

Abstract
Vision loss in diabetic retinopathy is due to macular edema characterized by increased vascular permeability, which involves phosphorylation associated with activation of protein kinase C (PKC) isoforms. Herein, we demonstrated PKC δ inhibition could prevent blood-retinal barrier breakdown in diabetic retinopathy. Increased vascular permeability of diabetic retina was accompanied by a decrease of zonula occludens (ZO)-1 and ZO-2 expression. In diabetic retina and advanced glycation end product-treated human retinal microvascular endothelial cells, vascular leakage and loss of ZO-1 and ZO-2 on retinal vessels were effectively restored or prevented with treatment of rottlerin, transfection of PKC-δ-DN, or siRNA for PKC δ. Interestingly, PKC δ translocated from cytosol to membrane in advanced glycation end product-treated human retinal microvascular endothelial cells, which was blocked by PKC δ inhibition. Taken together, PKC δ activation, related to its subcellular translocation, is involved in vascular permeability in response to diabetes, and inhibition of PKC δ effectively restores loss of tight junction proteins in retinal vessels. Therefore, we suggest that inhibition of PKC δ could be an alternative treatment to blood-retinal barrier breakdown in diabetic retinopathy.
Diabetic retinopathy (DR), a common and serious complication of diabetes, is one of the leading causes of blindness.1 Clinically, DR can be classified into two stages: nonproliferative and proliferative. With progression of retinal ischemia, nonproliferative DR progresses to proliferative DR, which is characterized by the growth of new blood vessels on the surface of the retina or the optic disk. These abnormal vessels are fragile to bleeding, resulting in vitreous hemorrhage and tractional retinal detachment.2 However, the principal cause of vision loss in diabetic patients is diabetic macular edema, which can occur at any stage of DR and is characterized by increased vascular permeability.3
Diabetes alters the structure and function of most cell types in the retina including the vasculature and neural network,4,5 which is closely related to the blood-retinal barrier (BRB) breakdown in the early stage of diabetic retinopathy.6,7 Therefore, BRB breakdown characterizes early stages of vascular dysfunction in DR.8 As our previous reports, the cellular interactions, regulating blood neural barrier by modulating both brain angiogenesis and tight junction formation,9 also play the critical role in retinal barrier genesis,10 and the barrier function in retinal vessels is modulated by the retinal endothelial junction structure.10,11 Specific junction molecules in retinal endothelial cells are requisite for the maintenance of barrier function. Recently, we have shown that zonula occludens (ZO)-1 and occludin are well-characterized components of the tight junction in retinal endothelial cells.10,12,13 ZO-1 is a cytoplasmic protein which links occludin to the other intracellular junction structures. Particularly, the level of ZO-1 expression is inversely related to permeability in blood-retinal barrier10,12–14 as well as that of occludin.15 However, the specific molecular pathogenesis for increased permeability has not been elucidated.
Increased vascular permeability in diabetes involves phosphorylation and reorganization of specific junction proteins.16 In diabetes, excess glucose is metabolized by glycolysis, which increases synthesis of intracellular diacylglycerol (DAG), the main endogenous activator of protein kinase C (PKC). The PKC superfamily is composed of three subfamilies including classical PKC (cPKC; α, β1, β2, and γ), novel PKC (nPKC; δ, ε, η, θ, and μ), and atypical PKC (aPKC: ζ, λ/ι).17 cPKC is activated by both calcium and DAG, nPKC is regulated by DAG, but not by calcium, and aPKC responds to neither calcium nor DAG.17 Since PKC activation appears to be due to increase of DAG, all isoforms sensitive to DAG are likely to be activated in diabetes. Actually, ruboxistaurin, a selective inhibitor for β isoforms, has shown the efficacy to ameliorate the vascular dysfunction in diabetes.18 Besides PKC β, some specific isoforms, PKC α and δ, are also crucial in diabetic microvascular complications.19
In the current study, we investigated that in diabetic retina, PKC δ activation is involved in decrease of tight junction proteins, particularly ZO-1 and ZO-2, which is followed by BRB breakdown. Moreover, our results suggest that PKC δ inhibition could prevent BRB breakdown in diabetic retinopathy.
Materials and Methods
Mice
C57BL/6 mice were purchased from Samtako (Korea). Care, use, and treatment of all animals in this study were in strict agreement with the ARVO statement for the Use of Animals in Ophthalmic and Vision Research. C57BL/6 mice were kept in standard 12-hour dark-light cycles and approximately 23°C room temperature.
Cell Culture
Human retina microvascular endothelial cells (HRMECs) were purchased from the Applied Cell Biology Research Institute and grown on attachment factor-coated plates in complete medium (Cell Systems, Kirkland, WA) or in M199 medium supplemented with 20% fetal bovine serum, 3 ng/ml basic fibroblast growth factor (Millipore, Bedford, MA), and 10 U/ml heparin (Sigma, St. Louis, MO). HRMECs used in this study were taken from passages 4 to 6. Advanced glycation end product (AGE) (Calbiochem, Darmstadt, Germany) treatment (10 μg/ml) was performed in cells cultured in serum-free M199 supplemented with 1% (v/v) penicillin-streptomycin.
Induction of Diabetes in Mice
To induce diabetes, 10-week-old male mice were intraperitoneally injected with 180 mg/kg streptozotocin (Sigma). If plasma glucose concentrations was >300 mg/dl at 24 hours after streptozotocin injection, mice were considered to be diabetic. To assess the anti-permeable activity of PKC δ inhibition, 10 μmol/L rottlerin (Sigma) or siPKC δ (50 nmol/L/1 μl), in 1 μl phosphate-buffered saline (PBS) or PBS only was intravitreously injected to diabetic mice of 7 days after streptozotocin injection.
Mice Retinal Tissue Preparation
At 2, 4, 6, and 8 days after streptozotocin injection with or without intravitreal injection of a PKC δ inhibitor, rottlerin (200 nmol/L/1 μl) or siPKC δ (50 nmol/L/1 μl), mice were carefully sacrificed and the eyes were enucleated and hemisected at the ora serrata. The retinas were gently teased off the sclera using a fine brush. Contamination by retinal pigment epithelial cells was reduced to a minimum. Whole retinal proteins were extracted with lysis buffer [50 mmol/L Tris (pH 7.6), 150 mmol/L NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate, protease inhibitor cocktail (Sigma), 1 mmol/L phenylmethylsulfonyl fluoride] on ice for 20 minutes, centrifuged at 14,000 rpm for 20 minutes, and then supernatants were harvested and stored at −80°C.
Leakage Assessment by Perfusion of Retinal Vessels with Fluorescein Isothiocyanate-Bovine Serum Albumin (FITC-BSA)
With some modifications of previous our description,20 at 8 days after streptozotocin injection with or without intravitreal injection of a PKC δ inhibitor, rottlerin (200 nmol/L/1 μl) or siPKC δ (50 nmol/L/1 μl), deeply anesthetized mice were perfused through the tail vein with FITC-BSA (Sigma) dissolved in PBS. For retinal flat mounting, after 1 hour of perfusion, the eyes were enucleated and fixed in 4% paraformaldehyde for 2 hours. The retinas were dissected, flat-mounted in Dako mounting medium (DakoCytomation, Glostrup, Denmark), and viewed by fluorescence microscopy (BX50, Olympus, Japan). For quantification of retinal vascular leakage, after 1 hour perfusion, the eyes were enucleated, embedded in OCT medium and immediately frozen in liquid nitrogen. The plasma was collected and assayed for fluorescence with an SPEX fluorescence spectrophotometer (Molecular Devices, Sunnyvale, CA) based on standard curves of FITC-BSA in normal mouse plasma. Frozen retinal sections (5 μm thick) collected every 30 μm were viewed with a fluorescence microscope (BX50, Olympus, Japan), and six images from nonvascular retina (200 μm2) in each section were collected. Quantification of FITC-BSA fluorescence intensity was calculated by computer software Q-win (Leica, Wetzlar, Germany) and normalized to plasma fluorescence intensity for each animal.
Western Blotting
Western blotting was performed using standard western blotting methods. The protein concentration was measured using a BCA protein assay kit (Pierce, Rockford, IL). Equal amounts of protein were separated by electrophoresis on 5–10% sodium dodecyl sulfate- polyacrylamide gel electrophoresis and transferred electrophoretically onto nitrocellulose membrane (Amersham, Little Chalfont, UK). The membranes were blocked for 30 minutes in 5% nonfat milk. The membranes after blocking were incubated overnight with anti-PECAM (1:1000, Santa Cruz, Santa Cruz, CA), anti-ZO-1 (1:1000, Zymed, San Francisco, CA), anti-ZO-2 (1:2000, Zymed) and anti-occludin (1:1000, Zymed), anti-PKC δ (1: 1000, Santa Cruz), and anti-c-myc (1: 1000, Santa Cruz) at 4°C. After they were washed with PBS-T, the membrane was incubated for 1 hour at room temperature with horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG (1:10,000, Pierce) in PBS-T and 1% nonfat milk. To ensure the equal loading of protein in each lane, the blots were stripped and reprobed with an antibody against β-actin. Intensity values were normalized relative to control values. The blots were scanned using a flatbed scanner and the band intensity analyzed using the TINA software program (Raytest, Staubenhardt, Germany).
Transient Transfection of Dominant-Negative PKC δ Construct
As our previous report,21 transient transfection of vector overexpressing dominant-negative PKC δ in HRMECs was performed using the FuGene 6 transfection reagent (Roche Applied Science, Indianapolis, IN) following the manufacturer's protocol. PKC-δ-DN [catalytic fragment dominant-negative mutant (K376R) of mouse PKC δ] plasmid was constructed by subcloning pMTH-PKC-δ-DN into pCS2 ± c-myc plasmid (Clontech, Palo Alto, CA). pMTH-PKC-δ-DN was provided by Dr. Yuspa (University of Pennsylvania). Briefly, HRMECs were seeded at 2 × 105 cells in a six-well plate 1 day before transfection. Cells were subsequently cultured for 48 hours to allow for detectable protein expression. Transfection of PKC-δ-DN was confirmed by Western blotting with anti-c-myc antibody.
siRNA Transfection
As our previous report,21 siRNA duplex (siRNA) targeting human PKC δ was chemically synthesized and purified in the 2-deprotected and desalted form (Dharmacon, Lafayette, CO). The sequences of PKC δ siRNA pair were 5′-GCUUCAAGGUUCACAACUAUU-3′ and 5′-UAGUUGUGAACCUUGAAGCUU-3′. Scrambled RNA (catalog no. D-001210–02) purchased from Pharmacon. Transfection of siRNA was performed using oligofectamine (Invitrogen, Rockville, MD), according to the manufacturer's instructions. Briefly, HRMECs were seeded at 2 × 105 cells in a six-well plate 1 day before transfection. The cells were transfected at the final concentration of 50 nmol/L siRNA duplex. The efficacy of knockdown was assessed using western blotting with anti-PKC δ antibody and reverse transcriptase-polymerase chain reaction (RT-PCR) using a PKC δ specific primer.
[3H]sucrose Permeability Assay
With some modifications of previous our description,22 HRMECs (1 × 105 cells) were plated onto a Transwell filter (Corning Costar, Cambridge, MA). After reaching confluence, rhVEGF (20 ng/ml, Sigma) or the inhibitors of PKC isoforms including a pan-PKC inhibitor (GF109203X, Sigma), a PKC β inhibitor (LY379196, Eli Lilly Company, Indianapolis, IN), rottlerin (Sigma) were treated for 6 hours in HRMECs with or without PKC-δ-DN plasmid, scrambled RNA or siPKC δ. [3H]sucrose, 50 μl (0.8 μCi/ml) (1 μCi/μl; Amersham Pharmacia, Bucks, UK), was added to the upper compartment. The amount of radioactivity that diffused into the lower compartment was determined after 30 minutes by liquid scintillation counter (Perkin Elmer/Wallac, Gaithersburg, MD).
RT-PCR Analysis
Total RNA from cells was isolated using Trizol reagent (Invitrogen, Rockville, MD), according to the manufacturer's instructions. First-stranded cDNA was synthesized with 3 μg of each DNA free total RNA and oligo-(dT) 16 primer by Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI). Equal amounts of cDNA were subsequently amplified by PCR in a 50-μl reaction volume containing 1X PCR buffer, 200 μmol/L dNTPs, 10 μmol/L specific primer for PKC-δ (5′-CAGGGATTAAAGTGTGAAG-3′ and 5′-TTCTTCTCGAAACCCTGATA-3′), β-actin (5′-GACTACCTCATGAAGATC-3′ and 5′-CACATCTGCTGGAA-3′) and 1.25 U TaqDNA polymerase (TaKaRa, Tokyo, Japan). PCR was performed with an initial denaturation step at 94°C for 5 minutes, followed by 25 (PKC-δ, β-actin) of denaturation (94°C for 1 minute), annealing (50°C [PKC-δ]; 55°C [β-actin] 1 minute), and extension (72°C for 1 minute). The PCR products (PKC-δ, 500 bp; β-actin, 550 bp) were separated on agarose gels and visualized using ethidium bromide staining under UV transillumination.
Cellular Fractionation
The cells were washed and pelleted after treatment, resuspended in 150 μl of hypotonic buffer (20 mmol/L Tris-HCl, 5 mmol/L EGTA, 2 mmol/L EDTA, 2 mmol/L β-mercaptoethanol) and left for 30 minutes at 4°C to swell. Cells were lysed using sonication for 5 seconds and centrifuged at 16,000 × g at 4°C for 30 minutes, and the supernatant was collected as the cytosol fraction. The pellet was re-suspended in the RIPA buffer (50 mmol/L Tris-HCl 150 mmol/L NaCl, 0.5 mmol/L EDTA, 1% NP-40, 0.5% DOC, 0.1% sodium dodecyl sulfate) and left for 30 minutes at 4°C. The suspension was centrifuged again at 16,000 × g at 4°C for 30 minutes and the supernatant was taken as the membrane fraction.
Immunohistochemistry
The enucleated mice eyes used for immunohistochemistry were immersion fixed in 4% paraformaldehyde and subsequently embedded in paraffin. Serial sections 4 mm thick were prepared from paraffin blocks. Sections were deparaffinized and hydrated by sequential immersion in xylene and graded alcohol solutions, treated with proteinase K for 5 minutes at 37°C and then treated with normal serum obtained from the same species in which the secondary antibody was developed for 10 minutes to block nonspecific staining. Slides were incubated overnight at 4°C with anti-ZO-1 (1:100, Zymed, San Francisco, CA) and anti-ZO-2 (1:100, Zymed), followed by a biotinylated goat anti-mouse antibody (Dako, Glostrup, Denmark), revealed by the avidin-biotin complex (Vectastain kit; Vector, Burlingame, CA) and the 3-amino-9-ethyl-carbazole chromogen. The slides were mounted Faramount aqueous mounting medium (DAKO) and observed under light microscopy (Carl Zeiss, Chester, VA).
Statistical Analysis
Statistical differences between groups were evaluated with the Student unpaired t-test (two-tailed). Mean ± SD is shown. P ≤ 0.05 was considered significant.
Results
Increased Vascular Permeability of Diabetic Retina Is Accompanied by Decrease of Tight Junction Proteins
To evaluate increased vascular permeability in diabetic retina, fluorescein angiography using FITC-BSA was performed. As shown in Figure 1A, fluorescein conjugated dextran easily infiltrates through the vessel wall and diffuses into the diabetic retina. Accordingly, increased vascular permeability was recognized as diffuse fluorescence all around the retina. FITC-BSA fluorescence intensity was measured by image analysis in serial retinal sections. The average retinal FITC-BSA fluorescence intensity was calculated and normalized to plasma fluorescence intensity. The retinal FITC-BSA fluorescence intensity at 8 days after induction of diabetes diabetic retina was significantly increased (3.41 ± 0.24 pixels) compared with in the control retina (0.35 ± 0.08, P < 0.05).
Figure 1.
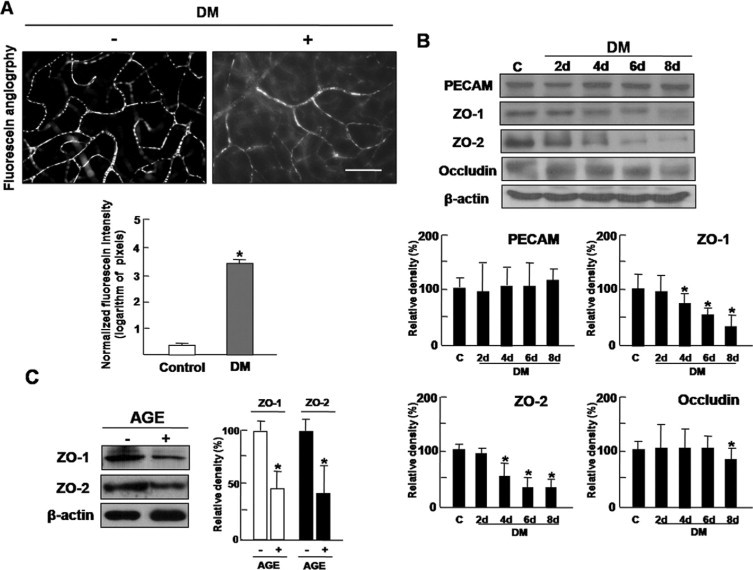
Increased vascular permeability of diabetic retina is accompanied by decrease of tight junction proteins. A: Increased vascular permeability was evaluated by fluorescein angiography using FITC-BSA. Whole-mount retinal preparation from 8 days after streptozotocin injection was performed after 1 hour perfusion of FITC-BSA. These experiments were repeated over three times with similar results. FITC-BSA fluorescence intensity was measured by image analysis in serial retinal sections. The average retinal FITC-BSA fluorescence intensity was calculated and normalized to plasma fluorescence intensity. Figures were selected as representative data from three independent experiments. Scale bars = 100 μm. *P < 0.005. DM, diabetes mellitus. B: At 2, 4, 6, and 8 days after streptozotocin injection, retinal proteins of diabetic mice were analyzed by Western blot analysis using PECAM, ZO-1, ZO-2, and occludin antibodies. β-Actin served as the loading control. DM, diabetes mellitus. C: HRMEC proteins from cells incubated with AGE treatment (10 μg/ml) for 12 hours were analyzed by Western blot analysis using ZO-1 and ZO-2 antibodies. β-Actin was served as the loading control. B and C: Quantitative analysis was performed by measuring protein expression relative to the control. Each point represents the mean (±SD) of three independent experiments, each performed in triplicate. *P < 0.005. Figures were selected as representative data from three independent experiments. DM, diabetes mellitus.
Given the correlation with between increased vascular permeability and loosening of the tight junctions in diabetic retinopathy, we investigated expression of an endothelial cell marker, PECAM, and tight junction proteins, ZO-1, ZO-2, and occludin in diabetic retina. While PECAM showed no significant change during 8 days of diabetes induction, marked decreases in ZO-1 and ZO-2 were detected even from 4 days after diabetes induction (Figure 1B). Occludin, whose phosphorylation was known to be requisite to vascular endothelial cell growth factor-induced endothelial permeability,23 decreased later than ZO-1 and ZO-2 did (Figure 1B).
To confirm whether expression of ZO-1 and ZO-2 in retinal endothelial cells decreases under diabetic condition, HRMECs were incubated with AGE treatment (10 μg/ml) for 12 hours and assessed for ZO-1 and ZO-2. With treatment of AGE, ZO-1 and ZO-2 were significantly reduced in HRMECs (Figure 1C). These results indicate that increased vascular permeability of diabetic retina is accompanied by decrease of tight junction proteins in retinal endothelial cells.
Inhibitors of PKC Isoforms Attenuate Loss of Tight Junction Proteins Under Diabetic Condition and Vascular Endothelial Growth Factor (VEGF)-Induced Hyperpermeability in HRMECs
Based on that VEGF-mediated vascular hyperpermeability in diabetic retinopathy could be regulated through activation of PKC in vivo,24,25 it was investigated whether anti-permeable activities of the inhibitors of PKC isoforms were related to regulation of tight junction proteins. As demonstrated in Figure 2A, loss of tight junction proteins in AGE-treated HRMECs was significantly inhibited by all inhibitors including a pan-PKC inhibitor, a PKC β inhibitor and rottlerin in dose-dependent manner. In addition, VEGF-mediated hyperpermeability in HRMECs was effectively prevented by inhibitors of PKC isoforms (Figure 2B). These results suggest that Inhibition of PKC isoforms could attenuate vascular leakage through regulation of tight junction proteins in retinal endothelial cells.
Figure 2.
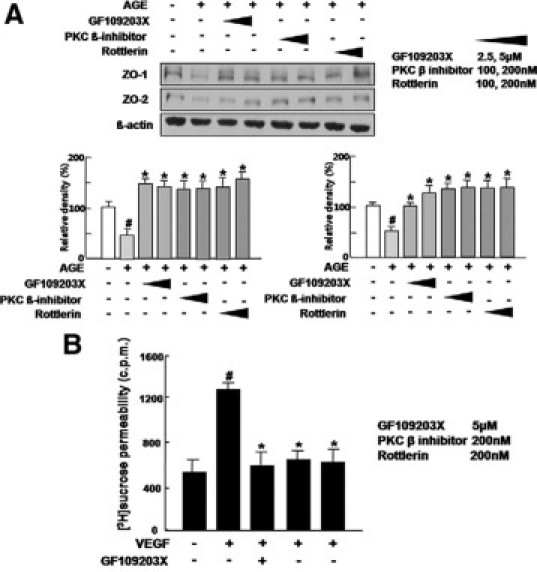
Inhibitors of PKC isoforms attenuate loss of tight junction proteins under diabetic condition and VEGF-induced hyperpermeability in HRMECs. A: HRMECs were incubated for 12 hours with or without the inhibitors of PKC isoforms including 5 μmol/L GF109203X), 200 nmol/L a PKC β inhibitor and 200 nmol/L rottlerin in AGE treatment and assayed for the expression of ZO-1 and ZO-2. ß-actin was served as the loading control. Figures were selected as representative data from three independent experiments. Quantitative analysis was performed by measuring protein expression relative to the control. Each point represents the mean (±SD) of three independent experiments, each performed in triplicate. *P < 0.05. #Comparison between control and AGE only treatment. *Comparison between AGE only treatment and AGE with PKC inhibitor treatment. #P, *P < 0.05. B: VEGF, 20 ng/ml, or the inhibitors of PKC isoforms including 5 μmol/L GF109203X), 200 nmol/L PKC β inhibitor, and 200 nmol/L rottlerin were treated for 6 hours in HRMECs. [3H]sucrose permeability assay in HRMECs treated with VEGF or the inhibitors was measured as counts per minute (c.p.m.). Each point represents the mean (±SD) of three independent experiments, each performed in triplicate. #Comparison between control and VEGF only treatment. *Comparison between VEGF only treatment and VEGF with PKC inhibitor treatment. #P, *P < 0.05.
Inhibition of PKC δ by PKC-δ-DN Attenuates VEGF-Induced Hyperpermeability and Loss of Tight Junction Proteins Under Diabetic Condition in HRMECs
To investigate the effect of PKC δ Inhibition on VEGF-induced hyperpermeability and the expression of tight junction proteins in retinal endothelial cells under diabetic condition, [3H]sucrose permeability assay in HRMECs and the expression of ZO-1 and ZO-2 in AGE-treated HRMECs were assessed with transfection of PKC-δ-DN plasmid. The efficacy of transfection for PKC-δ-DN was confirmed by c-myc expression. As shown in Figure 3A, PKC δ inhibition by PKC-δ-DN transfection effectively prevented hyperpermeability in VEGF-treated HRMECs (P < 0.05). Moreover, transfection of PKC-δ-DN significantly restored expression of ZO-1 and ZO-2 in AGE-treated HRMECs (Figure 3B).
Figure 3.
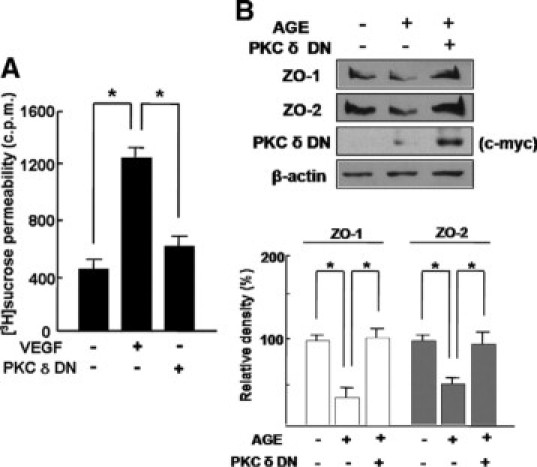
Inhibition of PKC δ by PKC-δ-DN attenuates VEGF-induced hyperpermeability and loss of tight junction proteins under diabetic condition in HRMECs. A: VEGF, 20 ng/ml, was treated for 6 hours in HRMECs with or without transfection of PKC-δ-DN plasmid. [3H]sucrose permeability assay in HRMECs was measured as counts per minute (c.p.m.). Each point represents the mean (±SD) of three independent experiments, each performed in triplicate. *P < 0.05. B: HRMECs were transfected by PKC-δ-DN plasmid. Cells were subsequently cultured for 48 hours to allow for detectable protein expression and were additionally incubated for 12 hours in AGE treatment. The expression of ZO-1 and ZO-2 was assessed, and transfection of PKC-δ-DN was confirmed by expression of c-myc. β-Actin was served as the loading control. Figures were selected as representative data from three independent experiments. Quantitative analysis was performed by measuring protein expression relative to the control. Each point represents the mean (±SD) of three independent experiments, each performed in triplicate. *P < 0.05.
Inhibition of PKC δ by siPKC δ Attenuates VEGF-Induced Hyperpermeability and Loss of Tight Junction Proteins under Diabetic Condition in HRMECs
Next, to confirm the effect of PKC δ Inhibition on VEGF-induced hyperpermeability and the expression of tight junction proteins in retinal endothelial cells under diabetic condition, it was investigated whether knockdown of PKC δ effectively inhibits hyperpermeability and loss of tight junction proteins in HRMECs. The efficacy of transfection for siPKC δ was confirmed by PKC δ expression. As demonstrated in Figure 4A, PKC δ inhibition by siPKC δ significantly blocked hyperpermeability in VEGF-treated HRMECs (P < 0.05), compared with no effect of scrambled RNA on hyperpermeability (P > 0.05). In addition, knockdown of PKC δ significantly prevented loss of ZO-1 and ZO-2 in AGE-treated HRMECs (Figure 4B).
Figure 4.

Inhibition of PKC δ by siPKC δ attenuates VEGF-induced hyperpermeability and loss of tight junction proteins under diabetic condition in HRMECs. A: VEGF, 20 ng/ml, was treated for 6 hours in HRMECs with or without transfection of scrambled RNA or siPKC δ. [3H]sucrose permeability assay in HRMECs was measured as counts per minute (c.p.m.). Each point represents the mean (±SD) of three independent experiments, each performed in triplicate. *P < 0.05; **P > 0.05. B: HRMECs were transfected with siPKC δ and assayed for the expression of ZO-1 and ZO-2 in AGE treatment. The efficacy of knockdown was assessed using western blotting with anti-PKC δ antibody. β-Actin served as the loading control. Figures were selected as representative data from three independent experiments. Quantitative analysis was performed by measuring protein expression relative to the control. Each point represents the mean (±SD) of three independent experiments, each performed in triplicate. *P < 0.05; **P > 0.05.
Inhibition of PKC δ by siPKC δ Attenuates Vascular Leakage in Diabetic Retina, Accompanied by Restoration of Tight Junction Proteins on Retinal Vessels
In particular, to investigate the effect of PKC δ inhibition on vascular permeability in diabetic retina, whole mount retinal preparation from 8 days after streptozotocin injection with intravitreal injection of siPKC δ was performed after 1 hour of perfusion of fluorescein conjugated dextran. As shown in Figure 5A, PKC δ inhibition by siPKC δ dramatically inhibited vascular leakage in diabetic retina. The normalized FITC-BSA fluorescence intensity was significantly decreased with the intravitreal injection of the siPKC δ in diabetic retina (P < 0.05). Compared with the control (0.43 ± 0.22 pixels), the FITC-BSA fluorescence intensity in diabetic retina was significantly increased (3.55 ± 0.42 pixels, P < 0.05) at 8 days after induction of diabetes. Intravitreal injection of scrambled RNA never reduced the FITC-BSA fluorescence intensity in diabetic retina (3.54 ± 0.38 pixels, P > 0.05), whereas the fluorescence intensity in eyes receiving intravitreal siPKC δ injection was effectively reduced (0.72 ± 0.34 pixels, P < 0.05).
Figure 5.
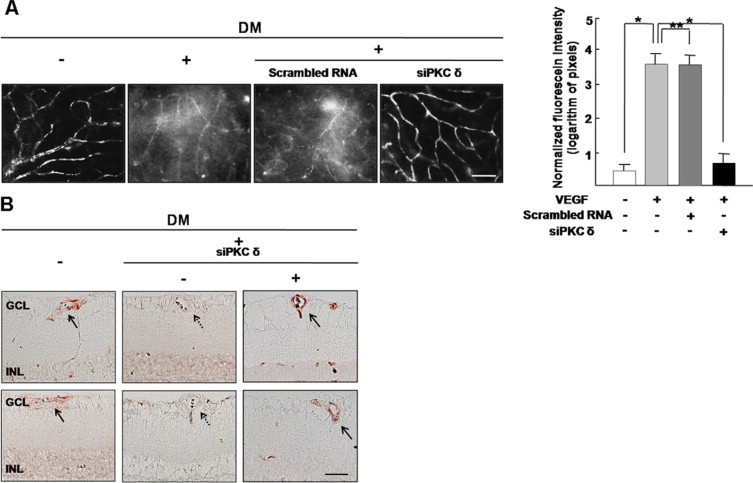
Inhibition of PKC δ by siPKC δ attenuates vascular leakage in diabetic retina, accompanied by restoration of tight junction proteins on retinal vessels. A: Vascular leakage in the retina was evaluated by fluorescein angiography using FITC-BSA. Whole-mount retinal preparation from 8 days after streptozotocin injection with or without intravitreal injection of siPKC δ was performed after 1 hour of perfusion of FITC-BSA. These experiments were repeated over three times with similar results. Figures were selected as representative data from three independent experiments. FITC-BSA fluorescence intensity was measured by image analysis in serial retinal sections. The average retinal FITC-BSA fluorescence intensity was calculated and normalized to plasma fluorescence intensity. Scale bars = 100 μm. *P < 0.05; **P > 0.05. DM, diabetes mellitus. B: Immunohistochemistry for ZO-1 and ZO-2 was performed in diabetic retina with or without intravitreal injection of siPKC δ. Arrows indicate ZO-1 (upper panel) or ZO-2 (lower panel) expression on retinal vessels, whereas arrow with dotted line indicates loss of ZO-1 (upper panel) or ZO-2 (lower panel). Figures were selected as representative data from three independent experiments. Scale bars = 50 μm. DM, diabetes mellitus; GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer.
To examine the effect of PKC δ inhibition on restoration of tight junction proteins accompanying decrease of vascular leakage in diabetic retina, Immunohistochemistry for ZO-1 and ZO-2 was performed in diabetic retina with intravitreal injection of siPKC δ. As shown in Figure 5B, ZO-1 and ZO-2 expression in diabetic retina was decreased compared with control, whereas it was recovered by knockdown of PKC δ.
Translocation of PKC δ from Cytosol to Membrane in HRMECs Is Significantly Increased under Diabetic Condition, but Not PKC δ Expression
Based on a previous report of differential, subcellular localization of PKC δ,22 we investigated the translocation of PKC δ in HRMECs under diabetic condition. As demonstrated in Figure 6, PKC δ expression in AGE-treated HRMECs did not change compared with that in control or PKC δ inhibition. Interestingly, PKC δ translocates from cytosol to membrane in AGE-treated HRMECs, whereas translocation of PKC δ was blocked by treatment of GF109203X, a pan-PKC inhibitor (Figure 6).
Figure 6.
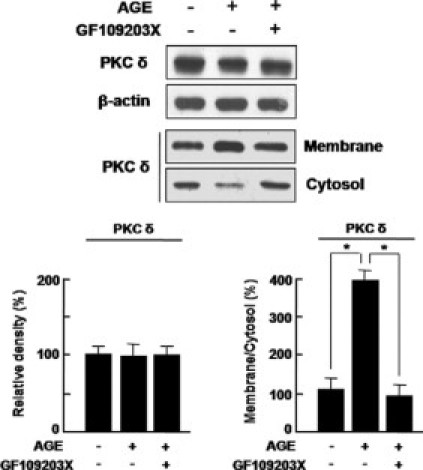
Translocation of PKC δ from cytosol to membrane in HRMECs was significantly increased under diabetic condition, but not PKC δ expression. HRMECs were incubated for 12 hours with or without a pan-PKC inhibitor, GF109203X (5 μmol/L) in AGE treatment. Cell extracts were fractionated as a membrane and a cytosol fraction. In each fraction, PKC δ expression was assessed by Western blotting. β-Actin served as the loading control. Figures were selected as representative data from three independent experiments. Quantitative analysis was performed by measuring protein expression relative to the control. Each point represents the mean (±SD) of three independent experiments, each performed in triplicate. *P < 0.05.
Discussion
Multiple diabetic complications including diabetic retinopathy have been associated with activation of PKC and its isoforms, which have been shown to cause vascular abnormalities in several different ways.19 Among various PKC isoforms, PKC β has been strongly emphasized in vascular dysfunction of diabetic retina.6,18 Given that PKC β exhibits a greater increase than other isoforms in vascular tissues,19 PKC β deserves to be considered as the critical player in diabetic retinopathy. Recent reports demonstrate that other PKC isoforms could also contribute to part of increased permeability in retinal endothelial cells.19,23 However, involvement of other specific isoform has not been fully characterized. Therefore, we herein provided the evidence that PKC δ inhibition could prevent BRB breakdown in diabetic retinopathy.
We demonstrated that PKC δ activation is involved in increased vascular permeability in response to diabetes, which is accompanied by decrease of tight junction proteins in retinal endothelial cells. Interestingly, expression of ZO-1 and ZO-2 was significantly decreased earlier than occludin in diabetic retina, which is supported by the indirect evidence that ZO-1 could provide the changes of permeability, for ZO-1 is closely linked to occludin, whose phosphorylation contributes to the regulation of permeability.26 ZO-1 and ZO-2 are basically junctional proteins associated with the cytoplasmic surface of the tight junction, which is localized to the points of membrane contact with the fibrils. They are involved in organizing tight junction and its cellular localization is closely related to the permeability property of vascular endothelial cells.27 In particular, we have suggested that ZO family is the well-characterized component of tight junction in retinal endothelial cells,10,12,13 whose expression is inversely related to permeability in blood-retinal barrier.10,12–14 Our results suggest that decrease of ZO-1 and ZO-2 expression might demonstrate the loosening of blood-retinal barrier in diabetic retina.
In the current study, we addressed the relation of PKC δ activation to expression of tight junction proteins by PKC δ inhibition. With treatment of rottlerin, transfection of PKC-δ-DN, and transfection of siRNA for PKC δ, loss of ZO-1 and ZO-2 was effectively restored or prevented. We however found out that PKC δ expression in diabetic retina didn’t change though PKC δ inhibition restored expression of ZO-1 and ZO-2. In addition, AGE-induced diabetic condition did not change PKC δ expression in HRMECs. Based on the previous report that PKC δ localizes to the cytoplasmic, subcellular organelles in unstimulated condition, and it is translocated to membrane with stimulation,28 subcellular location of PKC δ was investigated in HRMECs under diabetic condition. As expected, PKC δ translocated from cytosol to membrane in AGE-treated HRMECs, whereas translocation of PKC δ was blocked by PKC δ inhibition. These data provide that PKC δ activation, related to its subcellular translocation, is crucial in regulation of tight junction proteins, not PKC δ expression itself. In addition to the restoration of ZO-1 and ZO-2 in AGE-treated HRMECs, inhibition of PKC δ restored loss of tight junction proteins on vessels in diabetic retina.
In conclusion, PKC δ activation, related to its subcellular translocation, is involved in increased vascular permeability in response to diabetes. Inhibition of PKC δ effectively restores loss of tight junction proteins on vessels in diabetic retina as well as attenuates loss of tight junction proteins in AGE-treated HRMECs. Therefore, we suggest that PKC δ inhibition could be prevent BRB breakdown in diabetic retinopathy. Furthermore, inhibition of PKC δ could be further investigated for an alternative treatment to BRB breakdown in diabetic retinopathy.
Acknowledgements
We thank Dr. Yuspa for the gift of pMTH-PKC-δ-DN vector.
Footnotes
Supported by grant 03-2006-017-0 from the Seoul National University Hospital Research Fund and by the Bio-signal Analysis Technology Innovation Program (2009-0090895) of the Ministry of Science and Technology and Korea Science and Engineering Foundation.
J.-H.K. and J.H.K. contributed equally to this work.
References
- 1.Porta M, Bandello F. Diabetic retinopathy: an update. Diabetologia. 2002;45:1617–1634. doi: 10.1007/s00125-002-0990-7. [DOI] [PubMed] [Google Scholar]
- 2.Frank RN. Diabetic retinopathy. N Engl J Med. 2004;350:48–58. doi: 10.1056/NEJMra021678. [DOI] [PubMed] [Google Scholar]
- 3.Davidson JA, Ciulla TA, McGill JB, Kles KA, Anderson PW. How the diabetic eye loses vision. Endocrine. 2007;32:107–116. doi: 10.1007/s12020-007-0040-9. [DOI] [PubMed] [Google Scholar]
- 4.Mizutani M, Gerhardinger C, Lorenzi M. Muller cell changes in human diabetic retinopathy. Diabetes. 1998;47:445–449. doi: 10.2337/diabetes.47.3.445. [DOI] [PubMed] [Google Scholar]
- 5.Lorenzi M, Gerhardinger C. Early cellular and molecular changes induced by diabetes in the retina. Diabetologia. 2001;44:791–804. doi: 10.1007/s001250100544. [DOI] [PubMed] [Google Scholar]
- 6.Antonetti DA, Lieth E, Barber AJ, Gardner TW. Molecular mechanisms of vascular permeability in diabetic retinopathy. Semin Ophthalmol. 1999;14:240–248. doi: 10.3109/08820539909069543. [DOI] [PubMed] [Google Scholar]
- 7.Antonetti DA, Barber AJ, Bronson SK, Freeman WM, Gardner TW, Jefferson LS, Kester M, Kimball SR, Krady JK, LaNoue KF, Norbury CC, Quinn PG, Sandirasegarane L, Simpson IA, JDRF Diabetic Retinopathy Center Group Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes. 2006;55:2401–2411. doi: 10.2337/db05-1635. [DOI] [PubMed] [Google Scholar]
- 8.Cunha-Vaz JG, Faria de Abreu JR, Campos AJ, Figo GM. Early breakdown of the blood-retinal barrier in diabetes. Br J Ophthalmol. 1975;59:649–656. doi: 10.1136/bjo.59.11.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee SW, Kim WJ, Choi YK, Song HS, Son MJ, Gelman IH, Kim YJ, Kim KW. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med. 2003;9:900–906. doi: 10.1038/nm889. [DOI] [PubMed] [Google Scholar]
- 10.Choi YK, Kim JH, Kim WJ, Lee HY, Park JA, Lee SW, Yoon DK, Kim HH, Chung H, Yu YS, Kim KW. AKAP12 regulates human blood-retinal barrier formation by downregulation of hypoxia-inducible factor-1alpha. J Neurosci. 2007;27:4472–4481. doi: 10.1523/JNEUROSCI.5368-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JH, Kim JH, Park JA, Lee SW, Kim WJ, Yu YS, Kim KW. Blood-neural barrier: intercellular communication at glio-vascular interface. J Biochem Mol Biol. 2006;39:339–345. doi: 10.5483/bmbrep.2006.39.4.339. [DOI] [PubMed] [Google Scholar]
- 12.Kim JH, Kim JH, Yu YS, Min BH, Kim KW. The role of clusterin in retinal development and free radical damage. Br J Ophthalmol. 2007;91:1541–1546. doi: 10.1136/bjo.2007.115220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JH, Yu YS, Kim JH, Kim KW, Min BH. The role of clusterin in in vitro ischemia of human retinal endothelial cells. Curr Eye Res. 2007;32:693–698. doi: 10.1080/02713680701487871. [DOI] [PubMed] [Google Scholar]
- 14.Kim JH, Kim JH, Yu YS, Kim DH, Kim KW. The recruitment of pericytes and astrocytes is closely related to the formation of tight junction in developing retinal vessels. J Neurosci Res. 2009;87:653–659. doi: 10.1002/jnr.21884. [DOI] [PubMed] [Google Scholar]
- 15.Russ PK, Davidson MK, Hoffman LH, Haselton FR. Partial characterization of the human retinal endothelial cell tight and adherens junction complexes. Invest Ophthalmol Vis Sci. 1998;39:2479–2485. [PubMed] [Google Scholar]
- 16.Stasek JE, Jr, Patterson CE, Garcia JG. Protein kinase C phosphorylates caldesmon77 and vimentin and enhances albumin permeability across cultured bovine pulmonary artery endothelial cell monolayers. J Cell Physiol. 1992;153:62–75. doi: 10.1002/jcp.1041530110. [DOI] [PubMed] [Google Scholar]
- 17.Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J. 1998;332:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A, Bursell SE, Kern TS, Ballas LM, Heath WF, Stramm LE, Feener EP, King GL. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC β inhibitor. Science. 1996;272:728–731. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 19.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 20.Kim JH, Kim JH, Yu YS, Cho CS, Kim KW. Blockade of angiotensin II attenuates VEGF-mediated blood-retinal barrier breakdown in diabetic retinopathy. J Cereb Blood Flow Metab. 2009;29:621–628. doi: 10.1038/jcbfm.2008.154. [DOI] [PubMed] [Google Scholar]
- 21.Lee JW, Park JA, Kim SH, Seo JH, Lim KJ, Jeong JW, Jeong CH, Chun KH, Lee SK, Kwon YG, Kim KW. Protein kinase C-delta regulates the stability of hypoxia-inducible factor-1 alpha under hypoxia. Cancer Sci. 2007;98:1476–1481. doi: 10.1111/j.1349-7006.2007.00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Min JK, Cho YL, Choi JH, Kim Y, Kim JH, Yu YS, Rho J, Mochizuki N, Kim YM, Oh GT, Kwon YG. Receptor activator of nuclear factor (NF)-kappaB ligand (RANKL) increases vascular permeability: impaired permeability and angiogenesis in eNOS-deficient mice. Blood. 2007;109:1495–1502. doi: 10.1182/blood-2006-06-029298. [DOI] [PubMed] [Google Scholar]
- 23.Aiello LP, Bursell SE, Clermont A, Duh E, Ishii H, Takagi C, Mori F, Ciulla TA, Ways K, Jirousek M, Smith LE, King GL. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes. 1997;46:1473–1480. doi: 10.2337/diab.46.9.1473. [DOI] [PubMed] [Google Scholar]
- 24.Wang QJ, Bhattacharyya D, Garfield S, Nacro K, Marquez VE, Blumberg PE. Differential localization of protein kinase C delta by phorbol esters and related compounds using a fusion protein with green fluorescent protein. J Biol Chem. 1999;274:37233–37239. doi: 10.1074/jbc.274.52.37233. [DOI] [PubMed] [Google Scholar]
- 25.Harhaj NS, Felinski EA, Wolpert EB, Sundstrom JM, Gardner TW, Antonetti DA. VEGF activation of protein kinase C stimulates occludin phosphorylation and contributes to endothelial permeability. Invest Ophthalmol Vis Sci. 2006;47:5106–5115. doi: 10.1167/iovs.06-0322. [DOI] [PubMed] [Google Scholar]
- 26.Sakakibara A, Furuse M, Saitou M, Ando-Akatsuka Y, Tsukita S. Possible involvement of phosphorylation of occludin in tight junction formation. J Cell Biol. 1997;137:1393–1401. doi: 10.1083/jcb.137.6.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischer S, Wobben M, Marti HH, Renz D, Schaper W. Hypoxia-Induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res. 2002;63:70–80. doi: 10.1006/mvre.2001.2367. [DOI] [PubMed] [Google Scholar]
- 28.Kazi JU, Soh JW. Isoform-specific translocation of PKC isoforms in NIH3T3 cells by TPA. Biochem Biophys Res Commun. 2007;364:231–237. doi: 10.1016/j.bbrc.2007.09.123. [DOI] [PubMed] [Google Scholar]