

Abstract
Objective
Interferon-α (IFNα) is a heritable risk factor for systemic lupus erythematosus (SLE). Genetic variation near IRF7 is implicated in SLE susceptibility. SLE-associated autoantibodies can stimulate IFNα production through the Toll-like receptor/IRF7 pathway. This study was undertaken to determine whether variants of IRF7 act as risk factors for SLE by increasing IFNα production and whether autoantibodies are important to this phenomenon.
Methods
We studied 492 patients with SLE (236 African American, 162 European American, and 94 Hispanic American subjects). Serum levels of IFNα were measured using a reporter cell assay, and single-nucleotide polymorphisms (SNPs) in the IRF7/PHRF1 locus were genotyped.
Results
In a joint analysis of European American and Hispanic American subjects, the rs702966 C allele was associated with the presence of anti–double-stranded DNA (anti-dsDNA) antibodies (odds ratio [OR] 1.83, P = 0.0069). The rs702966 CC genotype was only associated with higher serum levels of IFNα in European American and Hispanic American patients with anti-dsDNA antibodies (joint analysis P = 4.1 × 10−5 in anti-dsDNA–positive patients and P = 0.99 in anti-dsDNA–negative patients). In African American subjects, anti-Sm antibodies were associated with the rs4963128 SNP near IRF7 (OR 1.95, P = 0.0017). The rs4963128 CT and TT genotypes were associated with higher serum levels of IFNα only in African American patients with anti-Sm antibodies (P = 0.0012). In African American patients lacking anti-Sm antibodies, an effect of anti-dsDNA–rs702966 C allele interaction on serum levels of IFNα was observed, similar to the other patient groups (overall joint analysis P = 1.0 × 10−6). In European American and Hispanic American patients, the IRF5 SLE risk haplotype showed an additive effect with the rs702966 C allele on IFNα level in anti-dsDNA–positive patients.
Conclusion
Our findings indicate that IRF7/PHRF1 variants in combination with SLE-associated autoantibodies result in higher serum levels of IFNα, providing a biologic relevance for this locus at the protein level in human SLE in vivo.
Systemic lupus erythematosus (SLE) is characterized by multisystem involvement commonly affecting the skin, renal, musculoskeletal, and hematopoietic systems. SLE is caused by interactions between susceptibility genes and environmental factors, which can include ultraviolet light, infections, and viruses, resulting in an irreversible loss of immunologic self tolerance (1). SLE incidence is highest in women of childbearing age (2); however, people of both sexes and all ages and ethnic backgrounds are susceptible. Disease features range from mild manifestations, such as rash or arthritis, to life-threatening end-organ manifestations, such as glomerulonephritis or thrombosis, and it is difficult to predict which manifestations will affect a given patient.
Interferon-α (IFNα) is a pleiotropic type I IFN with the potential to break self tolerance by activating antigen-presenting cells after uptake of self material (3). Serum IFNα levels are elevated in many SLE patients, and elevations may correlate with disease activity (4,5). SLE and lupus-like syndromes can develop when patients with chronic viral hepatitis and malignant diseases are treated with recombinant human IFNα (6). IFNα-induced SLE typically resolves after the IFNα is discontinued (7,8), suggesting that IFNα is causal. We have previously shown that high serum levels of IFNα are common in both healthy and affected members of SLE families as compared with healthy unrelated individuals (9). Additionally, serum IFNα activity is highest during the ages of peak SLE incidence in both patients and their healthy first-degree relatives (10). These data suggest that high serum IFNα activity is a heritable risk factor for SLE. The high IFNα trait in SLE families is inherited in a complex manner, suggesting polygenic inheritance, which is currently not fully characterized.
Interferon regulatory factor 5 (IRF-5) is a transcription factor that induces transcription of IFNα and IFNα-induced genes (11). Genetic association studies examining single-nucleotide polymorphisms (SNPs) in IRF5 have defined haplotypes which confer either susceptibility to or protection against SLE in individuals of European ancestry (12-14). These haplotypes are characterized by multiple functional genomic variants (13,14), and presumably, these functional variants alter IRF5-mediated transcription and subsequent risk of SLE. We have recently shown that an SLE risk haplotype of IRF5 is associated with high serum IFNα activity in vivo in SLE patients (15). The differential effect of the IRF5 genotype on serum levels of IFNα was most prominent in patients with either anti–double-stranded DNA (anti-dsDNA) or anti–RNA binding protein (anti-RBP) antibodies (15). In vitro models have shown that the addition of sera containing anti-dsDNA or anti-RBP antibodies to dendritic cells in culture results in brisk IFNα production (16). Nucleic acid contained within these autoantibody immune complexes could trigger endosomal Toll-like receptors (TLRs) after uptake into cells via Fc receptors, and IRF5 is activated downstream of endosomal TLRs. These data collectively support a model in which chronic stimulation of endosomal TLRs by endogenous autoantibody immune complexes is required for IRF5 risk variants to result in increased IFNα production.
IRF-7 is a transcription factor that can induce transcription of IFNα and IFNα-induced genes downstream of endosomal TLRs, similar to IRF5 (17). A SNP near IRF7 was found to be associated with SLE susceptibility in the recent International Consortium for SLE Genetics (SLEGEN) genome-wide association study (18). The associated SNP (rs4963128) was located 23 kb telomeric to IRF7 in a gene of unknown function named PHD and RING-finger domains 1 (PHRF1; also known as KIAA1542 or CTD-binding SR-like protein rA9). This SNP was in high linkage disequilibrium (r2 = 0.94) with the rs702966 SNP in IRF7 (18). The PHRF1 gene contains PHD-finger and RING-finger domains, and has not been functionally characterized to date. PHD and RING-finger domains both chelate zinc ions, and PHD domains are frequently found in proteins which mediate protein–protein interactions in the cell nucleus (19).
We hypothesized that the SLE-associated variant discovered in the IRF7/PHRF1 locus in the SLEGEN study (18) could be due to a polymorphism in IRF7 that predisposes to increased IFNα production. We tested this hypothesis by analyzing serum IFNα in SLE patients as a quantitative trait to determine associations with haplotype-tagging SNPs in the IRF7/PHRF1 locus. We studied SNPs in both genes, since the PHRF1 gene could also be the causal gene in this locus, and largescale followup studies refining this association have not yet been published. Multiple ethnic backgrounds were studied, and autoantibodies were incorporated into the analysis, given the importance of SLE-associated autoantibodies to the relationships we have previously demonstrated between IRF5 genotype and serum IFNα (15).
PATIENTS AND METHODS
Patients and samples
Serum and genomic DNA samples were obtained from the Translational Research Initiative in the Department of Medicine (TRIDOM) at the University of Chicago and Rush University Medical Center. Of the 492 SLE patients, 236 were African American, 162 were European American, and 94 were Hispanic American. African American controls (n = 140) from the TRIDOM registry were also genotyped, and these subjects were screened by medical record review for the absence of autoimmune or inflammatory disease by the same physician (LR). The study was approved by the institutional review board at each institution, and informed consent was obtained from all subjects.
Reporter cell assay for IFNα
The reporter cell assay for IFNα has been described in detail previously (9,20). Reporter cells were used to measure the ability of patient sera to cause IFN-induced gene expression. The reporter cells (WISH cells) (ATCC no. CCL-25; American Type Culture Collection, Manassas, VA) were cultured with 50% patient sera for 6 hours, and then lysed. Messenger RNA (mRNA) was purified from cell lysates, and complementary DNA was made from total cellular mRNA. Complementary DNA was then quantified using real-time polymerase chain reaction (PCR) using an Applied Biosystems 7900HT PCR machine with the SYBR Green fluorophore system. Forward and reverse primers for the genes MX1, PKR, and IFIT1, which are known to be highly and specifically induced by IFNα, were used in the reaction (9). GAPDH was amplified in the same samples to control for background gene expression.
Real-time PCR data analysis
The amount of PCR product of the IFNα-induced gene was normalized to the amount of product for the housekeeping gene GAPDH in the same sample. The relative expression of each of the 3 tested IFN-induced genes was calculated as the fold increase compared with its expression in WISH cells cultured with media alone. Results from the IFNα assay were standardized to a healthy multiethnic reference population as previously described, and a serum IFNα activity score was calculated based on the mean and SD of the reference population (9).
Measurement of autoantibodies
Antibodies to anti-Ro, anti-La, anti-Sm, and anti-RNP were measured in all samples by enzyme-linked immunosorbent assay methods in the University of Chicago clinical laboratory. Standard clinical laboratory cutoff points were used to categorize samples as positive or negative. Anti-dsDNA antibodies were measured using Crithidia luciliae immunofluorescence in the University of Chicago clinical laboratory. Samples with detectable fluorescence were considered to be positive for anti-dsDNA antibodies.
Genotyping
Individuals were genotyped at the rs12286521, rs4963128, rs702966, and rs2246614 SNPs in the IRF7/PHRF1 gene regions to tag haplotype blocks present in the Centre d’Etude du Polymorphisme Humain (CEPH) and Yoruba (YRI) HapMap reference data sets. These 4 SNPs capture >80% of common genetic variation (minor allele frequency >0.05) with an r2 value of 0.8 or greater in this 48-kb region in both the CEPH and YRI populations. This 48-kb region includes both IRF7 and PHRF1 genes and the haplotype block associated with SLE (18) and includes SNPs representing the haplotype blocks on either side of the SLE-associated block (SNPs 1 and 4) (Figure 1A). Diagrams of the D’ values estimating the degree of correlation between the 4 SNPs in each ancestral background were generated using Haploview 4.0 software (Figure 1B). Each of these SNPs conformed to Hardy-Weinberg equilibrium (P ≥ 0.001 for all markers for each ancestral background). Allele frequencies for each background are available from the corresponding author upon request. The IRF5 rs2004640, rs3807306, rs10488631, and rs2280714 SNPs were genotyped in the same subjects, which allowed for designation of an SLE risk haplotype (13). Genotyping was performed using ABI TaqMan Assay-By-Design primers and probes on an ABI 7900HT PCR machine.
Figure 1.
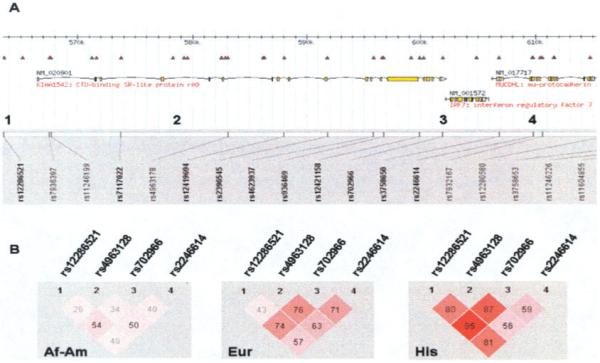
Diagram of IRF7/PHRF1 locus and haplotype structure in African American (Af-Am), European American (Eur), and Hispanic American (His) subjects. A, Diagram of the IRF7/PHRF1 locus with gene and single-nucleotide polymorphism (SNP) locations indicated on chromosome 11. The numbers 1–4 indicate the locations of SNPs genotyped in the study. B, Haplotype diagrams for the 4 genotyped SNPs in each ancestral background, as generated using Haploview software, version 4.0. D’ values for each pairwise comparison are shown in the boxes; darker shading indicates a higher D’ value.
Statistical analysis
Chi-square distribution was used to analyze categorical data, and the nonparametric Mann-Whitney U test was used to compare quantitative IFNα data between 2 genotype subgroups. P values shown are uncorrected for multiple comparisons. Logistic regression models were used to detect associations between autoantibodies and IRF7 gene region SNPs. Screening models incorporated each of the 4 SNPs with a single autoantibody trait in each ancestral background separately. In followup models, nonsignificant variables were discarded and potential interactions between significantly associated variables were assessed. Odds ratios (ORs) and 95% confidence intervals (95% CIs) were calculated.
In subjects of African American ancestry, the study had >80% power to detect an association between anti-Sm autoantibodies and IRF7 SNPs with an OR of ≥1.70 and an alpha level of 0.001 over the allele frequencies represented by these SNPs in an additive genetic model. In combined analysis of European American and Hispanic American subjects, the study had >80% power to detect an association between anti-dsDNA autoantibodies and IRF7 SNPs with an OR of ≥1.59 and an alpha level of 0.001 with the same parameters. Power calculations for autoantibody associations were performed using the CaTS Genetic Power Calculator (http://www.sph.umich.edu/csg/abecasis/CaTS/index.html) by Skol et al (21). In African American patients with SLE, there was 80% power to detect a variant which contributed >7.8% of the total variance in the serum IFNα trait in the setting of anti-Sm antibodies with an alpha level of 0.005. In combined analysis of European American and Hispanic American subjects, the study had 80% power to detect a variant which contributed >6.8% of the total variance in serum IFNα in the setting of anti-dsDNA antibodies with the same parameters. The proportion of variance in serum IFNα detectable due to IRF7 genotype given the sample size and stratifications performed in the study was calculated using the Genetic Power Calculator (http://pngu.mgh.harvard.edu/~purcell/gpc/#qcc_ins) as previously described (22).
RESULTS
Association of anti-dsDNA antibodies with the rs702966 C allele in European American patients with SLE
Logistic regression models were used to detect associations between autoantibodies and IRF7/PHRF1 gene region SNPs in European American and Hispanic American patients with SLE. In European American subjects, the rs702966 C SNP immediately downstream of the IRF7 gene was associated with the presence of anti-dsDNA antibodies (OR 1.81 [95% CI 1.04–3.15], P = 0.030). No other SNPs showed significant association with any of the other autoantibodies. No significant relationships were seen in patients of Hispanic ancestry; however, there was a trend toward an association between the rs702966 C allele and anti-dsDNA (OR 2.09 [95% CI 0.96–4.56], P = 0.062). The ORs observed between the rs702966 C allele and anti-dsDNA in European American and Hispanic American subjects were not significantly different (P = 0.93 by Breslow-Day test of homogeneity of ORs as corrected by Tarone [23]), and a combined analysis using a fixed-effects model resulted in a joint OR of 1.83 (95% CI 1.18–2.84) (P = 0.0069).
Association of the rs702966 C allele with higher serum levels of IFNα in European American and Hispanic American patients with anti-dsDNA antibodies
In European American and Hispanic American subjects, there was evidence of association of the rs702966 CC genotype with higher serum levels of IFNα in unstratified cohorts (P = 0.0052 and P = 0.0060, respectively) (Additional data are available from the corresponding author upon request). Given the association between the C allele and dsDNA antibodies, we next analyzed serum IFNα in patient subgroups stratified by the presence or absence of anti-dsDNA autoantibodies. The rs702966 CC genotype was only associated with higher serum levels of IFNα in European American and Hispanic American patients with anti-dsDNA antibodies (P = 0.0002 and P = 0.0004, respectively). (Additional data are available from the corresponding author upon request.) Both European American and Hispanic American subjects showed a similar pattern of serum IFNα activity in relation to the rs702966 CC genotype and anti-dsDNA antibodies, and the results of a joint analysis of both ethnic groups are shown in Figure 2. Higher serum levels of IFNα were observed in patients with the rs702966 CC genotype with anti-dsDNA antibodies than in patients with the rs702966 CG or GG genotype with anti-dsDNA antibodies (P = 4.1 × 10−5). There was no significant difference in serum levels of IFNα between patients with the CC genotype and patients with the CG or GG genotype who were anti-dsDNA negative (P = 0.99).
Figure 2.

Serum interferon-α (IFNα) activity in European American and Hispanic American patients with systemic lupus erythematosus, stratified by the presence or absence of anti–double-stranded DNA (anti-dsDNA) antibodies and rs702966 genotype. Rare minor allele homozygotes (GG) were combined with heterozygotes (CG) for analysis. The y-axis shows the serum IFNα activity score (see Patients and Methods for details). Bars show the median and interquartile range. P values were calculated using the Mann-Whitney U test.
Association of the rs4963128 T allele with anti-Sm antibodies in African American patients with SLE and association of this SNP/antibody combination with higher serum levels of IFNα
In African American patients, the rs4963128 T allele downstream of IRF7 was associated with the presence of anti-Sm antibodies (OR 1.95 [95% CI 1.29–2.97], P = 0.0017). The rs702966 C allele was not associated with anti-dsDNA in the African American subjects, and no other significant associations were seen in this group. African American patients stratified by genotype at rs4963128 did not show a significant difference in serum levels of IFNα (P = 0.20 for CC versus CT or TT genotypes) (Data are available from the corresponding author upon request). Given the association between this SNP and anti-Sm antibodies, we analyzed serum IFNα in African American patient groups stratified by rs4963128 genotype and the presence or absence of anti-Sm antibodies. In African American patients with anti-Sm antibodies, subjects with the rs4963128 CT and TT genotypes had higher IFNα levels than those with the CC genotype (P = 0.0012) (Figure 3). When the African American patients were stratified by rs702966 genotype and anti-dsDNA antibodies using the model for European American and Hispanic American patients described above, no significant differences in serum levels of IFNα were observed (data not shown).
Figure 3.

Serum interferon-α (IFNα) activity in African American patients with SLE, stratified by the presence or absence of anti-Sm antibodies and rs4963128 genotype. Minor allele homozygotes (TT) were combined with heterozygotes (CT) for analysis. The y-axis shows the serum IFNα score (see Patients and Methods for details). Bars show the median and interquartile range. P values were calculated using the Mann-Whitney U test.
Frequencies of rs4963128 alleles in African American patients with SLE and controls and HapMap populations demonstrate a strong influence of anti-Sm antibodies
Since the IRF7/PHRF1 locus has not been studied in the African American population, we genotyped 140 African American controls at the rs4963128 SNP to estimate the population frequency of this SNP in the African American population. The rs4963128 T allele showed a nonsignificant trend toward increased frequency in controls (T allele frequency 0.411 in cases and 0.454 in controls; P = 0.25) (Table 1). This was surprising, since we had observed a strong relationship between the rs4963128 T allele and anti-Sm antibodies, and anti-Sm antibodies are almost never found in the absence of SLE. Anti-Sm–positive patients with SLE did have a tendency toward a higher frequency of the rs4963128 T allele than did controls (0.505 versus 0.454; P = 0.27), and the anti-Sm–negative patients had a lower T allele frequency than controls (0.350 versus 0.454; P = 0.012).
Table 1.
SNP rs4963128 genotype and allele counts, and allele frequencies in African American cases and controls and HapMap reference populations*
| Controls | Cases | Anti-Sm− patients |
Anti-Sm+ patients |
CEPH | YRI | |
|---|---|---|---|---|---|---|
| CC genotype | 49 | 97 | 67 | 30 | – | – |
| CT genotype | 55 | 84 | 52 | 32 | – | – |
| TT genotype | 36 | 55 | 24 | 31 | – | – |
| C allele | 153 | 278 | 186 | 92 | 78 | 54 |
| T allele | 127 | 194 | 100 | 94 | 42 | 66 |
| T allele frequency | 0.454 | 0.411 | 0.350 | 0.505 | 0.350 | 0.550 |
Controls, cases, anti-Sm–negative patients, and anti-Sm–positive patients were all of African American ancestry and were from the Translational Research Initiative in the Department of Medicine registry at the University of Chicago and Rush University Medical Center. SNP = single-nucleotide polymorphism; CEPH = Centre d’Etude du Polymorphisme Humain (HapMap European ancestry population); YRI = Yoruba in Ibadan, Nigeria (HapMap African ancestry population); – = not applicable.
We explored this further by comparing allele frequencies in African American patients with SLE stratified by anti-Sm antibodies with allele frequencies found in the HapMap YRI and CEPH populations, which represent reference African and European populations, respectively (24). The rs4963128 T allele frequency in anti-Sm–positive African American patients with SLE was very similar to the YRI HapMap population. In African American patients without anti-Sm antibodies, the T allele frequency was strikingly similar to that in the CEPH HapMap population. Anti-Sm antibodies were 3.1 times more frequent in African American patients with SLE (40.7%) than in European American patients with SLE (13.0%) in our cohort, which is very similar to previously published data (25). The rs4963128 T allele was present at rates which could be expected due to ~20% European admixture in the controls and anti-Sm–positive patients (P = 0.18 and P = 0.92, respectively, for departure from expected allele frequencies assuming 20% European admixture using HapMap reference population data). The T allele was vastly underrepresented in the anti-Sm–negative patients compared with what would be expected due to European admixture in African American populations in the US (P = 1.0 × 10−4 for deviation from expected allele frequencies assuming 20% European admixture). Greater than 50% European admixture at this locus would be necessary to result in a nonsignificant departure from expected allele frequencies based on HapMap reference populations. The controls are derived from the same regional population as the cases, and an abnormally high proportion of overall European admixture in the African American patients with SLE in our cohort is not supported by either our regional controls or by genotyping data at other loci in these subjects (26,27).
Association of the rs702966 C allele with increased serum levels of IFNα in anti-Sm–negative African American patients with SLE with anti-dsDNA antibodies
Given the findings described above regarding allele frequencies in the African American patients stratified by the presence or absence of anti-Sm antibodies, we reanalyzed serum IFNα data in the anti-Sm–negative patients to determine whether the pattern of serum IFNα activity related to anti-dsDNA/rs702966 C would be present in this subgroup of African American patients with SLE. The rs702966 C allele was associated with higher serum levels of IFNα in anti-Sm–negative African American patients with SLE who had anti-dsDNA antibodies (P = 0.0021) (Data are available from the corresponding author upon request). This pattern of association, which was present in European American and Hispanic American patients with SLE, is also present in African American patients with SLE who lack anti-Sm antibodies. Inclusion of the anti-Sm–negative African American patients in a combined analysis with European American and Hispanic American patients showed that across the 3 ancestral backgrounds the rs702966 CC genotype is associated with increased serum levels of IFNα in subjects with anti-dsDNA antibodies. Anti-dsDNA–positive patients with the CC genotype had significantly higher serum levels of IFNα than did anti-dsDNA–positive patients with the CG or GG genotype (P = 1.0 × 10−6). There was no significant difference in serum levels of IFNα between anti-dsDNA–negative patients with the CC genotype and those with the CG or GG genotype (P = 0.76) (Figure 4).
Figure 4.

Combined analysis of serum interferon-α (IFNα) activity in anti-Sm–negative African American patients with SLE and European American and Hispanic American patients with SLE, stratified by the presence or absence of anti-dsDNA and rs702966 genotype. Rare minor allele homozygotes (GG) were combined with heterozygotes (CG) for analysis. The y-axis shows the serum IFNα score (see Patients and Methods for details). Bars show the median and interquartile range. P values were calculated using the Mann-Whitney U test.
The combination of IRF5 and IRF7/PHRF1 variants results in higher serum levels of IFNα in European American and Hispanic American patients with anti-dsDNA antibodies
We have previously shown that the SLE risk haplotype of IRF5 is associated with higher serum levels of IFNα in the presence of anti-dsDNA and anti-RBP antibodies in subjects of European and Hispanic ancestry (15). Given that the interaction of the IRF7 rs702966 SNP and anti-dsDNA has a similar effect on serum levels of IFNα, and the potential for functional overlap between IRF5 and IRF7 in the IFNα pathway, we examined our data for potential interactions between IRF5 and IRF7. Using logistic regression analysis, we did not observe any evidence of a statistically significant effect of the interaction between haplotype-tagging IRF5 SNPs and the IRF7 rs702966 C allele on the anti-dsDNA antibody trait (data not shown). When serum IFNα was analyzed in European American and Hispanic American patients with respect to both the IRF5 SLE risk haplotype (marked by the rs10488631 C allele) and the IRF7 rs702966 C allele, there was evidence of increasing serum levels of IFNα in subjects with anti-dsDNA antibodies as the numbers of risk alleles at these SLE risk loci increased (Figure 5). In the anti-dsDNA–positive subjects, rs702966 CC genotype was associated with higher serum levels of IFNα within IRF5 genotype subgroups. These data suggest an anti-dsDNA–dependent influence of IRF7/PHRF1 SNPs on serum IFNα, which is independent of IRF5 genotype.
Figure 5.

Serum interferon-α (IFNα) activity in European American and Hispanic American patients with SLE, stratified by genotype at IRF7 rs702966 and IRF5 SLE risk haplotype. For IRF7 rs702966, G–represents the CG and GG genotypes, and for the IRF5 haplotype, C– represents risk haplotype carriers or homozygous risk genotypes, and TT represents the homozygous nonrisk genotype. Patient distribution along the x-axis was performed using sequential stratification in the following order: anti-dsDNA, IRF5 genotype, and IRF7 rs702966 genotype. The y-axis shows the serum IFNα score (see Patients and Methods for details). Bars show the median and interquartile range. P values were calculated using the Mann-Whitney U test.
DISCUSSION
IRF7 plays an important physiologic role in viral infections, stimulating transcription of IFNα and IFNα-related genes downstream of endosomal TLR activation (17). SLE-associated autoantibodies can induce IFNα production in dendritic cells in culture (16), and it is likely that nucleic acid contained within immune complexes formed from these antibodies can activate the endosomal TLR pathway (28). These findings suggest a way in which normal viral immunity may be subverted by pathogenic immune complexes in SLE. SLE-associated autoantibodies are not sufficient for high serum levels of IFNα in vivo, however. We have previously shown that healthy mothers of neonatal lupus patients who had high titers of anti-Ro and/or anti-La autoantibodies did not have high serum levels of IFNα (29). High serum levels of IFNα were common in mothers of neonatal lupus patients who had SLE or Sjögren’s syndrome and either anti-Ro or anti-La antibodies (29). These findings suggest that background factors associated with autoimmune disease are also important to the relationship between SLE-associated autoantibodies and serum IFNα. This would also suggest that a “second hit” may be necessary in the endosomal TLR pathway to cause dysregulation of serum IFNα in vivo.
Previous work with IRF5 variants in SLE has supported this concept, since the SLE risk variant of IRF5 was associated with higher serum levels of IFNα only in the presence of particular autoantibodies (15). Thus, the SLE risk variant of IRF5 is likely one such background factor modulating the impact of SLE-associated autoantibodies on serum IFNα activity. In the present study, a similar model was observed, in which particular IRF7/PHRF1 variants were associated with increased serum levels of IFNα only in the setting of SLE-associated autoantibodies. We demonstrated differences in autoantibody associations by ancestry; however, in each ancestral background, a similar overall autoantibody–SNP–IFNα interaction was observed. The SNPs are genetically associated with the particular autoantibody that is required for higher serum IFNα within the SLE cohort, suggesting that these autoantibody–IRF7/PHRF1 SNP interactions play a causal role in disease. Functional elements in the IRF7 gene are not well mapped currently, but we hypothesize that the SNPs found to be associated with autoantibodies and serum IFNα in this study will be linked to causal functional elements in IRF7 that result in gain of function and increased IFNα production following endosomal TLR stimulation. It is also possible that functional variation in the PHRF1 gene results in the associations we have observed; however, PHRF1 gene function is not well described currently. The striking parallels we observe between the present study and our previous work with the IRF5 locus would make IRF7 a more likely causal candidate gene. Future large-scale mapping and sequencing in this region will help clarify the true causal genomic elements.
The difference in association we observed between ethnic groups was highly dependent on autoantibody profile. While the interaction between anti-dsDNA and the rs702966 C allele was observed to some degree in all backgrounds, a strong relationship between anti-Sm antibodies and the rs4963128 SNP was demonstrated in subjects of African American ancestry. The striking differences observed within the African American cohort separated by the presence or absence of anti-Sm antibodies suggest 2 independent patterns of association with IRF7/PHRF1 variants, which cannot be explained by European admixture at the locus. It is possible that the rs4963128 T allele marks a particular element in African-derived chromosomes that associates with anti-Sm antibodies and is not present in the other ancestral backgrounds. We hypothesize that the rs702966 C allele and elements in linkage with it may function similarly across all ancestral backgrounds, although the effect of the anti-Sm–rs4963128 T allele interaction on serum levels of IFNα in African Americans is independent of the effect at the rs702966 C allele.
Data from studies by our group and others support a role of IFNα in the initiation of SLE (3,6,7,9). Our findings suggest that particular SLE-associated autoantibody–SNP interactions result in higher serum levels of IFNα in SLE patients. SLE-associated autoantibodies are known to arise in the preclinical phase of SLE, often years before clinical diagnosis (30). Our data would fit a model of SLE pathogenesis in which SLE-associated autoantibodies that develop in the preclinical phase of disease interact with genetic variation within the endosomal TLR pathway (IRF7 and IRF5) to result in increased IFNα production and subsequent risk of SLE. It is also possible that the particular variants of IRF7/PHRF1 predispose to the generation of particular autoantibodies. If this is the case, then IRF7/PHRF1 variants could function as a positive feedback loop, first predisposing to specific autoantibodies, which then stimulate the TLR pathway and result in greater IFNα production in the setting of the same variant. These data provide biologic relevance for the IRF7/PHRF1 locus at the protein level in SLE, and support strong relevance of this gene within the IFNα pathway in SLE in vivo.
Acknowledgments
Dr. Utset’s work was supported by the Lupus Clinical Trials Consortium. Dr. Niewold’s work was supported by the NIH (National Institute of Allergy and Infectious Diseases grants K08-AI-083790 and AI-071651; and Clinical and Translational Science Award Consortium grants UL1-RR-024999 and RR-025000-02), and the Arthritis National Research Foundation Eng Tan Scholar Award.
REFERENCES
- 1.Harley JB, Kelly JA, Kaufman KM. Unraveling the genetics of systemic lupus erythematosus. Springer Semin Immunopathol. 2006;28:119–30. doi: 10.1007/s00281-006-0040-5. [DOI] [PubMed] [Google Scholar]
- 2.Lopez P, Mozo L, Gutierrez C, Suarez A. Epidemiology of systemic lupus erythematosus in a northern Spanish population: gender and age influence on immunological features. Lupus. 2003;12:860–5. doi: 10.1191/0961203303lu469xx. [DOI] [PubMed] [Google Scholar]
- 3.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFNα in systemic lupus erythematosus. Science. 2001;294:1540–3. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 4.Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. 1979;301:5–8. doi: 10.1056/NEJM197907053010102. [DOI] [PubMed] [Google Scholar]
- 5.Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-α pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005;52:1491–503. doi: 10.1002/art.21031. [DOI] [PubMed] [Google Scholar]
- 6.Ronnblom LE, Alm GV, Oberg KE. Possible induction of systemic lupus erythematosus by interferon-α treatment in a patient with a malignant carcinoid tumour. J Intern Med. 1990;227:207–10. doi: 10.1111/j.1365-2796.1990.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 7.Niewold TB, Swedler WI. Systemic lupus erythematosus arising during interferon-α therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin Rheumatol. 2005;24:178–81. doi: 10.1007/s10067-004-1024-2. [DOI] [PubMed] [Google Scholar]
- 8.Niewold TB. Interferon α-induced lupus: proof of principle. J Clin Rheumatol. 2008;14:131–2. doi: 10.1097/RHU.0b013e318177627d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-α activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8:492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niewold TB, Adler JE, Glenn SB, Lehman TJ, Harley JB, Crow MK. Age- and sex-related patterns of serum interferon-α activity in lupus families. Arthritis Rheum. 2008;58:2113–9. doi: 10.1002/art.23619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnes BJ, Moore PA, Pitha PM. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon α genes. J Biol Chem. 2001;276:23382–90. doi: 10.1074/jbc.M101216200. [DOI] [PubMed] [Google Scholar]
- 12.Sigurdsson S, Nordmark G, Goring HH, Lindroos K, Wiman AC, Sturfelt G, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76:528–37. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graham RR, Kyogoku C, Sigurdsson S, Vlasova IA, Davies LR, Baechler EC, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci U S A. 2007;104:6758–63. doi: 10.1073/pnas.0701266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38:550–5. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 15.Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-α activity in systemic lupus erythematosus patients. Arthritis Rheum. 2008;58:2481–2487. doi: 10.1002/art.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lovgren T, Eloranta ML, Bave U, Alm GV, Ronnblom L. Induction of interferon-α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004;50:1861–72. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 17.Barnes BJ, Richards J, Mancl M, Hanash S, Beretta L, Pitha PM. Global and distinct targets of IRF-5 and IRF-7 during innate response to viral infection. J Biol Chem. 2004;279:45194–207. doi: 10.1074/jbc.M400726200. [DOI] [PubMed] [Google Scholar]
- 18.Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–10. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bienz M. The PHD finger, a nuclear protein-interaction domain. Trends Biochem Sci. 2006;31:35–40. doi: 10.1016/j.tibs.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Hua J, Kirou K, Lee C, Crow MK. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti–RNA binding protein autoantibodies. Arthritis Rheum. 2006;54:1906–16. doi: 10.1002/art.21890. [DOI] [PubMed] [Google Scholar]
- 21.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006;38:209–13. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 22.Purcell S, Cherny SS, Sham PC. Genetic power calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149–50. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 23.Breslow NE. Statistics in epidemiology: the case-control study. J Am Stat Assoc. 1996;91:14–28. doi: 10.1080/01621459.1996.10476660. [DOI] [PubMed] [Google Scholar]
- 24.International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramos PS, Kelly JA, Gray-McGuire C, Bruner GR, Leiran AN, Meyer CM, et al. Familial aggregation and linkage analysis of autoantibody traits in pedigrees multiplex for systemic lupus erythematosus. Genes Immun. 2006;7:417–32. doi: 10.1038/sj.gene.6364316. [DOI] [PubMed] [Google Scholar]
- 26.Kariuki SN, Kirou KA, MacDermott EJ, Barillas-Arias L, Crow MK, Niewold TB. Cutting edge: autoimmune disease risk variant of STAT4 confers increased sensitivity to IFN-α in lupus patients in vivo. J Immunol. 2009;182:34–8. doi: 10.4049/jimmunol.182.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kariuki SN, Moore JG, Kirou KA, Crow MK, Utset TO, Niewold TB. Age-and gender-specific modulation of serum osteopontin and interferon α by osteopontin genotype in systemic lupus erythematosus. Genes Immun. 2009;10:487–94. doi: 10.1038/gene.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lovgren T, Eloranta ML, Kastner B, Wahren-Herlenius M, Alm GV, Ronnblom L. Induction of interferon-α by immune complexes or liposomes containing systemic lupus erythematosus autoantigen– and Sjögren’s syndrome autoantigen–associated RNA. Arthritis Rheum. 2006;54:1917–27. doi: 10.1002/art.21893. [DOI] [PubMed] [Google Scholar]
- 29.Niewold TB, Rivera TL, Buyon JP, Crow MK. Serum type I interferon activity is dependent on maternal diagnosis in anti-SSA/Ro–positive mothers of children with neonatal lupus. Arthritis Rheum. 2008;58:541–6. doi: 10.1002/art.23191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–33. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]