the latter, but it shares an identical catalytic zinc-binding sequence with ADAMTS1 (ie, HELGHVNMPHD),16,26 which suggested that both proteases might cleave the same substrates. Indeed, ADAMTS9 cleaves two ADAMTS1 proteoglycan substrates, aggrecan and versican,26 but it did not cleave TSP-1 and TSP2. Furthermore, unlike ADAMTS1, ADAMTS9 does not sequester exogenously added VEGF165. The proteolytic activity of ADAMTS9 is essential for its role, since the overexpression of active, but not inactive ADAMTS9 had effects opposite to those of knockdown, ie, it suppressed the formation of tube-like structures by EC. The unequivocal effects of siRNA knockdown in cultured EC strongly suggest that ADAMTS9 acts cell-autonomously in the endothelium. In contrast, in situ analyses suggest that although EC do not themselves express ADAMTS1,19,20 exogenous ADAMTS1 influences their behavior.16,24 Other differences between ADAMTS9 and ADAMTS1 include a propensity for ADAMTS1 binding to extracellular matrix,44 whereas ADAMTS9 preferentially binds to the cell surface.44 In terms of regulatory mechanisms, it is known that furin removes the ADAMTS1 propeptide within the secretory pathway to activate it, whereas furin processing of ADAMTS9 occurs at the cell surface and diminishes ADAMTS9 activity.38,39 Localization of ADAMTS9 at the cell surface26 and its inactivation by furin processing at the cell surface38 suggests that ADAMTS9 substrates are secreted or membrane proteins or proteoglycans that are cleaved within the secretory pathway or at the cell surface. Such a proteolytic mechanism is consistent with the putative EC-autonomous role proposed here, although the substrate(s) that mediate its anti-angiogenic function are presently unknown. On the basis of these differences, we suggest that rather than providing redundancy in angiogenesis, ADAMTS9 and ADAMTS1 operate via different mechanisms and may therefore each have unique roles in the vasculature.
A functional complementation approach showed that ADAMTS9 expression was down-regulated or lacking altogether in esophageal and nasopharyngeal carcinoma as a result of promoter hypermethylation.27,28 Furthermore, transfection of ADAMTS9 into nasopharyngeal carcinoma cell lines dramatically restored the colony-forming ability of the nasopharyngeal carcinoma lines, suggesting that ADAMTS9 was a tumor-suppressor gene in this cancer.28 The tumor suppressor effect of ADAMTS9 in these two tumors did not invoke a role for ADAMTS9 in suppression of angiogenesis. However, the data presented here strongly suggest that an anti-angiogenic role may confer an additional function to the observed tumor suppressor role. Furthermore, ADAMTS9 may be of broad relevance to all angiogenesis-dependent cancers through its novel and constitutive expression in capillary ECs and physiological anti-angiogenic role. It remains to be determined whether ADAMTS9 could have additional effects on neighboring cells such as pericytes, and tumor cells or target organs undergoing vascularization. In future studies it will be important to resolve these outstanding questions and to identify the ADAMTS9 substrate(s) in endothelial cells.
Acknowledgements
We thank Dr. Dietmar Vestweber for endomucin antibody and Dr. Luisa Iruela-Arispe for TSP1 neoepitope antibodies and ADAMTS1 plasmid. We thank Dr. Judy Mack for assistance with excisional skin wounding. Technical assistance from Rebecca Haney in tumor analysis is acknowledged.
Footnotes
Supported by NIH award AR49930 (to S. Apte).
None of the authors have an affiliation with any organization or entity having a direct financial or personal interest in the subject matter or materials discussed in the paper.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Current address of B.-H.K., Yonsei University, Seoul, Korea; current address of R.P.T.S., National Cancer Institute, Bethesda, Maryland; current address of M.E.Y., College of Medicine, The Ohio State University, Columbus, Ohio.
Web Extra Material
References
- 1.Folkman J. Fundamental concepts of the angiogenic process. Curr Mol Med. 2003;3:643–651. doi: 10.2174/1566524033479465. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 3.Roy R, Zhang B, Moses MA. Making the cut: protease-mediated regulation of angiogenesis. Exp Cell Res. 2006;312:608–622. doi: 10.1016/j.yexcr.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 4.Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97:1093–1107. doi: 10.1161/01.RES.0000191547.64391.e3. [DOI] [PubMed] [Google Scholar]
- 5.Genis L, Galvez BG, Gonzalo P, Arroyo AG. MT1-MMP: universal or particular player in angiogenesis? Cancer Metastasis Rev. 2006;25:77–86. doi: 10.1007/s10555-006-7891-z. [DOI] [PubMed] [Google Scholar]
- 6.van Hinsbergh VW, Engelse MA, Quax PH. Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler Thromb Vasc Biol. 2006;26:716–728. doi: 10.1161/01.ATV.0000209518.58252.17. [DOI] [PubMed] [Google Scholar]
- 7.Hamano Y, Zeisberg M, Sugimoto H, Lively JC, Maeshima Y, Yang C, Hynes RO, Werb Z, Sudhakar A, Kalluri R. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003;3:589–601. doi: 10.1016/s1535-6108(03)00133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heljasvaara R, Nyberg P, Luostarinen J, Parikka M, Heikkila P, Rehn M, Sorsa T, Salo T, Pihlajaniemi T. Generation of biologically active endostatin fragments from human collagen XVIII by distinct matrix metalloproteases. Exp Cell Res. 2005;307:292–304. doi: 10.1016/j.yexcr.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 9.Lee NV, Sato M, Annis DS, Loo JA, Wu L, Mosher DF, Iruela-Arispe ML. ADAMTS1 mediates the release of antiangiogenic polypeptides from TSP1 and 2. EMBO J. 2006;25:5270–5283. doi: 10.1038/sj.emboj.7601400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Apte SS. A disintegrin-like and metalloprotease (reprolysin type) with thrombospondin type 1 motifs: the ADAMTS family. Int J Biochem Cell Biol. 2004;36:981–985. doi: 10.1016/j.biocel.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 11.Porter S, Clark IM, Kevorkian L, Edwards DR. The ADAMTS metalloproteinases. Biochem J. 2005;386:15–27. doi: 10.1042/BJ20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colige A, Sieron AL, Li SW, Schwarze U, Petty E, Wertelecki W, Wilcox W, Krakow D, Cohn DH, Reardon W, Byers PH, Lapiere CM, Prockop DJ, Nusgens BV. Human Ehlers-Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am J Hum Genet. 1999;65:308–317. doi: 10.1086/302504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, Sarode R, Shurin SB, Chandrasekaran V, Stabler SP, Sabio H, Bouhassira EE, Upshaw JD, Jr, Ginsburg D, Tsai HM. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 14.Dagoneau N, Benoist-Lasselin C, Huber C, Faivre L, Megarbane A, Alswaid A, Dollfus H, Alembik Y, Munnich A, Legeai-Mallet L, Cormier-Daire V. ADAMTS10 mutations in autosomal recessive Weill-Marchesani Syndrome. Am J Hum Genet. 2004;75:801–806. doi: 10.1086/425231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuno K, Kanada N, Nakashima E, Fujiki F, Ichimura F, Matsushima K. Molecular cloning of a gene encoding a new type of metalloproteinase-disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J Biol Chem. 1997;272:556–562. doi: 10.1074/jbc.272.1.556. [DOI] [PubMed] [Google Scholar]
- 16.Vazquez F, Hastings G, Ortega MA, Lane TF, Oikemus S, Lombardo M, Iruela-Arispe ML. METH-1, a human ortholog of ADAMTS-1, and METH-2 are members of a new family of proteins with angio-inhibitory activity. J Biol Chem. 1999;274:23349–23357. doi: 10.1074/jbc.274.33.23349. [DOI] [PubMed] [Google Scholar]
- 17.Carpizo D, Iruela-Arispe ML. Endogenous regulators of angiogenesis–emphasis on proteins with thrombospondin–type I motifs. Cancer Metastasis Rev. 2000;19:159–165. doi: 10.1023/a:1026570331022. [DOI] [PubMed] [Google Scholar]
- 18.Luque A, Carpizo DR, Iruela-Arispe ML. ADAMTS1/METH1 inhibits endothelial cell proliferation by direct binding and sequestration of VEGF165. J Biol Chem. 2003;278:23656–23665. doi: 10.1074/jbc.M212964200. [DOI] [PubMed] [Google Scholar]
- 19.Thai SN, Iruela-Arispe ML. Expression of ADAMTS1 during murine development. Mech Dev. 2002;115:181–185. doi: 10.1016/s0925-4773(02)00115-6. [DOI] [PubMed] [Google Scholar]
- 20.Gunther W, Skaftnesmo KO, Arnold H, Bjerkvig R, Terzis AJ. Distribution patterns of the anti-angiogenic protein ADAMTS-1 during rat development. Acta Histochem. 2005;107:121–131. doi: 10.1016/j.acthis.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Mittaz L, Russell DL, Wilson T, Brasted M, Tkalcevic J, Salamonsen LA, Hertzog PJ, Pritchard MA. Adamts-1 is essential for the development and function of the urogenital system. Biol Reprod. 2004;70:1096–1105. doi: 10.1095/biolreprod.103.023911. [DOI] [PubMed] [Google Scholar]
- 22.Lee NV, Rodriguez-Manzaneque JC, Thai SN, Twal WO, Luque A, Lyons KM, Argraves WS, Iruela-Arispe ML. Fibulin-1 acts as a cofactor for the matrix metalloprotease ADAMTS-1. J Biol Chem. 2005;280:34796–34804. doi: 10.1074/jbc.M506980200. [DOI] [PubMed] [Google Scholar]
- 23.Shozu M, Minami N, Yokoyama H, Inoue M, Kurihara H, Matsushima K, Kuno K. ADAMTS-1 is involved in normal follicular development, ovulatory process and organization of the medullary vascular network in the ovary. J Mol Endocrinol. 2005;35:343–355. doi: 10.1677/jme.1.01735. [DOI] [PubMed] [Google Scholar]
- 24.Krampert M, Kuenzle S, Thai SN, Lee N, Iruela-Arispe ML, Werner S. ADAMTS1 proteinase is up-regulated in wounded skin and regulates migration of fibroblasts and endothelial cells. J Biol Chem. 2005;280:23844–23852. doi: 10.1074/jbc.M412212200. [DOI] [PubMed] [Google Scholar]
- 25.Blelloch R, Kimble J. Control of organ shape by a secreted metalloprotease in the nematode Caenorhabditis elegans. Nature. 1999;399:586–590. doi: 10.1038/21196. [DOI] [PubMed] [Google Scholar]
- 26.Somerville RP, Longpre JM, Jungers KA, Engle JM, Ross M, Evanko S, Wight TN, Leduc R, Apte SS. Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J Biol Chem. 2003;278:9503–9513. doi: 10.1074/jbc.M211009200. [DOI] [PubMed] [Google Scholar]
- 27.Lo PH, Leung AC, Kwok CY, Cheung WS, Ko JM, Yang LC, Law S, Wang LD, Li J, Stanbridge EJ, Srivastava G, Tang JC, Tsao SW, Lung ML. Identification of a tumor suppressive critical region mapping to 3p14.2 in esophageal squamous cell carcinoma and studies of a candidate tumor suppressor gene, ADAMTS9. Oncogene. 2007;26:148–157. doi: 10.1038/sj.onc.1209767. [DOI] [PubMed] [Google Scholar]
- 28.Lung HL, Lo PH, Xie D, Apte SS, Cheung AK, Cheng Y, Law EW, Chua D, Zeng YX, Tsao SW, Stanbridge EJ, Lung ML. Characterization of a novel epigenetically silenced, growth-suppressive gene, ADAMTS9, and its association with lymph node metastases in nasopharyngeal carcinoma. Int J Cancer. 2008;123:401–408. doi: 10.1002/ijc.23528. [DOI] [PubMed] [Google Scholar]
- 29.Silver DL, Hou L, Somerville R, Young ME, Apte SS, Pavan WJ. The secreted metalloprotease ADAMTS20 is required for melanoblast survival. PLoS Genet. 2008;4:e1000003. doi: 10.1371/journal.pgen.1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jungers KA, Le Goff C, Somerville RP, Apte SS. ADAMTS9 is widely expressed during mouse embryo development. Gene Expr Patterns. 2005;5:609–617. doi: 10.1016/j.modgep.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 31.Taylor KL, Oates RK, Grane R, Leaman DW, Borden EC, Lindner DJ. IFN-alpha1,8 inhibits tumor-induced angiogenesis in murine angiosarcomas. J Interferon Cytokine Res. 2006;26:353–361. doi: 10.1089/jir.2006.26.353. [DOI] [PubMed] [Google Scholar]
- 32.Baker DP, Lin EY, Lin K, Pellegrini M, Petter RC, Chen LL, Arduini RM, Brickelmaier M, Wen D, Hess DM, Chen L, Grant D, Whitty A, Gill A, Lindner DJ, Pepinsky RB. N-terminally PEGylated human interferon-beta-1a with improved pharmacokinetic properties and in vivo efficacy in a melanoma angiogenesis model. Bioconjug Chem. 2006;17:179–188. doi: 10.1021/bc050237q. [DOI] [PubMed] [Google Scholar]
- 33.Bauer JA, Morrison BH, Grane RW, Jacobs BS, Borden EC, Lindner DJ. IFN-alpha2b and thalidomide synergistically inhibit tumor-induced angiogenesis. J Interferon Cytokine Res. 2003;23:3–10. doi: 10.1089/10799900360520397. [DOI] [PubMed] [Google Scholar]
- 34.McCulloch DR, Goff CL, Bhatt S, Dixon LJ, Sandy JD, Apte SS. Adamts5, the gene encoding a proteoglycan-degrading metalloprotease, is expressed by specific cell lineages during mouse embryonic development and in adult tissues. Gene Expr Patterns. 2009;9:314–323. doi: 10.1016/j.gep.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brachtendorf G, Kuhn A, Samulowitz U, Knorr R, Gustafsson E, Potocnik AJ, Fassler R, Vestweber D. Early expression of endomucin on endothelium of the mouse embryo and on putative hematopoietic clusters in the dorsal aorta. Dev Dyn. 2001;222:410–419. doi: 10.1002/dvdy.1199. [DOI] [PubMed] [Google Scholar]
- 36.Farr AG, Berry ML, Kim A, Nelson AJ, Welch MP, Aruffo A. Characterization and cloning of a novel glycoprotein expressed by stromal cells in T-dependent areas of peripheral lymphoid tissues. J Exp Med. 1992;176:1477–1482. doi: 10.1084/jem.176.5.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Callahan MK, Williams KA, Kivisakk P, Pearce D, Stins MF, Ransohoff RM. CXCR3 marks CD4+ memory T lymphocytes that are competent to migrate across a human brain microvascular endothelial cell layer. J Neuroimmunol. 2004;153:150–157. doi: 10.1016/j.jneuroim.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Koo BH, Longpre JM, Somerville RP, Alexander JP, Leduc R, Apte SS. Cell-surface processing of pro-ADAMTS9 by furin. J Biol Chem. 2006;281:12485–12494. doi: 10.1074/jbc.M511083200. [DOI] [PubMed] [Google Scholar]
- 39.Koo BH, Longpre JM, Somerville RP, Alexander JP, Leduc R, Apte SS. Regulation of ADAMST9 secretion and enzymatic activity by its propeptide. J Biol Chem. 2007;282:16146–16154. doi: 10.1074/jbc.M610161200. [DOI] [PubMed] [Google Scholar]
- 40.Demircan K, Hirohata S, Nishida K, Hatipoglu OF, Oohashi T, Yonezawa T, Apte SS, Ninomiya Y. ADAMTS-9 is synergistically induced by interleukin-1beta and tumor necrosis factor alpha in OUMS-27 chondrosarcoma cells and in human chondrocytes. Arthritis Rheum. 2005;52:1451–1460. doi: 10.1002/art.21010. [DOI] [PubMed] [Google Scholar]
- 41.Traboulsi EI, Maumenee IH. Peters' anomaly and associated congenital malformations. Arch Ophthalmol. 1992;110:1739–1742. doi: 10.1001/archopht.1992.01080240079035. [DOI] [PubMed] [Google Scholar]
- 42.Iwao K, Inatani M, Matsumoto Y, Ogata-Iwao M, Takihara Y, Irie F, Yamaguchi Y, Okinami S, Tanihara H. Heparan sulfate deficiency leads to Peters anomaly in mice by disturbing neural crest TGF-beta2 signaling. J Clin Invest. 2009;119:1997–2008. doi: 10.1172/JCI38519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ambati BK, Nozaki M, Singh N, Takeda A, Jani PD, Suthar T, Albuquerque RJ, Richter E, Sakurai E, Newcomb MT, Kleinman ME, Caldwell RB, Lin Q, Ogura Y, Orecchia A, Samuelson DA, Agnew DW, St Leger J, Green WR, Mahasreshti PJ, Curiel DT, Kwan D, Marsh H, Ikeda S, Leiper LJ, Collinson JM, Bogdanovich S, Khurana TS, Shibuya M, Baldwin ME, Ferrara N, Gerber HP, De Falco S, Witta J, Baffi JZ, Raisler BJ, Ambati J. Corneal avascularity is due to soluble VEGF receptor-1. Nature. 2006;443:993–997. doi: 10.1038/nature05249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuno K, Matsushima K. ADAMTS-1 protein anchors at the extracellular matrix through the thrombospondin type I motifs and its spacing region. J Biol Chem. 1998;273:13912–13917. doi: 10.1074/jbc.273.22.13912. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.