

Abstract
The hexahistidine (His6)/Nickel (II)-Nitrilotriacetic Acid (Ni2+-NTA) system is widely used for affinity purification of recombinant proteins. The NTA group has many other applications, including the attachment of chromophores, fluorophores, or nano-gold to His6-proteins. Here we explore several applications of the NTA-derivative, (Ni2+-NTA)2-Cy3. This molecule binds our two model His6-proteins, N-ethylmaleimide Sensitive Factor (NSF) and O6-alklyguanine-DNA alkyltransferase (AGT), with moderate affinity (K ∼ 1.5 × 106 M-1) and no effect on their activity. Its high specificity makes (Ni2+-NTA)2-Cy3 ideal for detecting His6-proteins in complex mixtures of other proteins, allowing (Ni2+-NTA)2-Cy3 to be used as a probe in crude cell extracts and as a His6-specific gel stain. (Ni2+-NTA)2-Cy3 binding is reversible in 10 mM EDTA or 500 mM imidazole but in their absence, it exchanges slowly (kexchange ∼ 5 × 10-6 s-1 with 0.2 μM labeled protein in the presence of 1μM His6-peptide). Labeling with (Ni2+-NTA)2-Cy3 allows characterization of hydrodynamic properties by fluorescence anisotropy or analytical ultracentrifugation under conditions (e.g. high ADP absorbance) that prevent direct detection of protein. In addition, fluorescence resonance energy transfer (FRET) between (Ni2+-NTA)2-Cy3-labeled proteins and suitable donors/acceptors provides a convenient assay for binding interactions and for measurements of donor-acceptor distances.
Keywords: Ni2+-NTA, bis-NTA-Cy3, His6-tagged Proteins, Analytical Ultracentrifugation
Introduction
The utility of the Nickel (II)-Nitriloacetic Acid (Ni2+-NTA)-based methods for affinity purification of hexahistidine (His6)-containing recombinant proteins was first appreciated over twenty years ago [1]. Since that time, it has become an almost universal method for purifying recombinant proteins. Nickle ions are hexacoordinate. Four ligand sites can be occupied by NTA with the remaining two available for binding the histidines of a His6-tag. Because these interactions have high affinity and selectivity [2], and are easily reversed by treatment with imidazole, EDTA, or low pH, Ni2+-NTA-based methodologies have been adapted for a large number of applications [3]. Ni2+-NTA has been used to immobilize His6-tagged proteins on agarose beads [2], microtiter plates [4], and lipid surfaces [5 - 9]. It has also been exploited in protein-labeling schemes where Ni2+-NTA has been coupled to horseradish peroxidase [10], oligonucleotides [11; 12], biotin [13], and nano-gold particles [14; 15]. This plethora of applications of the Ni2+-NTA/His6-tag technology is a testament to the value and flexibility of this metal chelation system.
One particular use of Ni2+-NTA has been in linking it to fluorophores for site-specific labeling of His6-tagged proteins. Katayama and colleagues were one of the first groups to exploit this approach when they conjugated fluorescein isothiocyanate (FITC) to NTA and used the Ni2+-NTA-FITC to detect His6-tagged proteins [16]. Ebright and colleagues synthesized Ni2+-NTA-Cy3, Ni2+-NTA-Cy5, (Ni2+-NTA)2-Cy3, and (Ni2+-NTA)2-Cy5. They showed that the bis-NTA compounds had 10-100 fold greater affinity for His6-tags than did the monovalent derivatives [17]. Further increase in affinity has been achieved by synthesizing tris-NTA and tetra-NTA derivatives, although no additional binding advantage was observed for tetra-NTA derivatives [18]. To date, many different Ni2+-NTA-fluorophore compounds have been created, each with unique utility for both in vitro and in vivo labeling of His6-tagged proteins [19; 20].
In this report, we show how (Ni2+-NTA)2-Cy3 can be used as a probe for fluorescence-based analysis e.g. fluorescence resonance energy transfer (FRET) and anisotropy, and also for absorbance detection where the absorbance of solution components obscures the spectrum of an un-modified protein. Two model systems were chosen to illustrate the utility of (Ni2+-NTA)2-Cy3: the N-ethylmaleimide Sensitive Factor (NSF) ATPase, which is required for all intracellular vesicle trafficking [21] and the DNA repair enzyme O6-Alklyguanine-DNA alkyltransferase (AGT), which binds DNA cooperatively and repairs O6-alklyguanine residues [22]. We show that labeling with (Ni2+-NTA)2-Cy3 is specific, reversible, and of sufficient stability for hydrodynamic analyses. (Ni2+-NTA)2-Cy3-AGT binding to DNA could be monitored either by FRET using fluorescein-labeled DNA or by sedimentation equilibrium analysis where the protein's position was monitored by measuring absorbance at the λmax for Cy3 (550 nm). We further show the value of (Ni2+-NTA)2-Cy3 as a probe in analytical ultracentrifugation experiments performed in the presence of excess ADP or ATP. The high optical density of nucleotides at 260 nm severely hampers detection of unlabeled protein at 280-295 nm but does not interfere with detection of (Ni2+-NTA)2-Cy3-NSF at 550 nm. In summary, our data demonstrate the value of using (Ni2+-NTA)2-fluorophores as optical probes to monitor a specific protein in a complex mixture where direct detection, through more conventional methods, would not be possible.
Materials and Methods
(Ni2+-NTA)2-Cy3 Synthesis
Ni2+-loaded, bis-NTA-Cy3 ((Ni2+-NTA)2-Cy3) was synthesized using a modification of the method described in [17]. Nα,Nα-bis (carboxymethyl)-L-lysine hydrate (Sigma, St. Louis, MO) (26 mg, 100 μmol) was dissolved in 1.6 ml 0.1 M Na2CO3 (pH 11) and added to 1 vial of Cy3 bis-Reactive Dye (200 nmol, GE Healthcare, Piscataway, NJ). After 1 h at room temperature (RT) in the dark, the reaction mixture was passed through a Sep-Pak Plus C18 cartridge (Waters, Milford, MA) and the bound material was eluted with 60% methanol and dried. The mixture was dissolved in 10% methanol and separated on preparative thin layer chromatography plates (Uniplate Silica Gel G, Analtech, Newark, DE) using NH4OH:ethanol:water (33:21:6, v/v/v) as mobile phase. NTA2-Cy3 (Rf = 0.24) was eluted with H2O and dried. Its identity was confirmed by mass spectrometry (MALDI-TOF): m/z = 1205.51 (calculated 1205.46). Nickel ions were loaded into the NTA2-Cy3 as described [17] and the final product, (Ni2+-NTA)2-Cy3, was dissolved in H2O and stored at -80°C. The (Ni2+-NTA)2-Cy3 product had absorbance and fluorescence excitation maxima at 553 nm and an emission maximum at 563 nm, consistent with published values [17]. Dye concentrations were measured by absorbance at 550 nm (ε550 = 1.5 × 105 M-1 cm-1). The typical yield from the reaction was 24% relative to the starting Cy3 bis-Reactive Dye. (Ni2+-NTA)2-Cy5 has been made following this procedure [17] and the synthesis of other (Ni2+-NTA)2-Cy derivatives (i.e. Cy2, Cy5.5, Cy7) is not expected to require major modifications of this protocol.
Of note, there was a 2-fold increase in fluorescence intensity when (Ni2+-NTA)2-Cy3 was incubated with EDTA, suggesting that Ni2+ binding quenches fluorescence. More severe quenching was seen for the analogous (Ni2+-NTA)2-Cy5 compound (data not shown). Quenching by the coordinated Ni2+ has been reported for other Ni2+-NTA-conjugated fluorescent probes [16; 23; 24].
Protein, Polypeptide, and DNA Preparations
His6-NSF and His6-α-SNAP were expressed in the Rosetta pLacI E. coli cells (Novagen/EMD, Gibbstown, NJ), using the pProEx HT expression vector (Invitrogen, Carlsbad, CA), and purified as described [25]. Un-tagged NSF was prepared from His6-NSF by digestion with tobacco etch virus (TEV) protease and His6-containing fragments were removed with Ni2+-NTA-agarose beads [26]. The ATPase activity of (Ni2+-NTA)2-Cy3-His6-NSF or SNAP/SNARE binding and disassembly assays were performed as in [27]. The molecular weight of the intact NSF hexamer was ∼550 kDa as estimated by size exclusion chromatography (SEC). NSF and α-SNAP protein concentrations were measured by Bradford protein assay ([28], Bio-Rad, Hercules, CA). His6-human AGT (Mr = 21,519) and His6-AdaC were expressed in XL-1 Blue E. coli cells (Stratagene, La Jolla, CA). AGT was purified from cell lysates using Talon® (immobilized cobalt) chromatography as described in [29]. AGT samples were more than 95% pure as detected by SDS-PAGE (data not shown). AGT concentrations were measured by absorbance at 280 nm using ε280 = 3.93 × 104 M-1 cm-1 [30]. Hexahistidine polypeptide (His6-peptide) was obtained from Covance Research Products Inc. (Emeryville, CA). Single-stranded 26 nucleotide DNA, labeled at the 5′ end with 6-carboxy fluorescein (FAM) was obtained from Integrated DNA Technologies Inc. (Coralville, IA). The sequence of this DNA is: 5′-(FAM)-GAC TGA CTG ACT GAC TGA CTG ACT GA-3′. FAM-DNA samples tested by analytical ultracentrifugation and gel electrophoresis had apparent molecular weights and mobilities consistent with the monomeric state (data not shown). DNA concentrations were measured by absorbance at 260 nm using ε260 = 3.04 × 105 M-1 cm-1, estimated using the Oligo Calc online program [31].
(Ni2+-NTA)2-Cy3 Labeling
His6-NSF (2∼ 4 μM) was incubated with (Ni2+-NTA)2-Cy3 (1 mole of dye per mole of NSF hexamer) at 4°C for 30 min in Buffer A (50 mM HEPES/KOH, pH 7.4, 100 mM KCl, 1 mM MgCl2, 2 mM β-mecaptoethanol, 5% glycerol) supplemented with 0.5 mM nucleotide (ADP or AMP-PNP) as appropriate (see below). His6-AGT (155 μM) or His6-α-SNAP (46 μM) was incubated with (Ni2+-NTA)2-Cy3 (0.5 moles of dye per mole of protein) under similar conditions in Buffer B (10 mM Tris/HCl, pH 7.5; 150 mM KCl; 2 mM β-mercaptoethanol). Labeled proteins were resolved from low molecular weight reaction components by SEC on a column (0.7 × 30 cm) of Sephadex G-50 (Sigma) at 200 μl/min. Excluded fractions were collected and monitored by Bradford protein assay ([28], Bio-Rad, Hercules, CA) and by absorbance at 550 nm for Cy3. The degree of protein labeling (dye/protein) in the pooled fractions was confirmed by comparing the concentration of Cy3 with that of the protein. In all cases, labeling efficiency, in moles of (Ni2+-NTA)2-Cy3 per mole of protein, was within 20% of the starting ratio suggesting that the labeling procedure is ∼80% efficient.
Fluorescence Measurements
For fluorescence anisotropy measurements of (Ni2+-NTA)2-Cy3-His6-NSF, reaction mixtures (400 ul) were incubated in quartz cuvettes for 5 min (or for the indicated times) at 10°C. Fluorescence anisotropy was determined in a LS 55 Luminescence Spectrofluorometer (Perkin Elmer, Inc., Waltham, MA) with the slit widths set at 10 nm and the excitation and emission wavelengths set at 550 and 570 nm, respectively. Anisotropy was calculated from the ratio of polarized emission intensities (Equation 1).
| (1) |
Here IVV represents the intensity observed with vertically polarized excitation and emission, IVH the intensity observed with vertically polarized excitation and horizontally polarized emission and G = SV/SH, the ratio of detector sensitivity to vertically and horizontally polarized light [32].
For FRET measurements of (Ni2+-NTA)2-Cy3-His6-AGT or (Ni2+-NTA)2-Cy3-His6-α-SNAP and FAM-DNA, reaction mixtures were incubated in quartz cuvettes for 5 min at 10°C. In both cases FAM-DNA (0.5 μM) was held constant and the labeled protein was varied as indicated (0 – 12 μM). The slit widths were set at 4 nm and excitation was performed at 450 nm (to minimize direct excitation of Cy3) in a LS 55 Luminescence Spectrofluorometer. Emission spectra were recorded from 470 to 650 nm. Emission intensities of FAM-DNA alone (ID) or in the presence of (Ni2+-NTA)2-Cy3-His6-AGT (IDA) were measured at 516 nm (λem, max). Values of ID were corrected for a slight quenching observed with unlabeled AGT (<5% at the highest [AGT] used in these experiments). FRET efficiency was calculated using E = 1 - (IDA/ID). Equation 2 was used to fit the data and to estimate K0.5, the inverse of the free (Ni2+-NTA)2-Cy3-His6-AGT concentration at the mid-point of the binding transition.
| (2) |
We use K0.5 here, instead of the association constant (K) because the value of E does not scale in a simple way with AGT-DNA binding density. There are three reasons for this. First, weak binding of (Ni2+-NTA)2-Cy3 to His6-AGT predicts that some protein molecules that associate with DNA will not have dye bound. Second, free (Ni2+-NTA)2-Cy3 molecules make a small but measurable contribution to FRET efficiency (described below) that is not due to DNA binding by AGT. Finally, the relationship between binding density and FRET efficiency is complicated by the fact that as many as 6 AGT molecules can bind our test DNA. As a result, E should contain contributions from several (Ni2+-NTA)2-Cy3-His6-AGT molecules at different separations from the FAM label on the DNA, weighted by the 1/R6 dependence of Förster energy transfer [33]. Methods to evaluate and minimize the first two effects are discussed below.
Ultracentrifugation Analyses
The sedimentation velocity measurements of (Ni2+-NTA)2-Cy3-His6-NSF in Buffer A with either 0.5 mM ADP or AMP-PNP were performed at 10.0 ± 0.1°C in a Beckman XL-A analytical ultracentrifuge (Beckman, Fullerton, CA), using an An-60 Ti rotor at 15,000 rpm. Sample volumes were 300 μl with a starting OD550nm of between 0.15 and 0.17. The radial absorbance distributions during the experiment were recorded at 550 nm. The data were fit using a numerical solution of the Lamm equation implemented in the program SEDFIT [34; 35].
Sedimentation equilibrium measurements of (Ni2+-NTA)2-Cy3-His6-AGT (0.5 μM) incubated with FAM-DNA (14.5 μM) were performed at 4.0 ± 0.1°C and at 15,000, 22,500, and 30,000 rpm. After attainment of equilibrium at each speed, radial absorbance distributions were recorded at 260 nm and 550 nm. Because AGT binding to single-stranded DNA is positively cooperative, it can be described by the single-step mechanism nP + D ⇆ PnD. In this mechanism, free DNA (D) is in equilibrium with saturated complex (PnD) and intermediates with protein stoichiometries < n are not present at significant concentrations [36]. At sedimentation equilibrium, the radial distribution of absorbance is given by Equation 3.
| (3) |
In this expression A(r) is the absorbance at radial position ρ and αP and αD are absorbances of protein and DNA at the reference position, ro; the reduced molecular weights of AGT protein, DNA and protein-DNA complexes are given by σP = MP(1 - ν̄ P ρ)z2/(2RT), σD = MD(1 - ν̄ Dρ)z2/(2RT) and σPnD = (nMP + MD)(1 - ν̄PnD ρ)z2/(2RT). Here MP and MD are the molecular weights of protein and DNA, n is the protein:DNA ratio of the complex; ρ is the solvent density, z, the rotor angular velocity, R is the gas constant and T the temperature (Kelvin). The partial specific volumes of AGT and DNA were 0.744 mL/g and 0.550 mL/g, respectively [36]. The partial specific volume of the protein-DNA complex was estimated as previously described [37].
Results
Site-Specificity and Affinity of (Ni2+-NTA)2-Cy3 for His6-Tagged Proteins
His6-NSF hexamer was incubated with (Ni2+-NTA)2-Cy3 in 1:1 molar ratio, then analyzed by SEC using G-50 Sephadex (Figure 1A). The protein eluted in the excluded volume of the column (fraction 5-7), as demonstrated by Bradford protein assay of the fractions. Superimposed on this peak was the profile of absorbance at 550 nm, which corresponds to the presence of the Cy3 dye. When the His6-tag was removed by cleavage with TEV protease prior to incubation with (Ni2+-NTA)2-Cy3, no 550 nm absorbance was detected in the void volume with the protein; however, the dye was eluted in the included volume (fraction 10-11). When (Ni2+-NTA)2-Cy3-His6-NSF was rechromatographed, no free dye was detectable in the included volume suggesting that the interaction was stable and dissociation of the dye was slow (data not shown).
Figure 1. Specific interactions of (Ni2+-NTA)2-Cy3 with His6-proteins.
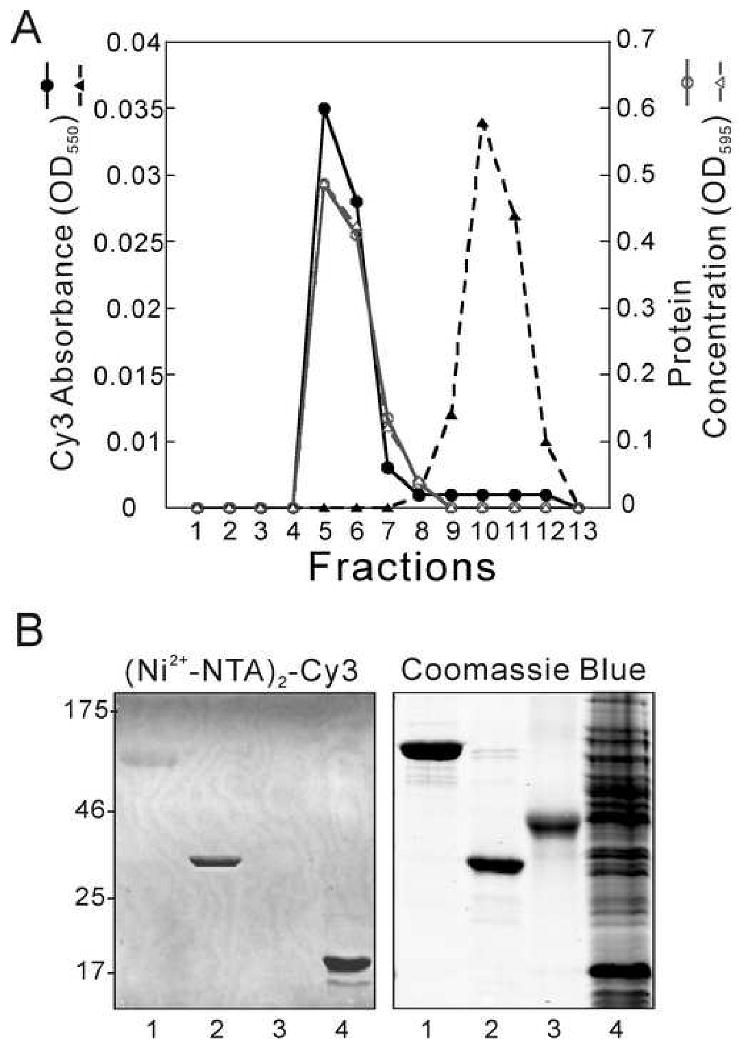
(A) Analytical chromatography on Sephadex G-50. His6-NSF or un-tagged NSF were pre-incubated with (Ni2+-NTA)2-Cy3 at a 1:1 dye:NSF hexamer ratio. Elution was monitored at 550 nm (black solid line for His6-NSF; black dashed line for NSF) and using the Bradford protein assay [28] at 595 nm (gray solid line for His6-NSF; gray dashed line for NSF). (B) Detection of His6-proteins after SDS-PAGE. Samples were prepared and electrophoresis was performed as described. Samples were His6-NSF (0.06 nmol, 5 μg, Lane 1), α-SNAP (0.23 nmol, 8 μg, Lane 2), ovalbumin (0.11 nmol, 5 μg, Lane 3) and E. coli cell extract containing His6-AdaC protein (total protein 50 μg, Lane 4). Left panel: fluorescence image of gel (λex = 532 nm, λem = 580 nm), obtained with a Typhoon 9400 imaging system. Right panel: visible light image of the same gel after staining with Coomassie Blue R-250. Relative molecular weights (kDa) of protein standards are indicated in the margin.
To further test its sensitivity and specificity, (Ni2+-NTA)2-Cy3 was used to stain His6-tagged and non-tagged proteins resolved in SDS-PAGE gels. Shown in Figure 1B are images of a gel in which the samples included His6-NSF (Lane 1), His6-α-SNAP (Lane 2), un-tagged ovalbumin (Lane 3) and a crude cell extract containing His6-AdaC protein [38] (Lane 4). The gel was stained without fixation for 5 min at room temperature in Buffer B containing 0.02 μM (Ni2+-NTA)2-Cy3. The fluorescent image, acquired with a Typhoon 9400 Imager (GE Healthcare, Piscataway, NJ; λex = 532 nm, λem = 580 nm, photomultiplier setting 450 V), showed clear bands for His6-α-SNAP and His6-AdaC with a weaker band for His6-NSF. The fluorescence intensities of the bands corresponding to His6-α-SNAP and His6-NSF were quantified using ImageQuant 5.2 software (GE Healthcare) and showed a ratio of 3.6 to 1 which is expected, based on the molar amounts of each protein loaded (0.23 vs. 0.06 nmoles, respectively). Significantly, no bands were detectable in the lane containing un-tagged ovalbumin, or in the regions populated by untagged cellular proteins in the sample containing crude cell extract (Lane 4). As a control, the same gel was re-stained with Coomassie Blue R-250 (right panel), revealing a large number of protein bands in the crude extract sample and single bands with appropriate monomer molecular weights for all purified proteins. Together, these results demonstrate the selective binding of (Ni2+-NTA)2-Cy3 to His6-proteins.
Binding of (Ni2+-NTA)2-Cy3 to His6-Tagged Proteins
Equilibrium constants for (Ni2+-NTA)2-Cy3 binding to His6-proteins were measured by fluorescence anisotropy. His6-NSF (0 – 8 μM) was added to a solution containing (Ni2+-NTA)2-Cy3 (50 nM) and incubated for 5 min at 10°C. The dependence of the anisotropy ratio (A – A0)/(Amax – A0) on NSF concentration is shown in Figure 2A. Here A is the anisotropy in the presence of His6-Protein, A0 is the anisotropy of free dye and Amax is the calculated anisotropy that is asymptotically approached at the highest protein concentrations. For a 1:1 complex, (A – A0)/(Amax – A0) is a measure of the fractional binding saturation and Equation 4 relates fractional saturation (Y) and association constant (K) for input concentrations of protein ([P]tot) and dye ([D]tot) [39].
Figure 2. Equilibria and kinetics of (Ni2+-NTA)2-Cy3 interaction with His6-proteins.
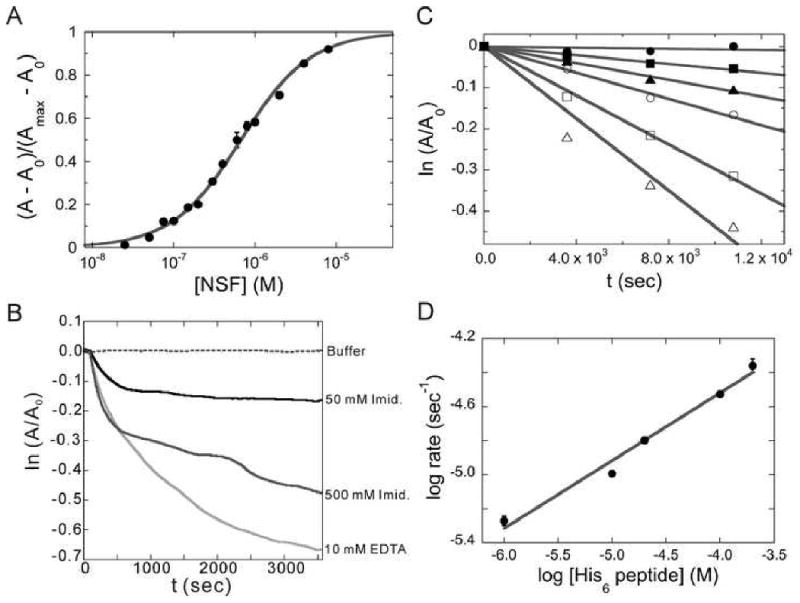
(A) Titration of 50 nM (Ni2+-NTA)2-Cy3 with increasing His6-NSF detected by fluorescence anisotropy. Reactions were carried out in (Buffer A + 0.5 mM ADP) at 10°C. The smooth curve is a fit of Equation 4 to the data. (B) Dissociation kinetics of the (Ni2+-NTA)2-Cy3-His6-NSF, monitored by fluorescence anisotropy. Reactions were carried out under the same conditions as (A) with the indicated concentration of EDTA or imidazole. (C) Kinetics of dye exchange between His6 groups. (Ni2+-NTA)2-Cy3-His6-NSF was incubated with 0-200 μM His6-peptide and fluorescence anisotropy was measured as a function of time. Concentrations of His6-peptide were 0μM (●), 1μM (■), 10μM (▲), 20μM (○), 100μM (□) and 200μM (△). The lines are linear fits to the data. (D) The dependence of log (initial rate) on log [His6-peptide] for dye exchange between His6 groups. The line is a linear fit of the data from panel C with a slope of 0.40 ± 0.03.
| (4) |
Fitting Equation 4 to the data in Figure 2A returns K = 1.54 ± 0.05 × 106 M-1. This compares well with values reported for other bis-NTA compounds and is larger than values reported for mono-Ni2+-NTA probes (0.05-0.1 × 106 M-1) [17; 18].
Dissociation of (Ni2+-NTA)2-Cy3 from His6-Tagged Proteins
The hallmark of Ni2+-NTA binding to His6-proteins is reversibility in the presence of EDTA or imidazole. This was explored by incubating dye-labeled proteins with either reagent, then monitoring the time evolution of fluorescence anisotropy (Figure 2B). The dissociation of dye from (Ni2+-NTA)2-Cy3-His6-NSF was rapid following addition of 10 mM EDTA (kd ∼ 2.0 × 10-4 s-1). Slower reactions were observed with 50 mM and 500 mM imidazole. In addition, the extent of dye release appeared to depend on the imidazole concentration as evidenced by the different limiting anisotropy values obtained at long reaction times. Closely similar results were obtained when dissociation of dye from (Ni2+-NTA)2-Cy3-His6-AGT was measured (data not shown).
The reversibility of (Ni2+-NTA)2-Cy3 binding to His6-tagged proteins raises the possibility that exchange might take place between different His6-tags. This has the potential to limit the utility of (Ni2+-NTA)2-Cy3 and related dyes in experiments containing several His6-tagged proteins. To determine whether this was likely to be a problem, the extent and rate of exchange was examined using 0.2 μM (Ni2+-NTA)2-Cy3-His6-NSF and various concentrations of His6-peptide. Addition of His6-peptide resulted in a time-dependent decrease in anisotropy, consistent with dissociation of (Ni2+-NTA)2-Cy3 from the NSF protein or with transfer of the dye to the lower molecular weight His6-peptide (Figure 2C). The apparent dissociation rate increased from 5.3 ± 0.3 × 10-6 s-1 to 4.4 ± 0.5 × 10-5 s-1 as peptide concentration increased from 1 μM to 200 μM (a 5-to-1,000-fold molar excess over His6-NSF); these rate constants correspond to half-lives of ∼50 h in a solution containing 1 μM His6-peptide and ∼6 h in a solution containing 200 μM His6-peptide. The kinetic order of the reaction in [His6-peptide] was determined from the dependence of log kd on log ([His6-peptide]) (Figure 2D). The fractional reaction order (0.40 ± 0.03) indicates that more than one mechanism contributes to this process. The simplest model consistent with these data is one in which ∼40% of the dissociation is attributable to a mechanism that is first order in His6-peptide and ∼60% is attributable to a peptide-independent process. These results indicate that dye-protein lifetimes can be extended by exclusion of chelators or competing His6 groups from reaction mixtures. They also demonstrate a method for determining complex lifetime in the presence of exchange and they show that at low His6 concentrations, the lifetime of the labeled species is long enough to be useful in transport experiments such as analytical ultracentrifugation or SEC.
(Ni2+-NTA)2-Cy3 as an Absorbance Probe
Many analytical methods rely on absorbance or fluorescence properties of amino acids for protein detection. Detection of tryptophan or tyrosine absorbance in the near UV (270-295 nm) or intrinsic tryptophan fluorescence (λex ∼295 nm, λem ∼340 nm) can be complicated by the presence of other chromophores such as nucleotides, nucleic acids, or other proteins that absorb in this wavelength range. The absorbance spectrum of (Ni2+-NTA)2-Cy3 (λmax (absorbance) ∼ 550 nm) makes it an ideal solution to this problem. This is demonstrated in two different experimental systems: a sedimentation velocity analysis of NSF in the presence of 0.5 mM ADP or AMP-PNP, and a sedimentation equilibrium study of AGT-DNA complexes.
1) Detecting protein during sedimentation velocity measurements in the presence of high nucleotide concentrations
Sedimentation velocity measurements of (Ni2+-NTA)2-Cy3-His6-NSF were made in Buffer A containing 0.5 mM ADP (Figure 3A). This nucleotide concentration corresponds to ∼ 7.7 A260 cm-1; this would mask the absorbance of a protein at 280 nm but not that of Cy3 at 550 nm. Analysis of the sedimentation velocity data yielded a sedimentation coefficient distribution, c(s), that was consistent with a single species of s20,w = 11.6 ± 1.5 (Figure 3B). This value is smaller than ones obtained by Fleming et al. by analytical ultracentrifugation with interference detection [40], although it accords well with earlier measurements made by glycerol gradient analysis [41].
Figure 3. Sedimentation velocity characterization of proteins in the presence of interfering substances.
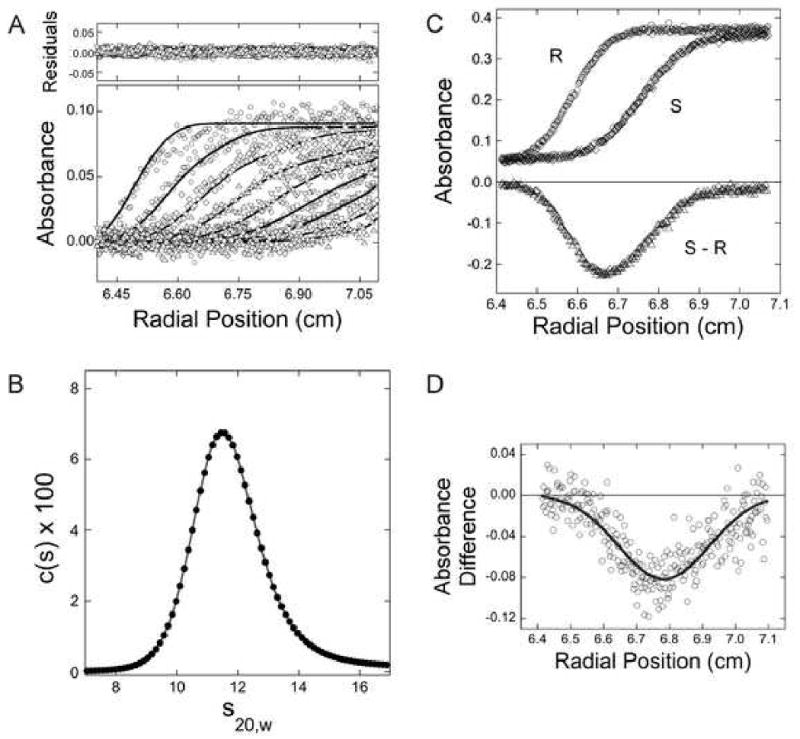
(A) Sedimentation velocity analysis of (Ni2+-NTA)2-Cy3-His6-NSF in the ADP-bound state. Samples contained (Ni2+-NTA)2-Cy3-His6-NSF dissolved in Buffer A containing 0.5 mM ADP. Sedimentation was performed at 15,000 rpm and 10°C. Radial absorbance scans separated by 16 min are shown; the smooth curves are fits of the c(s) model to this data, returning the spectrum of s-values shown in panel B. The small, symmetrical residuals demonstrate that this model is consistent with the data. (B) The c(s) spectrum derived from the data set shown in panel A; s-values have been corrected to reflect water density and viscosity at 20°C (s20,W) as described [49]. (C) Differential sedimentation to detect small differences in s-value: a schematic demonstrating the principle of the experiment. Absorbance profiles of samples differing in s-value and diffusion coefficient. If these were present in the sample (S) and reference (R) sectors as indicated, the difference profile (S-R) would be as indicated. (D) Differential sedimentation of the AMP-PNP and ADP forms of (Ni2+-NTA)2-Cy3-His6-NSF. Solutions placed in the sample and reference sectors were matched for OD550nm and volume. In addition to protein and buffer, the solution in the sample sector contained 0.5 mM AMP-PNP while that in the reference sector contained ADP. The scan, taken after 80 min of sedimentation, indicates that NSF in its AMP-PNP-bound form sediments more rapidly than the ADP-bound form.
In an additional experiment, we compared the sedimentation velocities of NSF in Buffer A containing AMP-PNP (0.5 mM) with that in the same buffer containing ADP (0.5 mM). NSF is expected to undergo a conformational switch upon substitution of ATP for ADP, but to date this has not been detected by hydrodynamic methods [40]. Shown in Figure 3D are the results of a differential sedimentation experiment [42; 43] in which a sample in buffer containing AMP-PNP (0.5 mM) was placed in the sample sector of the centrifuge cell and one containing ADP (0.5 mM) was placed in the reference sector. Samples were carefully matched in OD550nm and in volume. As shown in the representative schematic in Figure 3C, small differences in the s-value of the samples are expected to produce a difference peak in the absorbance profile; if the protein in the sample sector sediments more rapidly than that in the reference sector, the difference peak is negative in sign; if the protein in the sample sector sediments more slowly than that in the reference sector, the sign of the peak is positive. For the NSF samples examined here, the AMP-PNP-bound conformation sedimented more rapidly than the ADP-bound form (Figure 3D). The high absorbance of AMP-PNP and ADP solutions and their contributions to the refractive index of solutions complicate differential sedimentation measurements using ordinary absorbance or interferometric methods. As shown here, such measurements are straight-forward using absorbance optics at a wavelength where (Ni2+-NTA)2-Cy3 can be detected without interference from either nucleotide.
2) Detecting protein during sedimentation equilibrium experiments in the presence of DNA
Solutions containing (Ni2+-NTA)2-Cy3-His6-AGT and 26nt single-stranded DNA were brought to sedimentation equilibrium at 4 ± 0.1°C and at 15,000, 22,500, or 30,000 rpm. Radial absorbance distributions were recorded at 260 nm and 550 nm (Figure 4A and B, respectively). The smooth curves shown in panels A and B are fits of Equation 3 to the data, returning a stoichiometry of 6.1 ± 0.1 for data obtained at 260 nm and 5.9 ± 0.3 for data obtained at 550 nm. Both values are in good agreement with the expectation that AGT should form a 6:1 complex on 26nt DNA, based on its binding site size of 4nt/protein [36].
Figure 4. Detection of protein in the presence of DNA at sedimentation equilibrium.
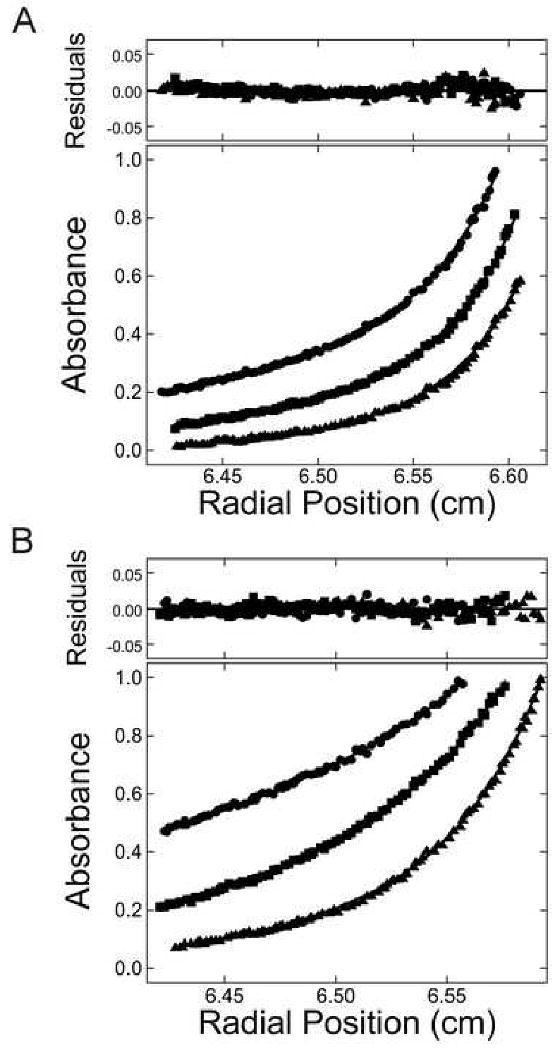
Sedimentation equilibrium data for solutions containing AGT and 26 nt DNA, taken at 4 ± 0.1 °C. (A) Data taken at 260 nm. (B) Data taken at 550 nm. Samples contained DNA (5 × 10-7 M) and AGT (1.45 × 10-5 M) in Buffer B. Radial scans acquired at 15,000 (▲), 22,500 (■), and 30,000 (●) rpm are shown. The smooth curves correspond to fits of Equation 3 to each data set. (For the 550 nm data (B), the term for free DNA in Equation 3 was set to zero.) The small residuals, symmetrically distributed about zero (upper panel), indicate that the cooperative nP + D ⇆ PnD model is consistent with the observed mass distributions in these samples.
(Ni2+-NTA)2-Cy3 as a FRET Acceptor
The AGT-DNA interaction was used to test the value of (Ni2+-NTA)2-Cy3 as a FRET acceptor in vitro, with 6-carboxyfluorescein (FAM) labeled DNA (FAM-DNA) as a donor (Figure 5). FAM and Cy3 are a well-characterized FRET pair with Förster distance R0 ∼ 63Å [44]. FAM (λex, max = 495 nm) was excited at 450 nm, to minimize direct excitation of Cy3, and emission spectra were recorded (Figure 5A). In the absence of (Ni2+-NTA)2-Cy3-His6-AGT, the spectrum of FAM-DNA had a strong emission maximum at 516 nm; emission was quenched slightly by addition of a saturating concentration (8 μM) of unlabeled His6-AGT. When excited at 450 nm, a solution containing 8 μM (Ni2+-NTA)2-Cy3-His6-AGT, but no FAM-DNA, exhibited no detectable fluorescence at 516 nm and only a small peak at the λmax for the Cy3 emission (566 nm). When samples contained both FAM-DNA and (Ni2+-NTA)2-Cy3-His6-AGT, the emission at 516 nm was strongly decreased while that at 566 nm increased, as expected for energy transfer from FAM to Cy3 (Figure 5B). However, because Cy3 emission was not efficient (discussed below), FRET was detected as decreased emission from FAM. The dependence of FRET on (Ni2+-NTA)2-Cy3-His6-AGT was modeled as the binding of free (Ni2+-NTA)2-Cy3-His6-AGT in solution (essentially no FRET) to DNA at a position close enough to the FAM moiety for FRET to occur. Analysis based on this model (Equation 2) gave K0.5 = 5.54 ± 0.54 × 105 M-1 (Figure 5C); this value is similar to association constants for AGT binding to single-stranded DNAs, measured under similar solution conditions by EMSA and analytical ultracentrifugation [36].
Figure 5. Detection of fluorescence resonance energy transfer between FAM-labeled DNA and (Ni2+-NTA)2-Cy3-His6-AGT.
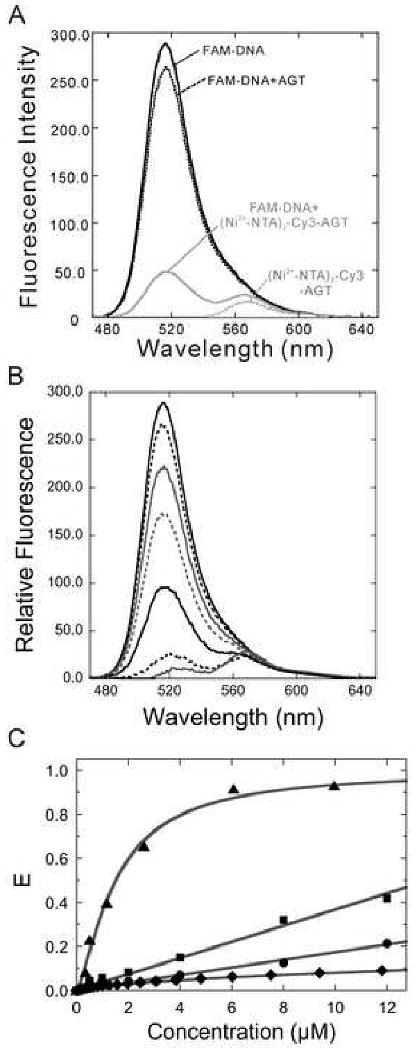
(A) FAM-DNA (0.5 μM) was incubated with (Ni2+-NTA)2-Cy3-His6-AGT (8 μM; solid grey) at 10°C for 5 min. FAM-DNA alone (solid black), FAM-DNA plus unlabeled AGT (8 μM; dashed black), and (Ni2+-NTA)2-Cy3-His6-AGT (8 μM; dashed grey) were used as controls. The samples were excited at 450 nm and the fluorescence intensities (470-650 nm) were recorded. (B) Titration of 0.5 μM FAM-DNA with (Ni2+-NTA)2-Cy3-His6-AGT (0-12 μM). Fluorescence intensities (470-650 nm) were recorded and background intensities (FAM-DNA plus unlabeled AGT) were subtracted. (C) Characterization of the AGT-DNA interaction. FRET efficiencies (E = 1 – IDA/ID) were determined as described in Methods. The dependence of E on the concentration of (Ni2+-NTA)2-Cy3-His6-AGT (▲) was fit using Equation 2. In control experiments, FAM-DNA (0.5 μM) was incubated with free (Ni2+-NTA)2-Cy3 (■) or (Ni2+-NTA)2-Cy3-His6-α-SNAP (●) at the indicated concentrations and the FRET efficiencies (E) are plotted. The free concentrations of (Ni2+-NTA)2-Cy3 were calculated for 1:1 dye-His6 protein mixtures using Equation 4, with K = 1.54 × 106 M-1 (see Figure 2). FRET efficiencies attributable to the free dye fraction were predicted using the experimental dependence of E on (Ni2+-NTA)2-Cy3 concentration (■) and are plotted as a function of the input (total, bound + free) dye concentration (◆).
To assess the specificity of the FRET measurements, two experiments were performed. First, we examined the energy transfer between FAM-DNA and free (Ni2+-NTA)2-Cy3 (Figure 5C). Although less efficient than that observed for samples containing (Ni2+-NTA)2-Cy3-His6-AGT, FRET amplitudes were significant when free dye concentrations were in the micromolar range, and reached E ∼ 0.45 at the highest concentrations tested (12 μM). However, a smaller FRET efficiency is predicted from the free dye in the presence of His6-containing protein because a substantial fraction of the dye is bound by protein. To show this, we used Equation 2 and the measured dye-His6 association constant (K = 1.54 ± 0.05 × 106 M-1) to calculate the concentrations of free (Ni2+-NTA)2-Cy3 for 1:1 dye-protein mixtures, as functions of protein concentration. We then used the concentration-dependence of the FRET efficiency observed for free dye to predict the FRET efficiency attributable to free dye in equilibrium with a protein that does not bind DNA. As shown in Figure 5C (◆), these values are much smaller than those observed for free (Ni2+-NTA)2-Cy3 (■), reaching only E ∼ 0.08 for an equilibrium mixture containing 12 μM dye and 12 μM His6-protein. To test the accuracy of this prediction we measured FAM-DNA fluorescence in the presence of (Ni2+-NTA)2-Cy3-His6-α-SNAP (●), a protein that is not expected to interact with DNA. FRET is detectable in these solutions as the concentration of (Ni2+-NTA)2-Cy3-His6-α-SNAP is increased, but the amplitude of this effect is only slightly greater than that predicted by our calculations for a non-DNA binding protein, reaching E ∼ 0.2 at 12μM protein. Together, these results suggest that the FRET observed for the system containing (Ni2+-NTA)2-Cy3-His6-AGT and FAM-DNA is unlikely to be dominated by contributions from unbound (Ni2+-NTA)2-Cy3 and FAM-DNA. While these control experiments show that chance interactions between fluorophores in solution can cause some level of energy transfer, the FRET efficiency obtained with FAM-DNA and (Ni2+-NTA)2-Cy3-His6-AGT cannot be accounted for by these random events and therefore must reflect DNA binding by the dye-protein complex.
(Ni2+-NTA)2-Cy3 Does Not Affect the Activities of the Model Proteins
The ATPase and SNAP/SNARE binding and disassembly activities of NSF were assayed, to determine whether labeling with (Ni2+-NTA)2-Cy3 affected NSF function. Compared to unlabeled protein, (Ni2+-NTA)2-Cy3-His6-NSF had similar basal and SNAP/SNARE-stimulated ATPase activities (Figure 6). In addition, the (Ni2+-NTA)2-Cy3 did not appear to affect NSF's ability to bind to SNAP/SNARE complexes in the presence of the non-hydrolyzable ATP analogue, AMP-PNP, or to disassemble them in the presence of ATP (data not shown). The DNA binding activities of (Ni2+-NTA)2-Cy3-AGT could also be compared with those of the unlabeled protein. The 6:1 stoichiometry detected at sedimentation equilibrium (Figure 4) is consistent with the saturating binding density of 4nt/protein observed with unlabeled protein [45], while analysis of binding detected by FRET between FAM-labeled DNA and (Ni2+-NTA)2-Cy3-AGT (Figure 5) returned an estimate K0.5 = 5.54 ± 0.54 × 105 M-1 that is in good agreement with the monomer-equivalent association constant measured for unlabeled protein [36]. Together, these results suggest that under appropriate conditions, labeling with (Ni2+-NTA)2-Cy3 has little effect on the hydrodynamics, ligand-binding properties, and enzymatic activities of target proteins.
Figure 6. Labeling with (Ni2+-NTA)2-Cy3 has little effect on NSF activities.
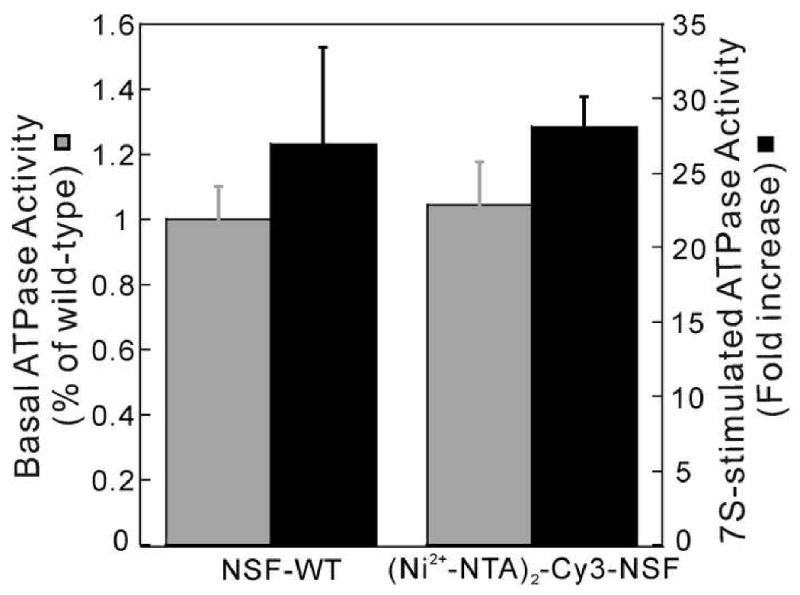
(A) ATPase activity of (Ni2+-NTA)2-Cy3-His6-NSF. Basal (gray bar) and SNAP/SNARE (7S)-stimulated-ATPase (black bar) activities of wild-type His6-NSF (NSF-WT) and (Ni2+-NTA)2-Cy3-His6-NSF were measured (n=2) and values normalized to that of the wild-type protein.
Discussion
Here we have shown that (Ni2+-NTA)2-Cy3 can be used as a chromophore to label His6-tagged proteins and to detect them under solution conditions in which the native absorbance and fluorescence signals of proteins are obscured. This approach should be particularly valuable for the analysis of individual protein components in complex mixtures by techniques such as analytical ultracentrifugation, SEC, or gel electrophoresis. The same label can be used for fluorescence-based assays that detect anisotropy or FRET ([17], this work) adding greatly to the utility of this strategy. The labeling procedure targets the His6-motif, widely used as a means of protein purification by immobilized metal affinity chromatography. The labeling reaction itself is gentle and, in the presented examples, does not appear to change the hydrodynamic, enzymatic, or interaction properties of the protein that is labeled. The very large number of proteins that have been purified under native conditions by immobilized metal affinity chromatography suggests that labeling with (Ni2+-NTA)2-Cy3 and related compounds will be innocuous in the majority of cases.
The binding of (Ni2+-NTA)2-Cy3 appears to be quite specific, as shown by stable complex formation with His6-tagged proteins, its easy removal from a solution of non-tagged protein by SEC (Figure 1A) and its inability to stain non-tagged proteins in an SDS-PAGE gel at a concentration of (Ni2+-NTA)2-Cy3 that labels His6-tagged proteins to saturation (Figure 1B). However, experience with Ni2+-NTA-affinity chromatography suggests that some proteins that lack an artificial His6 moiety may bind (Ni2+-NTA)2-Cy3 [46]. In particular, protein motifs rich in residues with known affinities for transition metals (e.g., His or Cys may retain (Ni2+-NTA)2-Cy3). These proteins represent one potential limitation on the applicability of these (Ni2+-NTA)2-chromophoric labels and thus justify the routine inclusion of the controls described in this paper. A second limitation, illustrated by our FRET experiments, is the relatively weak binding of (Ni2+-NTA)2-Cy3 to His6-motifs. This can result in significant concentrations of free (Ni2+-NTA)2-Cy3 dye and/or dye-free protein. The experiments shown in Figure 5C allow evaluation of the contributions of free dye to the FRET signal. They illustrate how Equation 4 can be used to calculate the fractional saturation of protein with dye from known dye and protein concentrations and the dye-protein equilibrium constant. We are currently evaluating strategies to improve the affinity of (Ni2+-NTA)2-Cy3 for His6-tagged proteins, including modest increases in solution pH and the substitution of other transition metals for Ni2+.
In the absence of exchange, (Ni2+-NTA)2-Cy3-complexes with His6-tagged proteins are kinetically stable. We detected no dissociation during a typical SEC run (duration ∼1 h) or during sedimentation velocity experiments (duration ∼8 h) and only a trace of free dye was detected at the end of a sedimentation equilibrium run with labeled AGT protein (duration 48 h). The stable binding of (Ni2+-NTA)2-Cy3 overcomes a major drawback of mono-NTA probes, which dissociate rapidly from His6-tags [17; 18]. Although binding stability may be increased further when > 2 Ni2+-NTA moieties are conjugated to fluorophores [18], the probability of protein crosslinking by multivalent NTA groups is also expected to increase. Thus the (Ni2+-NTA)2-linkage is a compromise that offers good labeling efficiency and few undesirable side-reactions. The slow exchange properties of the His6-(Ni2+-NTA)2-dye complex, especially at low concentrations of His6 groups, should make it possible to trace different proteins with several different (Ni2+-NTA)2-linked chromophores in one solution. Experiments are underway to test this possibility. However, because label-exchange rates depend, at least partially, on the concentration of His6-tag, very high concentrations of tagged proteins could result in exchange that is rapid enough to interfere with some methods of analysis.
The remarkable fluorescence quenching by coordinated Ni2+ is a feature of this labeling system that deserves further exploration. We noticed that fluorescence intensity was increased 2-fold when EDTA was incubated with (Ni2+-NTA)2-Cy3 and an even greater fluorescence enhancement (∼5-fold) was observed when EDTA was added to (Ni2+-NTA)2-Cy5 (data not shown). Since Ni2+ chelates can be FRET acceptors [23; 47], increasing the distance from NTA to fluorophore may improve quantum yield. Huang et al. put a PEG 2000 spacer between fluorescein and tri-NTA and found that the addition of Ni2+ caused only slight decrease in fluorescence intensity [48]. This feature must be taken into account when designing future fluorescent probes.
In conclusion, selective chromophore (or fluorophore)-labeling of His6-tagged proteins by (Ni2+-NTA)2-linked dyes promises to be a convenient method to detect proteins and to characterize their structures and interactions in solution. Because the current generation of (Ni2+-NTA)2-Cy3 or -Cy5 compounds absorb outside of the UV spectral envelopes of nucleotides, native proteins, and nucleic acids, they have the potential to allow study of interactions in solutions that approach biological complexity.
Acknowledgments
We acknowledge valuable advice on the synthesis of (Ni2+-NTA)2-Cy3 provided by Renhao Li (The University of Texas Health Science Center at Houston, Houston, TX, USA). This study was supported by National Institutes of Health (NIH) grants NS046242, HL56652, and HL091893 to SWW, Steckler Awards to ZC and LMH, and NIH grant GM070662 to MGF. Instruments used in this work are located in the Center for Structural Biology at the University of Kentucky and are supported by COBRE grant RR-03-014.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hochuli E, Dobeli H, Schacher A. New metal chelate adsorbent selective for proteins and peptides containing neighbouring histidine residues. J Chromatogr. 1987;411:177–84. doi: 10.1016/s0021-9673(00)93969-4. [DOI] [PubMed] [Google Scholar]
- 2.Schmitt J, Hess H, Stunnenberg HG. Affinity purification of histidine-tagged proteins. Mol Biol Rep. 1993;18:223–30. doi: 10.1007/BF01674434. [DOI] [PubMed] [Google Scholar]
- 3.Crowe J, Masone BS, Ribbe J. One-step purification of recombinant proteins with the 6×His tag and Ni-NTA resin. Mol Biotechnol. 1995;4:247–58. doi: 10.1007/BF02779018. [DOI] [PubMed] [Google Scholar]
- 4.Paborsky LR, Dunn KE, Gibbs CS, Dougherty JP. A nickel chelate microtiter plate assay for six histidine-containing proteins. Anal Biochem. 1996;234:60–5. doi: 10.1006/abio.1996.0050. [DOI] [PubMed] [Google Scholar]
- 5.Chikh GG, Li WM, Schutze-Redelmeier MP, Meunier JC, Bally MB. Attaching histidine-tagged peptides and proteins to lipid-based carriers through use of metal-ion-chelating lipids. Biochim Biophys Acta. 2002;1567:204–12. doi: 10.1016/s0005-2736(02)00618-1. [DOI] [PubMed] [Google Scholar]
- 6.Fischer NO, Blanchette CD, Chromy BA, Kuhn EA, Segelke BW, Corzett M, Bench G, Mason PW, Hoeprich PD. Immobilization of his-tagged proteins on nickel-chelating nanolipoprotein particles. Bioconjug Chem. 2009;20:460–5. doi: 10.1021/bc8003155. [DOI] [PubMed] [Google Scholar]
- 7.Patel JD, O'Carra R, Jones J, Woodward JG, Mumper RJ. Preparation and characterization of nickel nanoparticles for binding to his-tag proteins and antigens. Pharm Res. 2007;24:343–52. doi: 10.1007/s11095-006-9154-7. [DOI] [PubMed] [Google Scholar]
- 8.Gizeli E, Glad J. Single-step formation of a biorecognition layer for assaying histidine-tagged proteins. Anal Chem. 2004;76:3995–4001. doi: 10.1021/ac034855g. [DOI] [PubMed] [Google Scholar]
- 9.Altin JG, White FA, Easton CJ. Synthesis of the chelator lipid nitrilotriacetic acid ditetradecylamine (NTA-DTDA) and its use with the IAsys biosensor to study receptor-ligand interactions on model membranes. Biochim Biophys Acta. 2001;1513:131–48. doi: 10.1016/s0005-2736(01)00344-3. [DOI] [PubMed] [Google Scholar]
- 10.Jin L, Wei X, Gomez J, Datta M, Birkett A, Peterson DL. Use of alpha-N,N-bis[carboxymethyl]lysine-modified peroxidase in immunoassays. Anal Biochem. 1995;229:54–60. doi: 10.1006/abio.1995.1378. [DOI] [PubMed] [Google Scholar]
- 11.Shimada J, Maruyama T, Hosogi T, Tominaga J, Kamiya N, Goto M. Conjugation of DNA with protein using His-tag chemistry and its application to the aptamer-based detection system. Biotechnol Lett. 2008;30:2001–6. doi: 10.1007/s10529-008-9784-4. [DOI] [PubMed] [Google Scholar]
- 12.Goodman RP, Erben CM, Malo J, Ho WM, McKee ML, Kapanidis AN, Turberfield AJ. A facile method for reversibly linking a recombinant protein to DNA. Chembiochem. 2009;10:1551–7. doi: 10.1002/cbic.200900165. [DOI] [PubMed] [Google Scholar]
- 13.Reichel A, Schaible D, Al Furoukh N, Cohen M, Schreiber G, Piehler J. Noncovalent, site-specific biotinylation of histidine-tagged proteins. Anal Chem. 2007;79:8590–600. doi: 10.1021/ac0714922. [DOI] [PubMed] [Google Scholar]
- 14.Hainfeld JF, Liu W, Halsey CM, Freimuth P, Powell RD. Ni-NTA-gold clusters target His-tagged proteins. J Struct Biol. 1999;127:185–98. doi: 10.1006/jsbi.1999.4149. [DOI] [PubMed] [Google Scholar]
- 15.Tinazli A, Tang J, Valiokas R, Picuric S, Lata S, Piehler J, Liedberg B, Tampe R. High-affinity chelator thiols for switchable and oriented immobilization of histidine-tagged proteins: a generic platform for protein chip technologies. Chemistry. 2005;11:5249–59. doi: 10.1002/chem.200500154. [DOI] [PubMed] [Google Scholar]
- 16.Amano H, Ohuchi Y, Katayama Y, Maeda M. A new fluorescent reagent for the detection of proteins having histidine-tag (his-tag) Anal Sci. 2001;17 supplement:i1469–i1471. [Google Scholar]
- 17.Kapanidis AN, Ebright YW, Ebright RH. Site-specific incorporation of fluorescent probes into protein: hexahistidine-tag-mediated fluorescent labeling with (Ni(2+):nitrilotriacetic Acid (n)-fluorochrome conjugates. J Am Chem Soc. 2001;123:12123–5. doi: 10.1021/ja017074a. [DOI] [PubMed] [Google Scholar]
- 18.Lata S, Reichel A, Brock R, Tampe R, Piehler J. High-affinity adaptors for switchable recognition of histidine-tagged proteins. J Am Chem Soc. 2005;127:10205–15. doi: 10.1021/ja050690c. [DOI] [PubMed] [Google Scholar]
- 19.Soh N. Selective chemical labeling of proteins with small fluorescent molecules based on metal-chelation methodology. Sensors. 2008;8:1004–1024. doi: 10.3390/s8021004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J, Park HY, Ryu J, Kwon do Y, Grailhe R, Song R. Ni-nitrilotriacetic acid-modified quantum dots as a site-specific labeling agent of histidine-tagged proteins in live cells. Chem Commun (Camb) 2008:1910–2. doi: 10.1039/b719434j. [DOI] [PubMed] [Google Scholar]
- 21.Zhao C, Slevin JT, Whiteheart SW. Cellular functions of NSF: not just SNAPs and SNAREs. FEBS Lett. 2007;581:2140–9. doi: 10.1016/j.febslet.2007.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melikishvili M, Rasimas JJ, Pegg AE, Fried MG. Interactions of human O6-alkylguanine-DNA alkyltransferase (AGT) with short double-stranded DNAs. Biochemistry. 2008;47:13754–13763. doi: 10.1021/bi801666c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lata S, Gavutis M, Tampe R, Piehler J. Specific and stable fluorescence labeling of histidine-tagged proteins for dissecting multi-protein complex formation. J Am Chem Soc. 2006;128:2365–72. doi: 10.1021/ja0563105. [DOI] [PubMed] [Google Scholar]
- 24.Kim SH, Jeyakumar M, Katzenellenbogen JA. Dual-mode fluorophore-doped nickel nitrilotriacetic acid-modified silica nanoparticles combine histidine-tagged protein purification with site-specific fluorophore labeling. J Am Chem Soc. 2007;129:13254–64. doi: 10.1021/ja074443f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whiteheart SW, Rossnagel K, Buhrow SA, Brunner M, Jaenicke R, Rothman JE. N-ethylmaleimide-sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J Cell Biol. 1994;126:945–54. doi: 10.1083/jcb.126.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.May AP, Misura KM, Whiteheart SW, Weis WI. Crystal structure of the amino-terminal domain of N-ethylmaleimide-sensitive fusion protein. Nat Cell Biol. 1999;1:175–82. doi: 10.1038/11097. [DOI] [PubMed] [Google Scholar]
- 27.Zhao C, Matveeva EA, Ren Q, Whiteheart SW. Dissecting the N-Ethylmaleimide Sensitive Factor (NSF): Required Elements of the N and D1 Domains. J Biol Chem. 2009 doi: 10.1074/jbc.M109.056739. in press, online Nov 3, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 29.Daniels DS, Woo TT, Luu KX, Noll DM, Clarke ND, Pegg AE, Tainer JA. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat Struct Mol Biol. 2004;11:714–20. doi: 10.1038/nsmb791. [DOI] [PubMed] [Google Scholar]
- 30.Roy R, Shiota S, Kennel SJ, Raha R, von Wronski M, Brent TP, Mitra S. A comparative study of the biochemical properties of human and mouse recombinant O6-methylguanine-DNA methyltransferases. Carcinogenesis. 1995;16:405–411. doi: 10.1093/carcin/16.2.405. [DOI] [PubMed] [Google Scholar]
- 31.Kibbe WA. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res. 2007;35:W43–6. doi: 10.1093/nar/gkm234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lakowicz JR. Principles of Fluorescence Spectroscopy. Plenum; N.Y.: 1983. [Google Scholar]
- 33.Wu P, Brand L. Resonance energy transfer: methods and applications. Anal Biochem. 1994;218:1–13. doi: 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- 34.Schuck P. Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dam J, Schuck P. Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles. Methods Enzymol. 2004;384:185–212. doi: 10.1016/S0076-6879(04)84012-6. [DOI] [PubMed] [Google Scholar]
- 36.Rasimas JJ, Kar SR, Pegg AE, Fried MG. Interactions Of Human O6-Alkylguanine-DNA Alkyltransferase (AGT) With Short Single-Stranded DNAs. J Biol Chem. 2007;282:3357–3366. doi: 10.1074/jbc.M608876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daugherty MA, Fried MG. Protein-DNA Interactions at Sedimentation Equilibrium. In: Scott D, editor. Modern Analytical Ultracentrifugation: Techniques and Method. Royal Society of Chemistry; Oxford: 2005. pp. 195–209. [Google Scholar]
- 38.Crone TM, Kanugula S, Pegg AE. Mutations in the Ada O6-alkylguanine-DNA alkyltransferase conferring sensitivity to inactivation by O6-benzylguanine and 2,4-diamino-6-benzyloxy-5-nitrosopyrimidine. Carcinogenesis. 1995;16:1687–1692. doi: 10.1093/carcin/16.8.1687. [DOI] [PubMed] [Google Scholar]
- 39.LiCata VJ, Wowor AJ. Application of Fluorescence Anisotropy to the Study of Protein-DNA Interactions. Meth Cell Biol. 2008;84:243–262. doi: 10.1016/S0091-679X(07)84009-X. [DOI] [PubMed] [Google Scholar]
- 40.Fleming KG, Hohl TM, Yu RC, Muller SA, Wolpensinger B, Engel A, Engelhardt H, Brunger AT, Sollner TH, Hanson PI. A revised model for the oligomeric state of the N-ethylmaleimide-sensitive fusion protein, NSF. J Biol Chem. 1998;273:15675–81. doi: 10.1074/jbc.273.25.15675. [DOI] [PubMed] [Google Scholar]
- 41.Block MR, Glick BS, Wilcox CA, Wieland FT, Rothman JE. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc Natl Acad Sci U S A. 1988;85:7852–6. doi: 10.1073/pnas.85.21.7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richards EG, Schachman HK. A Differential Ultracentrifuge Technique For Measuring Small Changes in Sedimentation Coefficients. J Am Chem Soc. 1957;79:5324–5325. [Google Scholar]
- 43.Lohman TM, Wensley CG, Cina J, Burgess RR, Record MT., Jr Use of difference boundary sedimentation velocity to investigate nonspecific protein-nucleic acid interactions. Biochemistry. 1980;19:3516–22. doi: 10.1021/bi00556a016. [DOI] [PubMed] [Google Scholar]
- 44.Hong MH, Harbron EJ, O'Connor DB, Guo J, Barbara PF, Levin JG, Musier-Forsyth K. Nucleic Acid Conformational Changes Essential for HIV-1 Nucleocapsid Protein-mediated Inhibition of Self-priming in Minus-strand Transfer. J Mol Biol. 2003;325:1–10. doi: 10.1016/s0022-2836(02)01177-4. [DOI] [PubMed] [Google Scholar]
- 45.Rasimas JJ, Pegg AE, Fried MG. DNA-binding mechanism of O6-alkylguanine-DNA alkyltransferase. Effects of protein and DNA alkylation on complex stability. J Biol Chem. 2003;278:7973–7980. doi: 10.1074/jbc.M211854200. [DOI] [PubMed] [Google Scholar]
- 46.Janknecht R, de Martynoff G, Lou J, Hipskind RA, Nordheim A, Stunnenberg HG. Rapid and efficient purification of native histidine-tagged protein expressed by recombinant vaccinia virus. Proc Natl Acad Sci U S A. 1991;88:8972–6. doi: 10.1073/pnas.88.20.8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taraska JW, Puljung MC, Olivier NB, Flynn GE, Zagotta WN. Mapping the structure and conformational movements of proteins with transition metal ion FRET. Nat Methods. 2009;6:532–7. doi: 10.1038/nmeth.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang Z, Park JI, Watson DS, Hwang P, Szoka FC., Jr Facile synthesis of multivalent nitrilotriacetic acid (NTA) and NTA conjugates for analytical and drug delivery applications. Bioconjug Chem. 2006;17:1592–600. doi: 10.1021/bc0602228. [DOI] [PubMed] [Google Scholar]
- 49.Ralston G. Intruduction to Analytical Ultracentrifugation. Beckman Instruments, Inc; Fullerton: 1993. [Google Scholar]