

Abstract
Angiogenesis and lymphangiogenesis participate in many inflammatory diseases, and their reversal is thought to be beneficial. However, the extent of reversibility of vessel remodeling is poorly understood. We exploited the potent anti-inflammatory effects of the corticosteroid dexamethasone to test the preventability and reversibility of vessel remodeling in Mycoplasma pulmonis-infected mice using immunohistochemistry and quantitative RT-PCR. In this model robust immune responses drive rapid and sustained changes in blood vessels and lymphatics. In infected mice not treated with dexamethasone, capillaries enlarged into venules expressing leukocyte adhesion molecules, sprouting angiogenesis and lymphangiogenesis occurred, and the inflammatory cytokines tumor necrosis factor and interleukin-1 increased. Concurrent dexamethasone treatment largely prevented the remodeling of blood vessels and lymphatics. Dexamethasone also significantly reduced cytokine expression, bacterial burden, and leukocyte influx into airways and lungs over 4 weeks of infection. In contrast, when infection was allowed to proceed untreated for 2 weeks and then was treated with dexamethasone for 4 weeks, most blood vessel changes reversed but lymphangiogenesis did not, suggesting that different survival mechanisms apply. Furthermore, dexamethasone significantly reduced the bacterial burden and influx of lymphocytes but not of neutrophils or macrophages or cytokine expression. These findings show that lymphatic remodeling is more resistant than blood vessel remodeling to corticosteroid-induced reversal. We suggest that lymphatic remodeling that persists after the initial inflammatory response has resolved may influence subsequent inflammatory episodes in clinical situations.
Chronic inflammatory diseases such as asthma, chronic obstructive pulmonary disease, rheumatoid arthritis, Crohn's disease, and skin lesions in psoriasis are accompanied by a spectrum of remodeling changes in the microvasculature.1–5 In inflamed tissues, blood vessels undergo angiogenesis and remodeling to change their structure and function. Existing capillaries become leakier and abnormally enlarged in diameter and show venular features.6–8 The capillary-to-venule transformation increases the amount of vasculature capable of supporting leukocyte adhesion and migration in response to inflammation stimuli. Conventional sprouting angiogenesis also occurs, usually later than the capillary enlargement. Lymphatic vessels also proliferate from existing lymphatic endothelial cells by sprouting lymphangiogenesis and undergo remodeling to compensate for the extra need for drainage in the inflamed tissues and trafficking of leukocytes, thereby contributing to the development of pathophysiology.9–11
Whereas the remodeling and growth of vessels in inflammation has been documented in an increasing number of studies, the reversibility of vessel changes is not well understood. Relatively little is known about whether the newly grown lymphatics can regress after they have formed at sites of inflammation, and, if so, how quickly. Infection of the airways by the natural rodent respiratory tract pathogen Mycoplasma pulmonis results in persistent vessel changes and life-long airway inflammation.12,13 Similar airway vessel changes and chronic inflammation are also common symptomatic features found in human asthma and chronic bronchitis.11 In M. pulmonis infection, the robust growth and remodeling of blood vessels and lymphatics are driven by a cascade of immune responses to sustained bacterial infection.14 Gene profiling experiments have shown that many inflammatory molecules are up-regulated in M. pulmonis-infected lungs and that many interrelated pathways are likely to drive downstream endothelial cell remodeling.15–17 In this model, partial reversal of enlarged blood vessel diameter occurs after corticosteroid treatment for 1 week.7 Elimination of infecting bacteria with antibiotics for 4 weeks fully reverses the enlargement of blood vessels but results in only a partial reversal of the newly formed lymphatic network.10
The aim of this study was to further clarify the prevention and reversibility of all aspects of blood vessels and lymphatics associated with chronic airway inflammation after M. pulmonis infection. To achieve this purpose, we used the corticosteroid dexamethasone as a powerful tool to repress a wide array of inflammatory mediators, including chemokines, cytokines, growth factors, receptors, and adhesion molecules.18–20 In addition to its broad-spectrum anti-inflammatory function, dexamethasone can down-regulate the expression of vascular endothelial growth factor (VEGF)-A and VEGF-C.21,22 Dexamethasone can also reduce angiopoietin-2 expression in cultured endothelial cells.23 We reasoned that a study with a potent anti-inflammatory and anti-angiogenic agent would help in interpreting the maximum degree of prevention and reversibility and would be a useful basis for future studies with more selective agents.
We performed two treatment studies with dexamethasone, beginning either concurrently at the time of inoculation or after every aspect of vessel changes had already been established. In each study, we examined the time course and extent of vessel changes. We also examined the effects of dexamethasone treatment on the M. pulmonis-driven immune responses. We found that dexamethasone treatment prevented the vessel changes and the associated inflammatory responses induced by M. pulmonis infection more effectively than it reversed them. Delayed treatment reversed remodeled blood vessels almost to pathogen-free conditions and regressed angiogenic and lymphangiogenic sprouting. In contrast, newly formed lymphatics persisted and were remarkably resilient to regression. Furthermore, associated inflammatory responses were reduced, lymphocytes were eliminated, but neutrophils and macrophages were not.
Materials and Methods
Mice
Pathogen-free 8-week-old, female C57BL/6 mice (Charles River Laboratories, Inc., Wilmington, MA) were housed under barrier conditions before and during infection. Mice were anesthetized by intramuscular injection of ketamine (87 mg/kg) and xylazine (13 mg/kg) before all experimental procedures. The Institutional Animal Care and Use Committees of the University of California at San Francisco approved all experimental procedures.
M. pulmonis Infection
Mice were infected by intranasal inoculation of 50 μl of broth containing 106 colony-forming units of M. pulmonis organisms of strain CT7.24 The infection did not resolve over the time course after infection.
Dexamethasone Treatment
Mice received dexamethasone once daily (10 mg/kg i.p.; Phoenix Pharmaceuticals, Belmont, CA) or the same volume of sterile 0.9% NaCl as vehicle.7 A schematic diagram of prevention and reversal studies is shown in Supplemental Figure S1 (see http://ajp.amjpathol.org).
Immunohistochemistry
After anesthesia the vasculature was perfused as described previously.10 Lungs and bronchial lymph nodes were removed, blotted dry, and weighed. The weights were normalized to the final body weight of the mouse to estimate disease severity and degree of leukocyte trafficking. Tracheas and lungs were then immersed in fixative (1% paraformaldehyde in PBS, pH 7.4) for 1 hour and stained as sections or as whole mounts. The left lung and the two rostral-most cartilage rings of trachea from the same mouse were prepared for H&E staining of 3-μm-thick transverse sections. Tracheal whole mounts were stained using immunohistochemical procedures described previously.10 The following primary antibodies were used: endothelial cells (both vascular and lymphatic): platelet endothelial cell adhesion molecule (PECAM)-1 (CD31), (hamster anti-mouse, clone 2H8, Thermo Fisher Scientific, Waltham, MA; or rat anti-mouse, clone MEC 13.3, BD Biosciences, San Jose, CA) and vascular endothelial (VE)-cadherin, (rat anti-mouse, clone 11D4.1, BD Biosciences); lymphatics: lymphatic vessel endothelial cell receptor (LYVE)-1 (rabbit polyclonal, AngioBio, Del Mar, CA) (some leukocytes also expressed LYVE-1 and PECAM-125); venules: intercellular adhesion molecule (ICAM)-1 (CD54) (rat anti-mouse, clone YN1/1.7.4, eBioscience, San Diego, CA) and Ephrin receptor B4 (EphB4) (goat polyclonal, R&D Systems, Minneapolis, MN); neutrophils: S100A8 (goat polyclonal, R&D Systems); macrophages: ionized calcium-binding adapter molecule 1 (Iba1) (rabbit polyclonal, Wako Pure Chemicals Inc., Tokyo, Japan); T cells: CD3e (hamster anti-mouse, clone 500A2, BD Biosciences); and B cells: B220 (rat anti-mouse, clone RA3-6B2, eBioscience). When the epithelium stained strongly (ICAM-1 and EphB4) in the whole mount and prevented clear viewing of underlying vessels by a fluorescence microscope, we made Z-stack confocal projections of the mucosa and eliminated the slices containing the epithelium. Secondary antibodies were labeled with fluorescein isothiocyanate, Cy3 or Cy5 (Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Specimens were viewed with a Zeiss Axiophot fluorescence microscope or a Zeiss LSM-510 confocal microscope using AIM 4.0 confocal software.
Morphometric Measurements
Whole-mount tracheas were examined in real-time images and measured using a digitizer tablet linked to a video camera on a Zeiss Axiophot microscope. In some chronically infected tracheas, numerous leukocytes stained for PECAM-1 and interfered with accurate area density measurements of PECAM-1-labeled blood vessels. Therefore, we measured the diameters of capillaries. Capillary diameters were measured at the midpoints of PECAM-1-positive blood vessels over the cartilage rings at a final screen magnification of 600 and 5 vessels per ring, 10 rings per mouse.26 The number of capillaries per millimeter crossing the cartilage rings was assessed by counting PECAM-1-positive blood vessels at the center of cartilage rings at a final screen magnification of 760.26 Area densities (percentage of total tissue area) of LYVE-1 positive lymphatic vessels were measured by stereological point counting at a final screen magnification of 960.10 The abundance of neutrophils was determined by counting the number of S100A8-stained cells within each 0.1-mm2 area. Results were presented for regions of the mucosa overlying the cartilage rings, where the greatest changes occurred during infection.
Quantitative RT-PCR
Perfusion of mice followed by RNA extraction from tracheas and cDNA synthesis was performed as described previously.17 Samples of 1 ng of cDNA were subjected to quantitative (q) RT-PCR using SYBR GreenER qPCR SuperMix Universal (Invitrogen, Carlsbad, CA) and measured in duplicate with a Bio-Rad MyiQ detection system. Relative gene expression data were normalized to β-actin, and results were presented as fold changes versus pathogen-free controls. Because the increase in total RNA yield per trachea was largely due to the influx of leukocytes during infection, the expression of some genes may have been underestimated by qRT-PCR. Therefore, RNA copy numbers were additionally scaled to the total RNA yield of the corresponding trachea, except for the leukocyte markers and leukocyte-derived cytokines tumor necrosis factor (TNF)-α and interleukin (IL)-1β, for which the changes were very large. We chose markers for different leukocytes based on published reports. Our pilot experiments showed that the markers F4/80 and Gr-1 commonly used to identify macrophages and neutrophils in fluorescence-activated cell sorting analysis, but they did not work well for qRT-PCR. Iba1 immunoreactivity colocalizes with fms-enhanced green fluorescent protein expression in monocytes, macrophages, and microglial cells.27,28 In place of F4/80 we used the macrophage maker Iba1 for both qRT-PCR analysis and immunohistochemistry. S100A8 (also known as MRP-8 [migration inhibitory factor related protein-8] or calgranulin A) is highly expressed in neutrophils and is absent from macrophages.29 For similar reasons, we used S100A8 as a marker of neutrophils for both qRT-PCR analysis and immunohistochemistry. CD19 and B220 are surface markers for mature B cells. CD19 is well established for gene expression analysis30,31 and B220 is commonly used as a pan B-cell marker for immunohistochemical staining and fluorescence-activated cell sorting analysis.32,33 We used CD19 for qRT-PCR analysis because B220 did not work well for qRT-PCR. Our results showed that the expression profile of B220 immunoreactivity and CD19 mRNA were in agreement with each other in tracheas. Primer sequences are listed in Table 1.
Table 1.
Primer Sequences Used for qRT-PCR Analysis
| Name | Forward primer | Reverse primer |
|---|---|---|
| β-Actin | 5′-GGCTGTATTCCCCTCCATCG-3′ | 5′-CCAGTTGGTAACAATGCCATGT-3′ |
| TNF-α | 5′-CCCTCACACTCAGATCATCTTCT-3′ | 5′-GCTACGACGTGGGCTACAG-3′ |
| IL-1β | 5′-GCAACTGTTCCTGAACTCAACT-3′ | 5′-GGTCAAAGGTTTGGAAGCAG-3′ |
| IL-1Ra | 5′-CTGATGAAGCTCGTCAGGCAG-3′ | 5′-TGGTGCTGAGATAGTGTTTGTC-3′ |
| M. pulmonis 16S rRNA | 5′-CGGTACAGGAAACTGTTGCTAATACCG-3′ | 5′-CCATTTCAAAGTGAAGCAAACG-3′ |
| S100A8 | 5′-CCGTCTTCAAGACATCGTTTGA-3′ | 5′-GTAGAGGGCATGGTGATTTCCT-3′ |
| Iba1 | 5′-GCTGGAGGGGATCAACAAGC-3′ | 5′-TCTTCAGCTCTAGGTGGGTCT-3′ |
| CD19 | 5′-AGTGATTGTCAATGTCTCAGACC-3′ | 5′-CCCCACTATCCTCCACGTTCA-3′ |
| CD3e | Custom primers from SABiosciences (Frederick, MD) | Custom primers from SABiosciences |
| VEGF-A | 5′-ATCTTCAAGCCGTCCTGTGT-3′ | 5′-GCATTCACATCTGCTGTGCT-3′ |
| VEGF-C | Custom primers from SABiosciences | Custom primers from SABiosciences |
| VEGF-D | 5′-TTGAGCGATCATCCCGGTC-3′ | 5′-GCGTGAGTCCATACTGGCAAG-3′ |
| Angiopoietin-1 | 5′-AGCTACCAACAACAACAGCA-3′ | 5′-GCAAAGGCTGATAAGGTTATGA-3′ |
| Angiopoietin-2 | 5′-GTCACAATGACAAGGCCGTG-3′ | 5′-TCCCTGGATACTGGCTGCC-3′ |
| PDGF-B | 5′-GATCTCTCGGAACCTCATCG-3′ | 5′-GGCTTCTTTCGCACAATCTC-3′ |
Statistical Analysis
Values are presented as means ± SEM with four or more mice per group. Differences between means were assessed by analysis of variance followed by the Bonferroni test for multiple comparisons. P < 0.05 was considered significant.
Results
Time Course of Airway Vessel Changes during M. pulmonis Infection
Morphological changes of the airway microvasculature in tracheal whole mounts over the course of a 42-day infection are shown in Figure 1. The tracheal microvasculature of pathogen-free mice was organized in a repeating segmental pattern (Figure 1A). Arterioles, venules, and lymphatics were located in the mucosa between cartilage rings, whereas narrow capillaries formed a ladder-like pattern in regions overlying cartilage rings. Leukocytes were sparse under pathogen-free baseline conditions, and only a few scattered leukocytes in the mucosa were immunoreactive for LYVE-1 (Figure 1, A, G, and J). Seven days after inoculation, capillaries were enlarged in diameter with the greatest increase near the venous side, accompanied with the influx of leukocytes, some of which also stained for PECAM-1 (Figure 1, B and H). Lymphatics started to grow later than blood vessels. Lymphatic sprouts were first detectable at 7 days after inoculation (Figure 1, B and K). During the second week, the caliber of capillaries continued to increase, and lymphatics grew into the mucosa overlying the cartilage rings (Figure 1, C and D). Additional endothelial sprouts began to form from 14 days. At 28 days, blood vessels overlying the cartilages included both the preexisting vessels that had enlarged and the relatively thin, newly formed blood vessels with sprouts (Figure 1, E, I, and L). Newly formed lymphatic sprouts fused to form a continuous network over the cartilage rings (Figure 1, E and I). Finally, by 42 days, blood vessels and lymphatics were greatly remodeled and leukocytes were abundant (Figure 1F). However, the LYVE-1-immunoreactive leukocytes observed in pathogen-free mice were absent after M. pulmonis inoculation (Figure 1, J and K).
Figure 1.
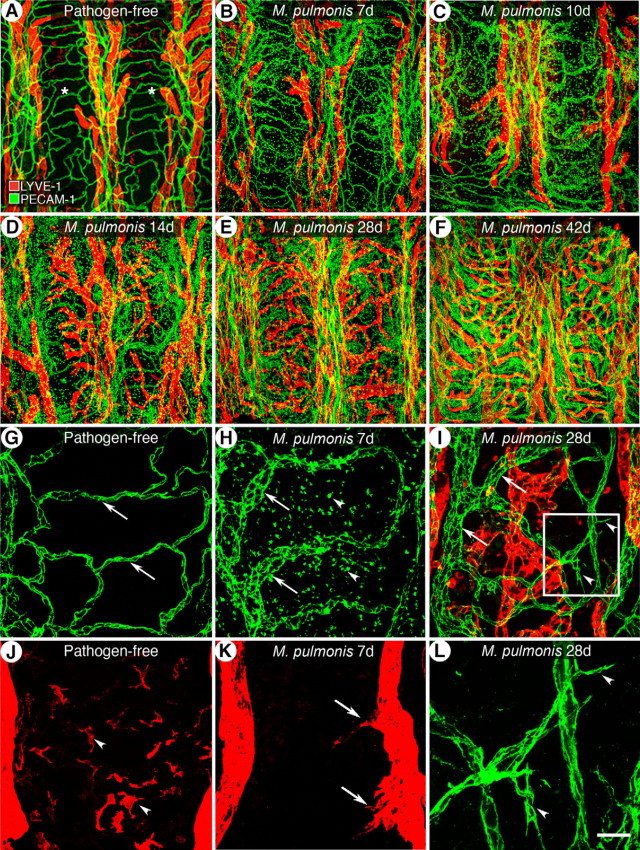
Time course of airway vessel changes during 42-day course of M. pulmonis infection. A–F: Overview of microvascular arcades stained for blood vessels (PECAM-1, green) and lymphatic vessels (LYVE-1, red) in a tracheal whole mount of pathogen-free mice and in mice infected for 7, 10, 14, 28, and 42 days. Asterisks indicate cartilage rings. G and J: In pathogen-free mice, capillaries of uniform caliber (G, arrows) transverse the cartilage rings but lymphatics do not (J). Some resident leukocytes stained for LYVE-1 (J, arrowheads). At 7 days of infection (H and K), blood vessels are enlarged (H, arrows) and accompanied by an influx of leukocytes, many showing PECAM-1 immunoreactivity (H, arrowheads). Initial lymphatics sprouts are present (K, arrows). I: By 28 days, blood vessels show both enlargement (arrows) and sprouting (arrowheads) and newly grown lymphatics fuse to a continuous network. L: Enlarged box in I shows the angiogenic sprouts (arrowheads). Scale bars: 200 μm in (A–F); 50 μm (G–K); 20 μm (L).
Prevention of Vessel Changes and Inflammatory Cytokine Expression
Starting dexamethasone treatment concurrently with infection (Supplemental Figure S1, see http://ajp.amjpathol.org) effectively blocked the remodeling of blood vessels and lymphatics induced by M. pulmonis infection. The vessel architecture was similar to that of pathogen-free mice (Figure 2, A–C). In addition, PECAM-1-immunoreactive leukocytes were much less abundant and LYVE-1-immunoreactive leukocytes returned (Figure 2C). Measurements confirmed that concurrent treatment prevented the enlargement of capillaries (Figure 2D). The diameter of the vehicle-treated capillaries decreased from 15.4 μm at 14 days to 11.9 μm at 28 days of the infection because of the greater abundance of narrow, newly formed blood vessels. Dexamethasone treatment also effectively prevented lymphatic growth (Figure 2E).
Figure 2.
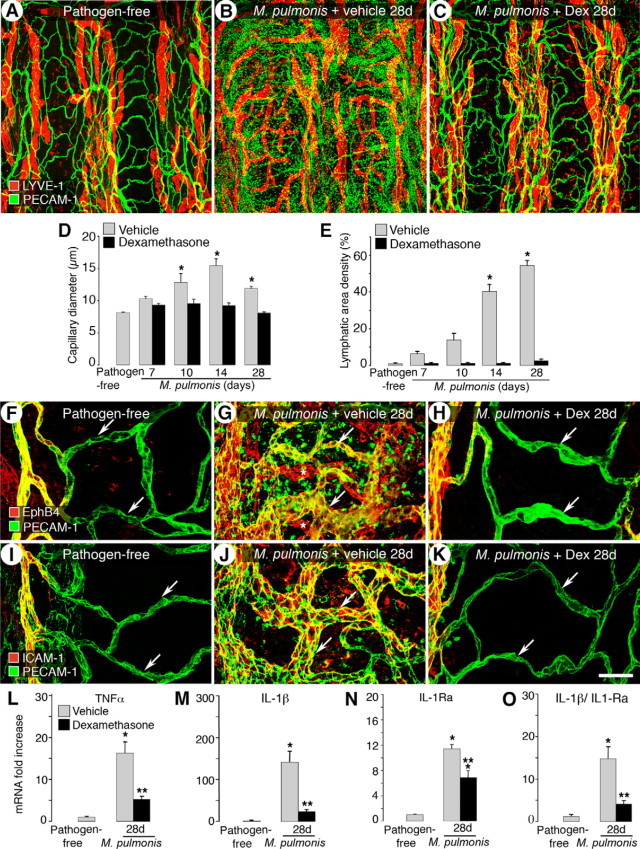
Prevention of M. pulmonis induced-vessel changes by dexamethasone. A–C: Confocal micrographs showing tracheal blood vessels (green) and lymphatics (red) in pathogen-free mice (A) and in 28-day infected mice treated concurrently with vehicle (B) or dexamethasone (C) for 28 days. D and E: Capillary diameters (D) and area density of lymphatics (E) showing prevention of infection-induced vessel changes by dexamethasone. F–K: Dexamethasone prevents transformation of capillary to venule phenotype. Venular endothelial markers EphB4 and ICAM-1 are shown in red and PECAM-1 is shown in green. In pathogen-free mice, EphB4 (F) and ICAM-1 (I) are restricted to the venules and neither is present in capillaries (arrows). EphB4 (G) and ICAM-1 (J) extend into remodeled capillaries (arrows) overlying cartilage rings after 28 days of infection. Scattered staining for EphB4 and ICAM-1 in other mucosal cells is also seen in the more remodeled whole-mount tracheas. The expansion is prevented by concurrent dexamethasone treatment (H and K, arrows). Lymphatics in G are indicated by asterisks. Scale bar: 200 μm in A–C; 60 μm in F–K. L–O: qRT-PCR measurement of TNF-α, IL-1β, IL-1Ra, and the ratio of IL-1β expression to IL-1Ra in tracheas of pathogen-free mice and mice infected and treated with vehicle or dexamethasone for 28 days. Values are shown as fold increase compared with pathogen-free levels. *P < 0.05 versus pathogen-free; **P < 0.05 versus corresponding infected, vehicle-treated group (n = 5 to 8 mice per group). Dex, dexamethasone.
Previous studies have shown that the enlargement of capillaries in infected airways is accompanied by a capillary-to-venule transformation that supports leukocyte adhesion.7 We next examined whether dexamethasone can also prevent this change in vessel phenotype. EphB4 and ICAM-1-immunoreactivity was absent in normal capillaries and was restricted to venules in pathogen-free mice (Figure 2, F and I). After 28 days of infection, remodeled capillaries stained for EphB4 and ICAM-1 (Figure 2, G and J). The expansion of these venule markers was prevented by dexamethasone treatment (Figure 2, H and K). EphB4 was also expressed in the lymphatics and the immunoreactivity was abundant in the newly grown lymphatics at 28 days (Figure 2G). Consistent with LYVE-1 immunoreactivity (Figure 2C), EphB4-immunoreactive lymphatics were not present after dexamethasone treatment (Figure 2H).
Among many up-regulated inflammatory cytokines, TNF-α and IL-1β are promising candidates responsible for the M. pulmonis-induced initial remodeling of blood vessels and/or lymphangiogenesis.17 qRT-PCR showed that TNF-α and IL-1β expression increased 16- and 140-fold, respectively, at 28 days of infection compared with pathogen-free baseline levels (Figure 2, L and M). Concurrent treatment with dexamethasone significantly reduced the increase (68 and 83% less, respectively). We also found that IL-1β and its naturally occurring functional antagonist, interleukin-1 receptor antagonist (IL-1Ra) had very similar baseline levels in pathogen-free tracheas (0.00015 RNA copy numbers relative to β-actin). However, after 28-day M. pulmonis infection, IL-1β mRNA expression increased 140-fold, whereas IL-1Ra expression increased a mere 11-fold (Figure 2, M and N). The ratio of IL-1β to IL-1Ra expression after infection increased 12-fold in favor of IL-1β signaling (Figure 2O). Concurrent dexamethasone treatment reduced the IL-1β/IL-1Ra expression ratio by 72%. These results confirmed that the expression of both cytokines was closely associated with the initiation of airway vessel remodeling.
Expression of mRNA for growth factors VEGF-A, C, and D at 28 days of infection was approximately three times the corresponding pathogen-free baseline values (Supplemental Figure S2, A–C, see http://ajp.amjpathol.org). Expression of VEGFs VEGF-A, C, and D was about the same as the pathogen-free values when the infection was continuously accompanied by dexamethasone treatment. At 28 days after infection, expression of angiopoietin-1 did not increase. In contrast, expression of angiopoietin-2 was about 4 times higher than the pathogen-free value (Supplemental Figure S2, D–E, see http://ajp.amjpathol.org). Concurrent dexamethasone treatment prevented the increase in angiopoietin-2 expression. Platelet-derived growth factor (PDGF)-B expression was about five times the pathogen-free value (Supplemental Figure S2F, see http://ajp.amjpathol.org). The increase was also effectively prevented by dexamethasone treatment.
Prevention of M. pulmonis Burden and Leukocyte Influx
Previous studies have shown that vessel changes in the infected airways are driven by the immune response to M. pulmonis infection.14 Therefore, we examined the effects of concurrent dexamethasone treatment on the immune responses with several readouts. As an index of infection severity, we measured M. pulmonis-specific 16S ribosomal RNA expression as a manifestation of bacterial burden within the airway. 16S rRNA expression was not detectable in pathogen-free tracheas but displayed a continual increase during infection (RNA copy number versus β-actin was 20 at 7 days, 23 at 14 days, and 75 at 28 days) (Figure 3A). Interestingly, the increase in 16S rRNA expression was greatly reduced (89% less at 7 days, 83% less at 14 days, and 94% less at 28 days) in dexamethasone-treated mice.
Figure 3.
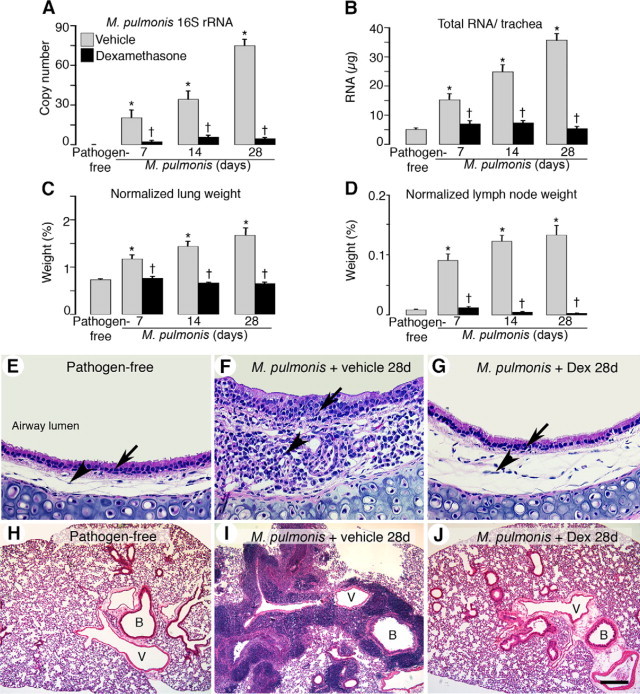
Prevention of M. pulmonis burden and leukocyte influx by dexamethasone. Bar graphs of expression of M. pulmonis 16S rRNA in tracheas by qRT-PCR (A), total RNA yield per trachea (B), and lung (C) and bronchial lymph node weights (D) from pathogen-free mice and mice infected for 7, 14, or 28 days with concurrent treatment with vehicle or dexamethasone. Lung and bronchial lymph node weights are normalized to the corresponding final body weights. *P < 0.05 versus pathogen-free; †P < 0.05 versus the corresponding infected, vehicle-treated controls (n = 5 to 8, mice per group). E–J: H&E-stained sections of mouse tracheas (E–G) and mouse left lungs (H–J) in pathogen-free mice (E and H) or in infected mice with concurrent treatment with vehicle (F and I) or dexamethasone (G and J) for 28 days. Arrows indicate leukocyte influx in tracheal epithelial layers, and arrowheads mark leukocyte influx in tracheal mucosa. Dex, dexamethasone; B, bronchiole; V, blood vessel. Scale bars: 100 μm in (E–G); 400 μm (H–J).
We then quantified the disease-related phenotypes occurring in the tracheas, lungs, and bronchial lymph nodes during infection. Measurement of total RNA yield per trachea, reflecting a change in total tracheal mass, showed that pathogen-free trachea yielded 5 μg, whereas infected tracheas increased threefold at 7 days of infection, fivefold at 14 days, and sevenfold to 36 μg at 28 days (Figure 3B). Infected lung weights also increased approximately twofold at 28 days (Figure 3C). Both phenomena were effectively prevented by dexamethasone prevention treatment. Furthermore, bronchial lymph nodes became hypertrophic during infection and resulted in a 15-fold increase in weight at 28 days. This increase was completely prevented by dexamethasone treatment (Figure 3D). In fact, after 28-day dexamethasone treatment, some bronchial lymph nodes were even smaller than those in pathogen-free mice (ie, <1 mg) and were therefore difficult to detect.
H&E-stained sections revealed that the normal pathogen-free tracheal mucosa was smooth and thin and contained very few leukocytes (Figure 3E). In contrast, the inflamed tracheas contained many leukocytes in both the mucosa (about three times as thick) and epithelial layer of 28-day infected mice (Figure 3F). In comparison, concurrent administration of dexamethasone resulted in a level of leukocyte infiltration and mucosa thickness resembling that in the pathogen-free mice (Figure 3G). The huge influx of leukocytes in the inflamed tracheal mucosa may account for much of the prominent increase in tracheal total RNA yield.
Dexamethasone treatment also blocked leukocyte influx into the lungs (Figure 3, H–J). In infected lungs, leukocytes predominantly accumulated in the interstitium around large blood vessels and bronchi, contributing in part to the increase in lung weight (Figure 3I). Lungs from infected mice treated concurrently for 28 days with dexamethasone were mostly free of leukocytes (Figure 3J).
Differential Prevention of Leukocyte Influx into Infected Airways
To further dissect the kinetics of leukocyte influx in mice infected and concurrently treated with either vehicle or dexamethasone, we used two strategies to determine the influx between different cell types in the infected airways. First we assessed the overall influx of four leukocyte markers in whole-mount tracheas by immunohistochemical staining: neutrophil marker S100A8, macrophage marker Iba1, T-cell marker CD3e, and B-cell marker B220. Second we analyzed the corresponding gene expression in tracheas by qRT-PCR.
Pathogen-free tracheas were nearly free of S100A8-immunoreactive neutrophils, but some Iba1-immunoreactive resident macrophages were present in the mucosa between cartilage rings (Figure 4A). The amount and intensity of both S100A8 and Iba1 staining had clearly increased at 28 days of infection (Figure 4, B and G). The increased number of Iba1-immunoreactive cells was especially evident in the mucosa overlying the cartilage rings where the remodeled capillaries are located. Compared with vehicle-treated controls, the concurrent dexamethasone treatment of infected mice greatly decreased but did not fully prevent the infiltration of both S100A8-labeled neutrophils and Iba1-labeled macrophages into the airways (Figure 4C). In pathogen-free tracheas, both CD3e- and B220-immunoreactive cells were generally absent (Figure 4D). After 28 days of infection, B220-immunoreactive cells often formed tightly patched clusters, whereas CD3e-immunoreactive cells were abundant throughout the trachea (Figure 4, E and H). In contrast to that of macrophages and neutrophils, the infiltration of B cells and T cells was effectively prevented by dexamethasone treatment (Figure 4F).
Figure 4.
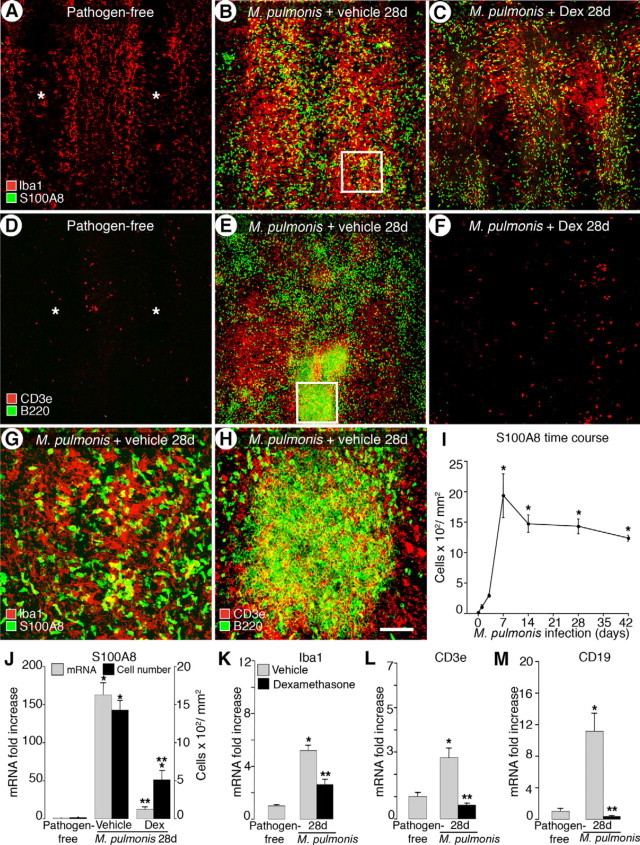
Differential prevention of leukocyte influx into infected airways by dexamethasone. Confocal microscopic images of tracheas stained for markers of macrophages (Iba1, red in A–C) and neutrophils (S100A8, green in A–C) or T cells (CD3e, red in D–F) and B cells (B220, green in D–F) in pathogen-free mice (A and D) and mice infected for 28 days with concurrent treatment with vehicle (B and E) or dexamethasone (C and F). Asterisks indicate cartilage rings in A and D. Boxes in B and E are enlarged in G and H, respectively. Scale bars: 200 μm in (A–F); 50 μm (G–H). I: Time course of influx of S100A8-immunoreactive cells during infection. J: Number of S100A8-immunoreactive cells and qRT-PCR analysis of S100A8 mRNA expression in pathogen-free mice and after infection for 28 days with concurrent treatment with vehicle or dexamethasone. K–M: qRT-PCR measurement for Iba1, CD3e, and CD19 (B-cell marker). *P < 0.05 versus pathogen-free; **P < 0.05 versus the corresponding infected, vehicle-treated controls (n = 4 to 9 mice per group).
We used qRT-PCR to measure the extent of the four different leukocyte populations in airways. Our attempts to count cell numbers in immunohistochemically stained specimens were unsuccessful except for S100A8-immunoreactive neutrophils, largely because of the abundance of infiltrated leukocytes overlying one another. The number of S100A8-immunoreactive cells increased within 1 day of infection, peaked at 7 days, decreased 24% at 14 days, and then remained relatively stable up to 42 days (Figure 4I). The qRT-PCR analysis showed that the expression of S100A8 at 28 days of infection increased 118-fold compared with a 96-fold increase in cell number (Figure 4J). Dexamethasone treatment reduced S100A8 cell number and mRNA expression by 65 and 90%, respectively (Figure 4J). Infection induced a 4.4-fold increase in Iba1 expression at 28 days, and Iba1 expression was still increased by nearly 50% over pathogen-free levels in the dexamethasone-treated group (Figure 4K). Expression of CD3e increased approximately twofold and that of CD19 increased 10-fold at 28 days of infection (Figure 4, L and M). However, with concurrent dexamethasone treatment levels of CD3e and CD19 were not significantly different from their respective pathogen-free levels. Taken together, our findings show that dexamethasone treatment largely prevented leukocyte influx into infected tracheas and specifically blocked the influx of lymphocytes more effectively than the influx of neutrophils and macrophages.
Reversibility of Vessel Changes and Inflammatory Cytokine Expression
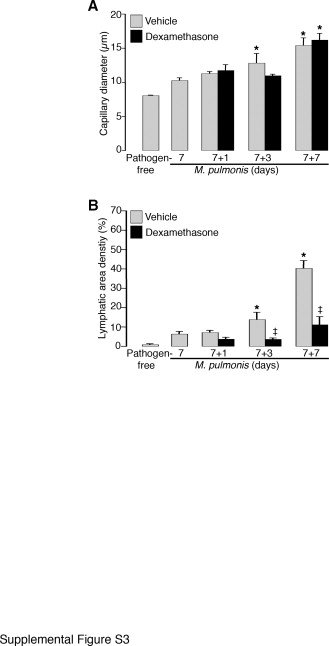
Previous studies have shown that treatment with the antibiotic oxytetracycline to eliminate M. pulmonis reversed blood vessel remodeling to pathogen-free conditions within 4 weeks but not lymphatic remodeling.10 Consistent with these findings, the present study showed that lymphatic remodeling was more resistant than blood vessel remodeling to dexamethasone-induced reversal (Figure 5, A–G). In a preliminary study, in which 7 days of infection were followed by a short 7-day treatment with dexamethasone (Supplemental Figure S1, see http://ajp.amjpathol.org), reversal of blood vessel enlargement did not occur (Supplemental Figure S3A, see http://ajp.amjpathol.org). However, further growth of the initial lymphatic sprouts beyond the 7-day infected baseline level was prevented (Supplemental Figure S3B, see http://ajp.amjpathol.org). Moreover, when infection was allowed to progress untreated for 14 days to let the remodeled lymphatics become better established, 28 days of prolonged treatment with dexamethasone differentially reversed blood vessel versus lymphatic remodeling (Figure 5, A–C and Supplemental Figure S1, see http://ajp.amjpathol.org). Enlarged blood vessel diameters reversed to near the pathogen-free baseline (60% narrower compared with the 14-day infected baseline, 1.4-fold wider than the pathogen-free baseline) (Figure 5D). In addition, angiogenic sprouts were absent, and the number of capillaries crossing the cartilage midline was essentially identical to the pathogen-free levels (Figure 5E and Supplemental Figure S4, A–C, see http://ajp.amjpathol.org). On the other hand, although lymphatic sprouts were absent after the 28-day dexamethasone treatment, the surviving newly formed lymphatic vessels were only slightly reduced in area (20% less compared with the 14-day infected baseline) and were still 40-fold higher than the pathogen-free baseline (Figure 5F and Supplemental Figure S4, D-F, see http://ajp.amjpathol.org). Dexamethasone-treated mice had some small “island-like” lymphatic segments that were separated from the main lymphatics (Figure 5C) and that others have termed “lymphatic islands.”34,35 However, such observations were relatively rare in both pathogen-free mice and untreated infected mice. Quantification showed a sevenfold increase in the number of LYVE-1-positive lymphatic islands in dexamethasone treatment mice compared with pathogen-free mice (Figure 5G). We confirmed that these discontinuous lymphatic islands were not artifacts of incomplete imaging or of the down-regulation of LYVE-1 immunoreactivity by observation with a conventional fluorescence microscope (data not shown) and by staining of tracheal whole mounts with another endothelial cell marker, VE-cadherin (Supplemental Figure S4F, see http://ajp.amjpathol.org).
Figure 5.
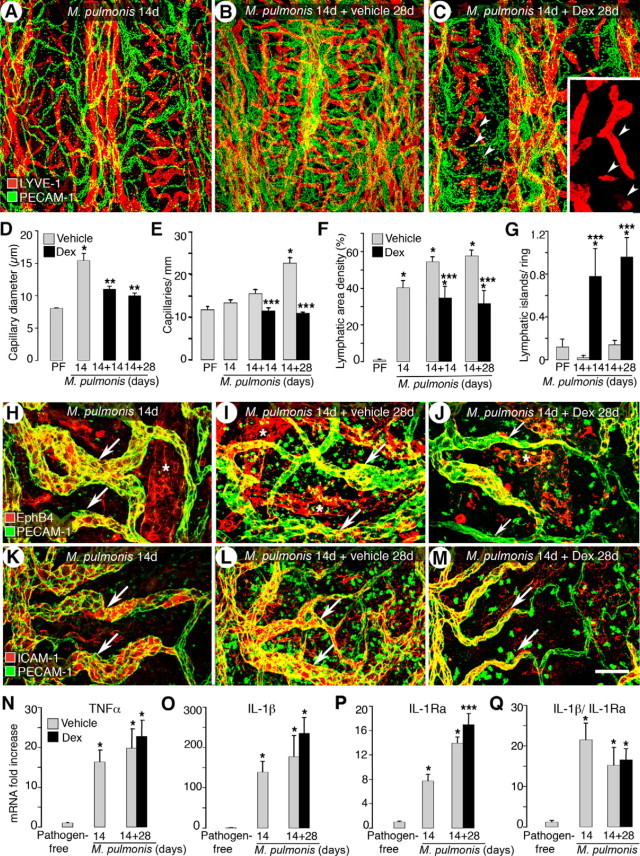
Reversibility of M. pulmonis-induced vessel changes by dexamethasone. A–C: Confocal images of tracheas stained for LYVE-1 (red) and PECAM-1 (green) immunoreactivity in mice infected for 14 days (A) and infected for 42 days and treated with vehicle (B) or with dexamethasone (C) for the last 28 days. Lymphatic islands (arrowheads) are enlarged in inset in C. D–G: Bar graphs show changes of blood vessels and lymphatics in mucosa overlying cartilage rings in reversal studies, as reflected by capillary diameter (D), number of capillaries crossing cartilage midline (E), area density of lymphatics (F), and number of lymphatic islands (G). H–M: Effects of delayed dexamethasone treatment on distribution of venular endothelial markers EphB4 and ICAM-1 (red) and PECAM-1 (green). Remodeled capillaries (arrows) overlying cartilage rings in 14-day (H and K) and 42-day (I and L) infected mice are strongly stained for EphB4 and ICAM-1. Scattered staining for EphB4 and ICAM-1 in other mucosal cells is also seen in the more remodeled whole-mount tracheas. Some EphB4 and ICAM-1 immunoreactivity is still present in the capillaries (arrows) after 28 days of dexamethasone treatment (J and M). Asterisks indicate EphB4-positive lymphatics in H–J. Scale bars: 200 μm in (A–C); 60 μm (H–M). N–Q: qRT-PCR measurement of TNF-α, IL-1β, IL-1Ra, and the ratio of IL-1β expression to IL-1Ra. *P < 0.05 versus pathogen-free mice; **P < 0.05 versus 14-day infected baseline group before treatment; ***P < 0.05 versus corresponding infected, vehicle-treated controls (n = 5 to 8 mice per group).
We next examined the phenotype of the dexamethasone-treated blood vessels regarding the venular markers EphB4 (Figure 5, H–J) and ICAM-1 (Figure 5, K–M). Remodeled blood vessels became much thinner and showed reduced expression of EphB4 and ICAM-1 after treatment (Figure 5, J and M). EphB4 immunoreactivity was also expressed in lymphatics but was still abundant in lymphatics after dexamethasone treatment for 28 days (Figure 5J).
TNF-α mRNA expression increased 15-fold at 14 days and 19-fold at 42 days after infection. IL-1β increased 138-fold at 14 days and 176-fold at 42 days (Figure 5, N and O). In contrast to the almost complete reversal in blood vessels, 28-day dexamethasone treatment did not stop the further increase of cytokines at 42 days of infection. Similar effects of dexamethasone on gene expression of IL-1Ra were also observed (7-fold increase at 14 days, 13-fold increase at 42 days, and 16-fold increase after treatment) (Figure 5P). The ratio of IL-1β to IL-1Ra after dexamethasone treatment was still significantly increased compared with pathogen-free levels and remained similar to that of the corresponding vehicle-treated control group (Figure 5Q).
Expression levels of growth factors VEGF-A, C, and D at 14 days of infection were two to three times the corresponding pathogen-free baseline values (Supplemental Figure S5, A–C, see http://ajp.amjpathol.org). Values increased slightly at 42 days. Notably, delayed dexamethasone treatment reduced the extent of this increase and produced particularly significant reductions in expression of VEGF-C and VEGF-D. Angiopoietin-1 expression remained relatively flat even at 42 days of infection (Supplemental Figure S5D, see http://ajp.amjpathol.org). In contrast, expression of angiopoietin-2 and PDGF-B was three to four times the corresponding pathogen-free values (Supplemental Figure S5, E–F, see http://ajp.amjpathol.org). This increase was also significantly reduced after dexamethasone treatment.
Reversibility of M. pulmonis Burden and Leukocyte Influx
The same readouts used in the prevention studies were used in the reversal studies to determine the effects of delayed dexamethasone treatment on bacterial burden and leukocyte influx. During infection, increases in M. pulmonis 16S rRNA expression and total tracheal RNA yield reached a plateau at 28 days and remained elevated at 42 days (Figure 6, A and B). Dexamethasone treatment starting at 14 days after infection resulted in a reduction at 28 days that was maintained at 42 days of infection (69 versus 64% less for 16S rRNA and 56 versus 50% less for tracheal RNA yield) compared with the corresponding vehicle-treated controls. Infected lung weights continued to increase to 2.5-fold more than the pathogen-free baseline at 42 days (Figure 6C). A 28-day dexamethasone treatment resulted in a 17% reduction in lung weights compared with the infected baseline controls. In contrast, hypertrophy of the infected bronchial lymph nodes was greatly reduced after dexamethasone treatment and lymph node weights returned to pathogen-free levels at 42 days of infection (Figure 6D).
Figure 6.
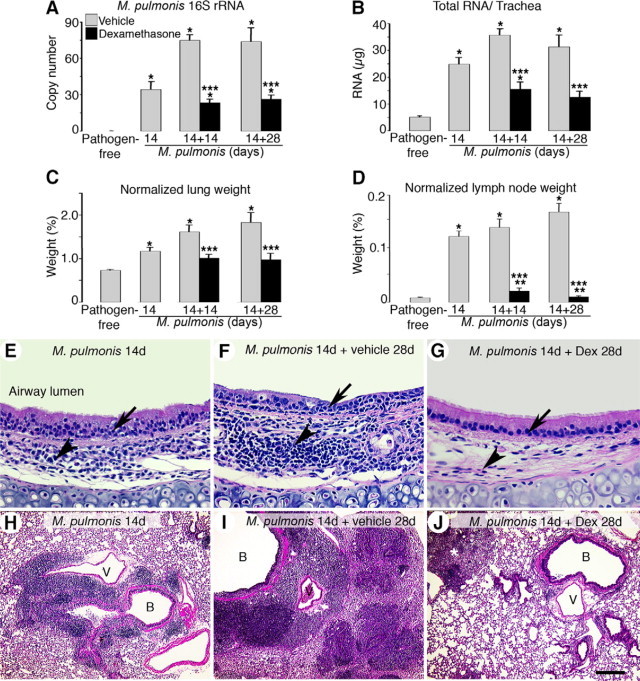
Reversibility of M. pulmonis burden and leukocyte influx by dexamethasone. Bar graphs of expression of M. pulmonis 16S rRNA in tracheas by qRT-PCR (A), total RNA yield per trachea (B), lung weight (C), and bronchial lymph node weight (D) from pathogen-free mice and mice infected for 14 days and untreated or infected for 14 days followed by treatment with vehicle or with dexamethasone for 14 days or 28 days. Lung and bronchial lymph node weights are normalized to the corresponding final body weights *P < 0.05 versus pathogen-free mice; **P < 0.05 versus 14-day infected baseline group before dexamethasone treatment; ***P < 0.05 versus corresponding infected, vehicle-treated controls (n = 10 to 15 mice per group). E–J: H&E-stained sections of mouse tracheas (E–G) and mouse left lungs (H–J) in mice infected for 14 days (E and H) and in mice infected for 42 days and treated with vehicle (F and I) or dexamethasone (G and J) for the last 28 days. Arrows mark leukocyte influx in tracheal epithelial layers, and arrowheads mark leukocyte influx in tracheal mucosa. Asterisk in J marks leukocyte clusters remaining in lungs after 28 days of dexamethasone treatment. Dex, dexamethasone; B, bronchiole; V, blood vessel. Scale bars: 100 μm in (E–G); 400 μm (H–J).
H&E-stained tracheas and lungs showed that the leukocyte influx was also reduced after dexamethasone treatment (Figure 6, E–J). Compared with the 14-day infected tracheas (Figure 6E), many leukocytes were present in the epithelial layer and formed clear lymphoid-like tissues in the mucosa of the 42-day infected mice (Figure 6F). Mice treated with dexamethasone for 28 days had fewer leukocytes in the mucosa and almost none in the epithelium (Figure 6G). Leukocytes predominantly accumulated in peribronchial and perivascular areas of the infected lungs at 14 days of infection (Figure 6H). Sometimes the entire left lung or individual lobes of the right lung were packed solid with numerous leukocytes at 42 days of infection (Figure 6I). After dexamethasone treatment, the aggregates of leukocytes surrounding the bronchi and blood vessels were greatly reduced, but some small clusters of cells remained in lung parenchyma (Figure 6J).
Differential Reversal of Leukocyte Influx into Infected Airways
Dexamethasone treatment reduced overall leukocyte influx into tracheal mucosa of the H&E-stained sections, yet the response of individual cell types was different. Iba1-immunoreactive macrophages and S100A8-immunoreactive neutrophils were more abundant at 14 days than at 42 days of infection (Figure 7, A, B, and G). Dexamethasone treatment for 28 days slightly reduced the amount of both cell types compared with the vehicle-treated controls (Figure 7C). Conversely, both CD3e and B220-immunoreactive lymphocytes were most abundant at 42 days of infection, the longest time point assessed within the reversal study (Figure 7, D, E, and H). Clusters of B220-immunoreactive cells were first seen at 14 days of infection and gradually increased in size and number over the 42-day period of infection. Dexamethasone treatment greatly reduced the number of both types of lymphocytes compared with the 14-day infected baseline group (Figure 7F).
Figure 7.
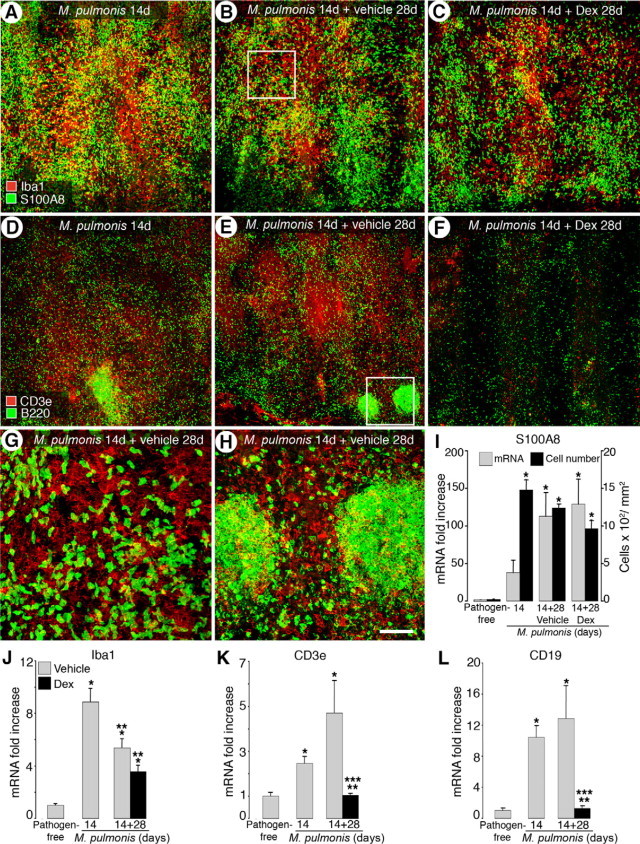
Differential reversal of leukocyte influx into infected airways by dexamethasone. Confocal microscopic images of tracheas stained for macrophages (Iba1, red in A–C) and neutrophils (S100A8, green in A–C) or T cells (CD3e, red in D–F) and B-cells (B220, green in D–F) in 14-day infected untreated mice (A and D) and mice infected for 14 days followed by treatment with vehicle (B and E) or dexamethasone (C and F) for 28 days. G–H: Boxed areas enlarged in B and E, respectively. Scale bars: 200 μm in (A–F); 50 μm (G and H). I: qRT-PCR analysis of S100A8 mRNA and number of S100A8-immunoreactive cells in pathogen-free mice, mice infected for 14 days, and mice infected for 14 days followed by 28-day treatment with vehicle or dexamethasone. J–L: qRT-PCR measurements for Iba1, CD3e, and CD19 (B-cell marker) in tracheas of pathogen-free mice, 14-day infected baseline group, and mice infected for 14 days followed by treatment with vehicle or dexamethasone for 28 days. *P < 0.05 versus pathogen-free; **P < 0.05 versus 14-day infected baseline before dexamethasone treatment; ***P < 0.05 versus corresponding infected, vehicle treated controls (n = 4 to 9 mice per group).
Counts of S100A8-immunoreactive cells confirmed that neutrophil influx was somewhat more abundant at 14 days than at 42 days (1476 versus 1236 cells/mm2) (Figure 7I). Dexamethasone treatment slightly reduced the number of S100A8-immunoreactive cells (by 22%) compared with the corresponding vehicle-treated controls at 42 days. On the other hand, qRT-PCR measurement showed a continued increase in S100A8 expression during infection (a 110-fold increase at 42 days versus a 37-fold increase at 14 days) (Figure 7I). Expression of S100A8 in the dexamethasone-treated group stayed at a level similar to that in corresponding vehicle-treated controls. Taken together, both the cell counts and S100A8 expression profiling indicate that delayed dexamethasone treatment did not reverse neutrophil influx.
Consistent with the immunohistochemical staining results, Iba1 gene expression was higher at 14 days than at 42 days of infection (an 8-fold increase versus a 4.4-fold increase) (Figure 7J). Compared with the vehicle-treated controls, dexamethasone treatment reduced Iba1 expression by 34% at 42 days of infection, but the reduction was not significant.
In contrast to S100A8 and Iba1, gene expression of both lymphocyte markers continued to increase steadily after initial infection and was highest at 42 days (a 4-fold increase for CD3e and a 12-fold increase for CD19) (Figure 7, K and L). Dexamethasone treatment for 28 days resulted in an almost complete reversal of the increased CD3e and CD19 mRNA expression back to their corresponding pathogen-free levels similar to the immunochemical staining results of CD3e and B220. Collectively, these findings showed that dexamethasone effectively reversed lymphocyte influx but not influx of neutrophils and macrophages.
Discussion
Infection of mouse airways by M. pulmonis caused a sequential response involving blood vessel enlargement, sprouting angiogenesis, and lymphangiogenesis. Blood vessels enlarged within the first week of infection, whereas lymphatic growth and angiogenic sprouting began during the second week. The major findings of this study are shown schematically in Figure 8. Vessel remodeling was more effectively prevented than reversed by treatment with dexamethasone, and remodeling of lymphatics was more resistant than blood vessel changes to corticosteroid-induced reversal.
Figure 8.
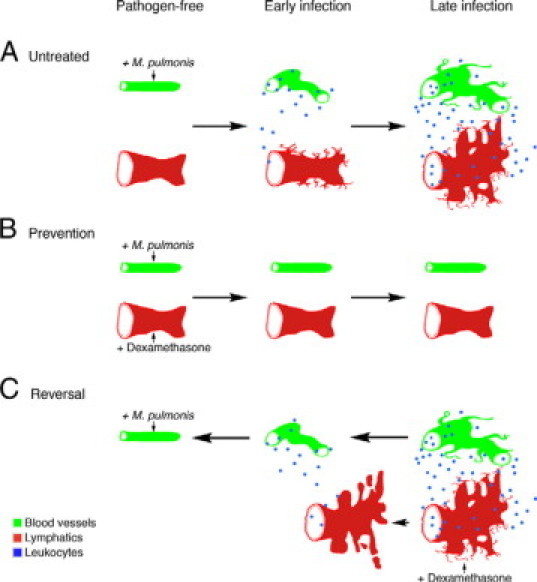
Schematic diagram of prevention and reversibility of blood vessel and lymphatic remodeling in M. pulmonis-infected airways. Blood vessels are shown in green, lymphatics in red, and leukocytes in blue in three conditions: infected and untreated (A); dexamethasone-prevention studies (B); and dexamethasone-reversal studies (C). In pathogen-free airways, vessels show normal baseline appearance with little or no leukocyte influx. A: In infected and untreated airways, uniform, thin capillaries enlarge into venules that support leukocyte recruitment, followed later by angiogenic sprouting. Leukocyte influx increases as infection continues. The normal lymphatic vessel network expands first by sprouting and then by enlargement and growth and fusion of newly formed lymphatics. B: In prevention studies, when dexamethasone is given concurrently with infection, blood vessels and lymphatics do not undergo extensive remodeling but remain similar to the pathogen-free state with little or no leukocyte influx. C: In reversal studies, delayed dexamethasone treatment reverses enlargement of blood vessels and growth of angiogenic sprouts almost to pathogen-free values. Leukocyte influx is reduced, as the blood vessels are less remodeled. In contrast to blood vessels, newly formed lymphatics are much more resistant to reversal, except for the disappearance of fine filopodia and formation of some disconnected lymphatic islands.
Prevention and Reversal of M. pulmonis-Induced Vessel Changes
Prevention of angiogenesis by corticosteroids is well established,18,36 but little is known of the potential of dexamethasone to reverse vascular changes. Our previous studies on the reversal of blood vessel changes in M. pulmonis-infected airways were confined to C3H mice, which do not show conventional angiogenic sprouting in response to infection.24 Here we used C57BL/6 mice, which display both vascular enlargement and sprouting angiogenesis. Both of these processes have been reported in asthma and chronic bronchitis.37,38 Our results clearly showed that both vascular enlargement and sprouting were preventable and almost fully reversible.
Lymphatic vessels seemed to be less active after delayed dexamethasone treatment because the surface of the lymphatic endothelial cells was smoother and showed fewer filopodia and cellular projections. The number of lymphatic islands also increased, suggesting that preexisting lymphatics may have regressed slightly, but at a much slower rate than that of blood vessels, and broken up into islands. Persistent hyperplasia may be a typical characteristic of lymphatic vessels and is not restricted to the results of the present dexamethasone study. Similar observations have been reported in M. pulmonis-infected airways in which the pathogen has been eliminated with antibiotic for 12 weeks10 and in adult mouse skin 6 months after cessation of transgenic VEGF-C expression.39 It is well known that initial lymphatics in many tissues40–43 including airways17 do not have a prominent covering of pericytes and basement membrane. Because pericytes and basement membrane are generally believed to stabilize newly formed blood vessels in tumors44,45 and in inflamed airways,46 the reason than newly formed lymphatics are resistant to regression despite their lack of pericyte and basement membrane coverage remains unknown.
Blood and lymphatic endothelial cells showed distinct reactions to M. pulmonis infection and to dexamethasone treatment, suggesting that the factors responsible for growth and maintenance are distinct from each other. Several factors are likely to be involved in the immune response-driven events and their roles in M. pulmonis-infected airways are just beginning to be elucidated. Growth of blood vessels and lymphatics was prevented to a similar extent, suggesting that growth factors driving the initial vessel changes are sensitive to dexamethasone treatment. TNF-α signaling directly promotes vascular enlargement and may drive lymphagiogenesis indirectly by inducing VEGF-C/D signaling via VEGF receptor (VEGFR)-3.10,17 Similar expression kinetics for IL-1β during the course of both dexamethasone-treated and untreated infection suggests that it may play a role similar to TNF-α in driving vessel remodeling. On the other hand, it seems that TNF-α and IL-1β are not essential for maintenance of remodeled blood vessels, because dexamethasone reversed the vascular changes but not TNF-α and IL-1β expression.
Inhibition of VEGF-A signaling causes regression of a few capillaries in normal airways.47 Overexpression of VEGF-A causes sprouting angiogenesis, and removal of VEGF-A induces rapid regression of newly formed blood vessels in airways of VEGF-A transgenic mice.48 As shown by the qRT-PCR analysis, expression of VEGF-A was sensitive to both concurrent and reversal dexamethasone treatment. This result is consistent with the potential role of VEGF-A driving sprouting angiogenesis and maintaining the newly grown blood vessels. On the other hand, overexpression of angiopoietins induces microvascular enlargement,49 and inhibition of angiopoietin/Tie2 signaling reduces the enlargement induced by infection (J. Fuxe, personal communication). Our qRT-PCR study suggests that angiopoietin-2 rather than angiopoietin-1 is likely to promote vessel remodeling and that neither molecule seems to be a factor responsible for maintenance of newly formed lymphatics. Further studies of the role of the angiopoietin/Tie2 pathway in mediating vessel changes in M. pulmonis-infected airways are underway.
PDGF-B plays an important role in the recruitment of pericytes to newly formed vessels,50 and loss of pericytes is a prominent characteristic of diabetic retinopathy associated with vascular regression.51,52 Our qRT-PCR studies showed an up-regulation of PDGF-B expression in infection, suggesting the possibility of an increase in pericyte recruitment to the remodeled blood vessels. Dexamethasone treatment effectively prevented and reversed the increase in PDGF-B expression, consistent with the concept that remodeling of blood vessels is preventable and reversible.
VEGF-C/D signaling via VEGFR-3 is the dominant stimulus for lymphangiogenesis in inflammatory diseases10,53 and is required for maintenance of lymphatics during embryonic development.54 Later blockade of VEGFR-3 signaling in airways of adult mice does not result in regression of lymphatics.10 Our qRT-PCR studies suggest that the newly formed lymphatics may have become similarly independent of VEGFR-3 signaling for survival despite the finding that expression of VEGF-C/D was down-regulated by 28 days of delayed treatment. It remains to be seen which maintenance factors are responsible for the survival of the newly grown lymphatics.
ICAM-1 and EphB4 are typical markers of venules that support leukocyte adhesion and extravasation.55 Remodeled capillaries expressed ICAM-1 and EphB4, consistent with previous reports of capillary-to-venule transformation in response to infection.7 The reduction in these markers after delayed dexamethasone treatment favors the reversal of remodeled blood vessels and may promote reductions in leukocyte influx.
Prevention and Reversal of M. pulmonis Burden
By 28 days of infection, M. pulmonis bacterial burden in the airways stabilized, suggesting that a state of equilibrium was reached for the rates of bacterial replication and clearance and that the host immune responses were not able to eliminate the organisms in chronically infected airways. Concurrent dexamethasone treatment almost prevented the increase in M. pulmonis burden, whereas delayed treatment significantly reduced but did not reverse bacterial burden. These findings are consistent with previous reports of reduced burden of Mycoplasma pneumoniae in lungs of mice pretreated with fluticasone56 and are consistent with the consensus that corticosteroids do not directly kill infecting bacteria or rhinoviruses.56,57 How then could corticosteroids reduce bacterial burden? Two possibilities are by inhibiting bacterial adhesion and by reducing nutrient availability. Corticosteroids decrease the expression of adhesion molecules on bacteria and/or receptors on host epithelial cells57 and reduce vascular leakage, which in turn may reduce the amount of nutrient-rich edema fluid.58,59
Differential Prevention and Reversal of Leukocyte Influx into Infected Airways
Gene expression profiling in lungs and in bronchoalveolar lavage fluid has been widely used in studying the molecular mechanisms underlying pulmonary inflammation. Here we assessed vascular remodeling and measured the kinetics of leukocyte influx and inflammatory cytokines in the same tissue because the cell populations and cytokine profiles in bronchoalveolar lavage fluid may not accurately reflect those in inflamed lung tissues or in airways themselves.60,61 The increased total yield of RNA mainly due to the large leukocyte influx into infected airways could have diluted some genes in our qRT-PCR studies. Therefore, some increases in gene expression may have been underestimated in highly remodeled airways.
Leukocytes have been recognized as the source of growth factors mediating vessel changes during infection, but the roles of individual leukocyte types in M. pulmonis-induced vessel changes have only just begun to be elucidated.14 Four different types of leukocytes were recruited with different kinetics during infection and showed differential sensitivity to dexamethasone. Neutrophils, which are capable of secreting a wide spectrum of inflammatory cytokines,62 were among the earliest responders. However, the presence of neutrophils was not limited to the acute phase of untreated inflammation, in contrast to many other inflammatory situations in which neutrophils are recruited early and then rapidly undergo apoptosis. Macrophages are another potential source of inflammatory cytokines and growth factors in M. pulmonis-infected airways.10,14,17 The relative resistance of neutrophils and macrophages to dexamethasone treatment observed in this study is consistent with other reports in mice with asthmatic-like pulmonary inflammation, such as allergic-like mouse models and acute exacerbations in chronic asthma models61,63 and may account for the elevated expression of TNF-α and IL-1β in the reversal studies.
As for lymphocytes, characteristic lymphoid follicular-like structures similar to bronchus-associated lymphoid tissue developed after 14 days of infection, suggesting that some remodeled blood vessels had features of high endothelial venules that favor lymphocyte entry. It was evident that systemic administration of dexamethasone significantly prevented and reversed the influx of both T and B lymphocytes, which is consistent with prior reports that dexamethasone suppresses recruitment and survival of lymphocytes and reduces lymph node hypertrophy.64,65 Further studies are required to separate the multiple actions of dexamethasone on endothelial cells from other cells to better understand which factors are responsible for the differential reversal of blood vessel and lymphatic remodeling.
Clinical Implications
Lymphatics drain tissue fluid and serve as routes for antigen and leukocyte transit from airways to lymph nodes.66 Lymphangiogenesis is recognized as a feature of airway inflammation, but the physiological consequences of these newly formed lymphatics are poorly understood. Previous studies suggest that newly formed lymphatics in the inflamed airways may be defective, resulting in bronchial lymphedema.10 Our observation that lymphatics did not regress once they had formed suggests that remodeled, preexisting lymphatics may modulate subsequent rounds of inflammatory responses in chronically re-occurring infections. It remains to be seen whether the chronic changes of lymphatic vessels exaggerate or improve the course of the disease on recurrent inflammation.
Infections with M. pneumoniae or Chlamydia pneumoniae are the human counterparts of M. pulmonis infection of rodent airways.67,68 Accumulating evidence has suggested that such bacterial and viral infections play a significant role as triggers of both acute and chronic asthma.69,70 In contrast to the chronic inflammation induced by M. pulmonis infection, M. pneumoniae infection of BALB/c mice induces transient symptoms, including bronchial hyperresponsiveness and lung inflammation.56 The reversal aspects of our present study have particular clinical relevance because the extent of reversibility of vessel remodeling is poorly understood. Although corticosteroids are the first choice of treatment for asthma, relatively little is known about the potential of corticosteroids or, for that matter, of any anti-inflammatory agents to reverse vessel remodeling. In fact, compared with many prevention studies, few studies have addressed the reversal of inflammation in airways.63,71
In conclusion, this study showed that changes in blood vessels and lymphatics are easier to prevent than to reverse in chronic inflamed airways. Delayed dexamethasone treatment decreased the inflammatory stimulus, reduced leukocyte influx, and allowed blood vessels to reverse to the almost pathogen-free state, but newly formed lymphatics were remarkably resistant to reversal. A better understanding of the role of distinct growth and maintenance factors that first induce the growth of blood and lymphatic vessels and then protect them from regression in chronic inflammatory disease may provide insight to the vascular contribution to the course of inflammatory disease.
Acknowledgements
We thank Kristoffer Larsen, Beverley Falcon, Ryan Naylor, Barbara Sennino, and Prescott Woodruff for helpful discussion and DongJi Zhang and Nigel Killeen for production of M. pulmonis stock.
Footnotes
Supported in part by the National Heart, Lung, and Blood Institute (grants HL-24136 and HL-59157) and from the National Cancer Institute (CA-82923).
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Contributor Information
Li-Chin Yao, Email: li-chin.yao@ucsf.edu.
Donald M. McDonald, Email: Donald.McDonald@ucsf.edu.
Web Extra Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Halin C, Detmar M. Chapter 1. Inflammation, angiogenesis, and lymphangiogenesis. Methods Enzymol. 2008;445:1–25. doi: 10.1016/S0076-6879(08)03001-2. [DOI] [PubMed] [Google Scholar]
- 2.Jackson JR, Seed MP, Kircher CH, Willoughby DA, Winkler JD. The codependence of angiogenesis and chronic inflammation. FASEB J. 1997;11:457–465. [PubMed] [Google Scholar]
- 3.Walters EH, Reid D, Soltani A, Ward C. Angiogenesis: a potentially critical part of remodelling in chronic airway diseases? Pharmacol Ther. 2008;118:128–137. doi: 10.1016/j.pharmthera.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Paredi P, Barnes PJ. The airway vasculature: recent advances and clinical implications. Thorax. 2009;64:444–450. doi: 10.1136/thx.2008.100032. [DOI] [PubMed] [Google Scholar]
- 5.Van Kruiningen HJ, Colombel JF. The forgotten role of lymphangitis in Crohn's disease. Gut. 2008;57:1–4. doi: 10.1136/gut.2007.123166. [DOI] [PubMed] [Google Scholar]
- 6.Thurston G, Baluk P, Hirata A, McDonald DM. Permeability-related changes revealed at endothelial cell borders in inflamed venules by lectin binding. Am J Physiol. 1996;271:H2547–H2562. doi: 10.1152/ajpheart.1996.271.6.H2547. [DOI] [PubMed] [Google Scholar]
- 7.Thurston G, Maas K, Labarbara A, McLean JW, McDonald DM. Microvascular remodelling in chronic airway inflammation in mice. Clin Exp Pharmacol Physiol. 2000;27:836–841. doi: 10.1046/j.1440-1681.2000.03342.x. [DOI] [PubMed] [Google Scholar]
- 8.Ezaki T, Baluk P, Thurston G, La Barbara A, Woo C, McDonald DM. Time course of endothelial cell proliferation and microvascular remodeling in chronic inflammation. Am J Pathol. 2001;158:2043–2055. doi: 10.1016/S0002-9440(10)64676-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, Vestweber D, Corada M, Molendini C, Dejana E, McDonald DM. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med. 2007;204:2349–2362. doi: 10.1084/jem.20062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baluk P, Tammela T, Ator E, Lyubynska N, Achen MG, Hicklin DJ, Jeltsch M, Petrova TV, Pytowski B, Stacker SA, Yla-Herttuala S, Jackson DG, Alitalo K, McDonald DM. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest. 2005;115:247–257. doi: 10.1172/JCI22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonald DM. Angiogenesis and remodeling of airway vasculature in chronic inflammation. Am J Respir Crit Care Med. 2001;164:S39–S45. doi: 10.1164/ajrccm.164.supplement_2.2106065. [DOI] [PubMed] [Google Scholar]
- 12.Lindsey JR, Baker HJ, Overcash RG, Cassell GH, Hunt CE. Murine chronic respiratory disease. Significance as a research complication and experimental production with Mycoplasma pulmonis. Am J Pathol. 1971;64:675–708. [PMC free article] [PubMed] [Google Scholar]
- 13.Lindsey JR, Cassell H. Experimental Mycoplasma pulmonis infection in pathogen-free mice. Models for studying mycoplasmosis of the respiratory tract. Am J Pathol. 1973;72:63–90. [PMC free article] [PubMed] [Google Scholar]
- 14.Aurora AB, Baluk P, Zhang D, Sidhu SS, Dolganov GM, Basbaum C, McDonald DM, Killeen N. Immune complex-dependent remodeling of the airway vasculature in response to a chronic bacterial infection. J Immunol. 2005;175:6319–6326. doi: 10.4049/jimmunol.175.10.6319. [DOI] [PubMed] [Google Scholar]
- 15.Sun X, Jones HP, Hodge LM, Simecka JW. Cytokine and chemokine transcription profile during Mycoplasma pulmonis infection in susceptible and resistant strains of mice: macrophage inflammatory protein 1β (CCL4) and monocyte chemoattractant protein 2 (CCL8) and accumulation of CCR5+ Th cells. Infect Immun. 2006;74:5943–5954. doi: 10.1128/IAI.00082-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis CC, Yang JY, Huang X, Banerjee SK, Blackburn MR, Baluk P, McDonald DM, Blackwell TS, Nagabhushanam V, Peters W, Voehringer D, Erle DJ. Disease-specific gene expression profiling in multiple models of lung disease. Am J Respir Crit Care Med. 2008;177:376–387. doi: 10.1164/rccm.200702-333OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baluk P, Yao LC, Feng J, Romano T, Jung SS, Schreiter JL, Yan L, Shealy DJ, McDonald DM. TNF-α drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice. J Clin Invest. 2009;119:2954–2964. doi: 10.1172/JCI37626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Folkman J, Ingber DE. Angiostatic steroids. Method of discovery and mechanism of action. Ann Surg. 1987;206:374–383. doi: 10.1097/00000658-198709000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barnes PJ. Molecular mechanisms and cellular effects of glucocorticosteroids. Immunol Allergy Clin North Am. 2005;25:451–468. doi: 10.1016/j.iac.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Horvath G, Wanner A. Inhaled corticosteroids: effects on the airway vasculature in bronchial asthma. Eur Respir J. 2006;27:172–187. doi: 10.1183/09031936.06.00048605. [DOI] [PubMed] [Google Scholar]
- 21.Yano A, Fujii Y, Iwai A, Kageyama Y, Kihara K. Glucocorticoids suppress tumor angiogenesis and in vivo growth of prostate cancer cells. Clin Cancer Res. 2006;12:3003–3009. doi: 10.1158/1078-0432.CCR-05-2085. [DOI] [PubMed] [Google Scholar]
- 22.Ristimäki A, Narko K, Enholm B, Joukov V, Alitalo K. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J Biol Chem. 1998;273:8413–8418. doi: 10.1074/jbc.273.14.8413. [DOI] [PubMed] [Google Scholar]
- 23.Villeneuve J, Galarneau H, Beaudet MJ, Tremblay P, Chernomoretz A, Vallieres L. Reduced glioma growth following dexamethasone or anti-angiopoietin 2 treatment. Brain Pathol. 2008;18:401–414. doi: 10.1111/j.1750-3639.2008.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thurston G, Murphy TJ, Baluk P, Lindsey JR, McDonald DM. Angiogenesis in mice with chronic airway inflammation: strain-dependent differences. Am J Pathol. 1998;153:1099–1112. doi: 10.1016/S0002-9440(10)65654-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baluk P, McDonald DM. Markers for microscopic imaging of lymphangiogenesis and angiogenesis. Ann NY Acad Sci. 2008;1131:1–12. doi: 10.1196/annals.1413.001. [DOI] [PubMed] [Google Scholar]
- 26.Baffert F, Thurston G, Rochon-Duck M, Le T, Brekken R, McDonald DM. Age-related changes in vascular endothelial growth factor dependency and angiopoietin-1-induced plasticity of adult blood vessels. Circ Res. 2004;94:984–992. doi: 10.1161/01.RES.0000125295.43813.1F. [DOI] [PubMed] [Google Scholar]
- 27.Bulloch K, Miller MM, Gal-Toth J, Milner TA, Gottfried-Blackmore A, Waters EM, Kaunzner UW, Liu K, Lindquist R, Nussenzweig MC, Steinman RM, McEwen BS. CD11c/EYFP transgene illuminates a discrete network of dendritic cells within the embryonic, neonatal, adult, and injured mouse brain. J Comp Neurol. 2008;508:687–710. doi: 10.1002/cne.21668. [DOI] [PubMed] [Google Scholar]
- 28.Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007;55:412–424. doi: 10.1002/glia.20468. [DOI] [PubMed] [Google Scholar]
- 29.Sasmono RT, Ehrnsperger A, Cronau SL, Ravasi T, Kandane R, Hickey MJ, Cook AD, Himes SR, Hamilton JA, Hume DA. Mouse neutrophilic granulocytes express mRNA encoding the macrophage colony-stimulating factor receptor (CSF-1R) as well as many other macrophage-specific transcripts and can transdifferentiate into macrophages in vitro in response to CSF-1. J Leukoc Biol. 2007;82:111–123. doi: 10.1189/jlb.1206713. [DOI] [PubMed] [Google Scholar]
- 30.Sakhinia E, Byers R, Bashein A, Hoyland J, Buckle AM, Brady G. Gene expression analysis of myeloid and lymphoid lineage markers during mouse haematopoiesis. Br J Haematol. 2006;135:105–116. doi: 10.1111/j.1365-2141.2006.06254.x. [DOI] [PubMed] [Google Scholar]
- 31.Ishii T, Sootome H, Shan L, Yamashita K. Validation of universal conditions for duplex quantitative reverse transcription polymerase chain reaction assays. Anal Biochem. 2007;362:201–212. doi: 10.1016/j.ab.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Schuhmann B, Dietrich A, Sel S, Hahn C, Klingenspor M, Lommatzsch M, Gudermann T, Braun A, Renz H, Nockher WA. A role for brain-derived neurotrophic factor in B cell development. J Neuroimmunol. 2005;163:15–23. doi: 10.1016/j.jneuroim.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 33.Monteith CE, Chelack BJ, Davis WC, Haines DM. Identification of monoclonal antibodies for immunohistochemical staining of feline B lymphocytes in frozen and formalin-fixed paraffin-embedded tissues. Can J Vet Res. 1996;60:193–198. [PMC free article] [PubMed] [Google Scholar]
- 34.Ji RC, Kato S. Enzyme-histochemical study on postnatal development of rat stomach lymphatic vessels. Microvasc Res. 1997;54:1–12. doi: 10.1006/mvre.1997.2016. [DOI] [PubMed] [Google Scholar]
- 35.Ohtani Y, Ohtani O. Postnatal development of lymphatic vessels and their smooth muscle cells in the rat diaphragm: a confocal microscopic study. Arch Histol Cytol. 2001;64:513–522. doi: 10.1679/aohc.64.513. [DOI] [PubMed] [Google Scholar]
- 36.Hori Y, Hu DE, Yasui K, Smither RL, Gresham GA, Fan TP. Differential effects of angiostatic steroids and dexamethasone on angiogenesis and cytokine levels in rat sponge implants. Br J Pharmacol. 1996;118:1584–1591. doi: 10.1111/j.1476-5381.1996.tb15578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson JW, Kotsimbos T. Airway vascular remodeling in asthma. Curr Allergy Asthma Rep. 2003;3:153–158. doi: 10.1007/s11882-003-0028-3. [DOI] [PubMed] [Google Scholar]
- 38.Siafakas NM, Antoniou KM, Tzortzaki EG. Role of angiogenesis and vascular remodeling in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2007;2:453–462. [PMC free article] [PubMed] [Google Scholar]
- 39.Lohela M, Helotera H, Haiko P, Dumont DJ, Alitalo K. Transgenic induction of vascular endothelial growth factor-C is strongly angiogenic in mouse embryos but leads to persistent lymphatic hyperplasia in adult tissues. Am J Pathol. 2008;173:1891–1901. doi: 10.2353/ajpath.2008.080378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leak LV, Burke JF. Fine structure of the lymphatic capillary and the adjoining connective tissue area. Am J Anat. 1966;118:785–809. doi: 10.1002/aja.1001180308. [DOI] [PubMed] [Google Scholar]
- 41.Veikkola T, Lohela M, Ikenberg K, Makinen T, Korff T, Saaristo A, Petrova T, Jeltsch M, Augustin HG, Alitalo K. Intrinsic versus microenvironmental regulation of lymphatic endothelial cell phenotype and function. FASEB J. 2003;17:2006–2013. doi: 10.1096/fj.03-0179com. [DOI] [PubMed] [Google Scholar]
- 42.Karpanen T, Makinen T. Regulation of lymphangiogenesis—from cell fate determination to vessel remodeling. Exp Cell Res. 2006;312:575–583. doi: 10.1016/j.yexcr.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 43.Tammela T, Petrova TV, Alitalo K. Molecular lymphangiogenesis: new players. Trends Cell Biol. 2005;15:434–441. doi: 10.1016/j.tcb.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 44.Baluk P, Hashizume H, McDonald DM. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev. 2005;15:102–111. doi: 10.1016/j.gde.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 45.Mancuso MR, Davis R, Norberg SM, O'Brien S, Sennino B, Nakahara T, Yao VJ, Inai T, Brooks P, Freimark B, Shalinsky DR, Hu-Lowe DD, McDonald DM. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest. 2006;116:2610–2621. doi: 10.1172/JCI24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okazaki T, Ni A, Baluk P, Ayeni OA, Kearley J, Coyle AJ, Humbles A, McDonald DM. Capillary defects and exaggerated inflammatory response in the airways of EphA2-deficient mice. Am J Pathol. 2009;174:2388–2399. doi: 10.2353/ajpath.2009.080949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baffert F, Le T, Sennino B, Thurston G, Kuo CJ, Hu-Lowe D, McDonald DM. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am J Physiol Heart Circ Physiol. 2006;290:H547–H559. doi: 10.1152/ajpheart.00616.2005. [DOI] [PubMed] [Google Scholar]
- 48.Baluk P, Lee CG, Link H, Ator E, Haskell A, Elias JA, McDonald DM. Regulated angiogenesis and vascular regression in mice overexpressing vascular endothelial growth factor in airways. Am J Pathol. 2004;165:1071–1085. doi: 10.1016/S0002-9440(10)63369-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim KE, Cho CH, Kim HZ, Baluk P, McDonald DM, Koh GY. In vivo actions of angiopoietins on quiescent and remodeling blood and lymphatic vessels in mouse airways and skin. Arterioscler Thromb Vasc Biol. 2007;27:564–570. doi: 10.1161/01.ATV.0000256458.82320.be. [DOI] [PubMed] [Google Scholar]
- 50.Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005;7:452–464. doi: 10.1215/S1152851705000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hirschi KK, D'Amore PA. Pericytes in the microvasculature. Cardiovasc Res. 1996;32:687–698. [PubMed] [Google Scholar]
- 52.Enge M, Bjarnegard M, Gerhardt H, Gustafsson E, Kalen M, Asker N, Hammes HP, Shani M, Fassler R, Betsholtz C. Endothelium-specific platelet-derived growth factor-B ablation mimics diabetic retinopathy. EMBO J. 2002;21:4307–4316. doi: 10.1093/emboj/cdf418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005;438:946–953. doi: 10.1038/nature04480. [DOI] [PubMed] [Google Scholar]
- 54.Karpanen T, Wirzenius M, Makinen T, Veikkola T, Haisma HJ, Achen MG, Stacker SA, Pytowski B, Yla-Herttuala S, Alitalo K. Lymphangiogenic growth factor responsiveness is modulated by postnatal lymphatic vessel maturation. Am J Pathol. 2006;169:708–718. doi: 10.2353/ajpath.2006.051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thurston G, Baluk P, McDonald DM. Determinants of endothelial cell phenotype in venules. Microcirculation. 2000;7:67–80. [PubMed] [Google Scholar]
- 56.Chu HW, Campbell JA, Rino JG, Harbeck RJ, Martin RJ. Inhaled fluticasone propionate reduces concentration of Mycoplasma pneumoniae, inflammation, and bronchial hyperresponsiveness in lungs of mice. J Infect Dis. 2004;189:1119–1127. doi: 10.1086/382050. [DOI] [PubMed] [Google Scholar]
- 57.Papi A, Papadopoulos NG, Degitz K, Holgate ST, Johnston SL. Corticosteroids inhibit rhinovirus-induced intercellular adhesion molecule-1 up-regulation and promoter activation on respiratory epithelial cells. J Allergy Clin Immunol. 2000;105:318–326. doi: 10.1016/s0091-6749(00)90082-4. [DOI] [PubMed] [Google Scholar]
- 58.Boschetto P, Rogers DF, Fabbri LM, Barnes PJ. Corticosteroid inhibition of airway microvascular leakage. Am Rev Respir Dis. 1991;143:605–609. doi: 10.1164/ajrccm/143.3.605. [DOI] [PubMed] [Google Scholar]
- 59.Bowden JJ, Schoeb TR, Lindsey JR, McDonald DM. Dexamethasone and oxytetracycline reverse the potentiation of neurogenic inflammation in airways of rats with Mycoplasma pulmonis infection. Am J Respir Crit Care Med. 1994;150:1391–1401. doi: 10.1164/ajrccm.150.5.7524980. [DOI] [PubMed] [Google Scholar]
- 60.Nagata N, Takayama K, Nikaido Y, Yokosaki Y, Kido M. Comparison of alveolar septal inflammation to bronchoalveolar lavage in interstitial lung diseases. Respiration. 1996;63:94–99. doi: 10.1159/000196525. [DOI] [PubMed] [Google Scholar]
- 61.Ito K, Herbert C, Siegle JS, Vuppusetty C, Hansbro N, Thomas PS, Foster PS, Barnes PJ, Kumar RK. Steroid-resistant neutrophilic inflammation in a mouse model of an acute exacerbation of asthma. Am J Respir Cell Mol Biol. 2008;39:543–550. doi: 10.1165/rcmb.2008-0028OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scapini P, Lapinet-Vera JA, Gasperini S, Calzetti F, Bazzoni F, Cassatella MA. The neutrophil as a cellular source of chemokines. Immunol Rev. 2000;177:195–203. doi: 10.1034/j.1600-065x.2000.17706.x. [DOI] [PubMed] [Google Scholar]
- 63.Kim J, McKinley L, Siddiqui J, Bolgos GL, Remick DG. Prevention and reversal of pulmonary inflammation and airway hyperresponsiveness by dexamethasone treatment in a murine model of asthma induced by house dust. Am J Physiol Lung Cell Mol Physiol. 2004;287:L503–L509. doi: 10.1152/ajplung.00433.2003. [DOI] [PubMed] [Google Scholar]
- 64.Evans-Storms RB, Cidlowski JA. Regulation of apoptosis by steroid hormones. J Steroid Biochem Mol Biol. 1995;53:1–8. doi: 10.1016/0960-0760(95)00034-w. [DOI] [PubMed] [Google Scholar]
- 65.Chapman MS, Askew DJ, Kuscuoglu U, Miesfeld RL. Transcriptional control of steroid-regulated apoptosis in murine thymoma cells. Mol Endocrinol. 1996;10:967–978. doi: 10.1210/mend.10.8.8843413. [DOI] [PubMed] [Google Scholar]
- 66.Cueni LN, Detmar M. The lymphatic system in health and disease. Lymphat Res Biol. 2008;6:109–122. doi: 10.1089/lrb.2008.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martin RJ, Chu HW, Honour JM, Harbeck RJ. Airway inflammation and bronchial hyperresponsiveness after Mycoplasma pneumoniae infection in a murine model. Am J Respir Cell Mol Biol. 2001;24:577–582. doi: 10.1165/ajrcmb.24.5.4315. [DOI] [PubMed] [Google Scholar]
- 68.Martin RJ. Infections and asthma. Clin Chest Med. 2006;27:87–98. doi: 10.1016/j.ccm.2005.10.004. vi. [DOI] [PubMed] [Google Scholar]
- 69.Dougherty RH, Fahy JV. Acute exacerbations of asthma: epidemiology, biology and the exacerbation-prone phenotype. Clin Exp Allergy. 2009;39:193–202. doi: 10.1111/j.1365-2222.2008.03157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Venarske DL, Busse WW, Griffin MR, Gebretsadik T, Shintani AK, Minton PA, Peebles RS, Hamilton R, Weisshaar E, Vrtis R, Higgins SB, Hartert TV. The relationship of rhinovirus-associated asthma hospitalizations with inhaled corticosteroids and smoking. J Infect Dis. 2006;193:1536–1543. doi: 10.1086/503809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uller L, Lloyd CM, Rydell-Tormanen K, Persson CG, Erjefalt JS. Effects of steroid treatment on lung CC chemokines, apoptosis and transepithelial cell clearance during development and resolution of allergic airway inflammation. Clin Exp Allergy. 2006;36:111–121. doi: 10.1111/j.1365-2222.2006.02396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.