

Summary
Polyglutamine expansions within different proteins are associated with nine different neurodegenerative diseases. There is growing interest in understanding the roles of flanking sequences from disease-relevant proteins on the intrinsic conformational and aggregation properties of polyglutamine. We report results, from atomistic simulations and circular dichroism experiments that quantify the effect of the N-terminal 17-residue (Nt17) segment of the huntingtin protein on polyglutamine conformations and intermolecular interactions. We show that the Nt17 segment and polyglutamine domains become increasingly disordered as polyglutamine length (N) increases in Nt17-QN constructs. Hydrophobic groups within Nt17 become sequestered in intramolecular, inter-domain interfaces. We also show that the Nt17 segment suppresses the intrinsic propensity of polyglutamine aggregation. This inhibition arises from the incipient micellar structures adopted by monomeric forms of the peptides with Nt17 segments. The degree of intermolecular association increases with increasing polyglutamine length and is governed mainly by associations between polyglutamine domains. Comparative analysis of intermolecular associations for different polyglutamine containing constructs leads to clearer interpretations of recently published experimental data. Our results suggest a framework for fibril formation, and identify roles for flanking sequences in modulating polyglutamine aggregation.
Keywords: Polyglutamine, Huntingtin, N-terminal domain, Simulations, Aggregation
Introduction
There are at least nine different neurodegenerative diseases that are associated with the expansion of polyglutamine domains within specific disease proteins.1 In these diseases, the ages-of-onset and severities of symptoms are inversely correlated with polyglutamine expansion lengths.2 The sequences flanking the polyglutamine expansions in host proteins share no similarity, although there is an apparent preference for low sequence complexity (see Table S1 in the supplementary material for details).
Huntingtin is the protein that is involved in Huntington's disease (HD). The gene's first exon (exon1) contains the CAG repeat that leads to the polyglutamine expansion. Of particular interest is the 17-residue stretch MATLEKLMKAFESLKSF that is directly N-terminal to the polyglutamine expansion. We refer to this stretch as the Nt17 segment. Short N-terminal sequence constructs encompassing exon1 have been used to demonstrate several specific protein-protein interactions in Drosophila3 and some of the interaction partners were confirmed to be genetic modifiers of the disease phenotype. N-terminal sequence constructs of varying length mediate the association with mitochondria and a pathogenic mechanism related to axonal transport and Ca2+ homeostasis was suggested.4; 5 The presence of the Nt17 segment alters the aggregation behavior of fragments with polyglutamine expansions in vivo and causes a relocalization from nuclei to mitochondria.4 Consistent with the idea of Nt17 acting as a cytosolic retention signal, SUMOylation of lysine residues within the Nt17 stretch promotes both nuclear localization and subsequent interference with transcriptional regulation when compared to non-SUMOylated substrates.6 All of these results suggest that sequence context plays an important role in modulating the interactions and toxicity associated with fragments of exon1 of huntingtin. Additional evidence for the influence of sequence context on polyglutamine conformations and aggregation comes from studies of proteins implicated in different spinocerebellar ataxias. As an example, in spinocerebellar ataxia 1, the AXH domain of ataxin-1 enhances the aggregation of mutant forms of this protein even though the polyglutamine region lies outside the AXH domain.7
Polyglutamine molecules are intrinsically disordered8; 9; 10 and long polyglutamine domains can have destabilizing effects when tethered to a stable protein.11; 12 This in turn makes proteins containing long polyglutamine expansions susceptible to proteolysis.13 Products of proteolysis can retain polyglutamine stretches because these regions are resistant to degradation.14; 15; 16; 17 Proteolytic analysis of mutant huntingtin has revealed that proteolytic products form a heterogeneous population of peptides of differing length.18; 19 As a starting point, we focus on the effects of the Nt17 segment on polyglutamine conformation and spontaneous intermolecular association. This sequence is likely to be a part of disease-relevant constructs because it is directly N-terminal to the polyglutamine expansion.
Considerable effort has been dedicated to biophysical characterization of synthetic, homopolymeric, constructs of polyglutamine.8; 20; 21; 22; 23; 24; 25; 26; 27; 28; 29; 30; 31 Systematic biophysical studies on the effects of flanking sequences on polyglutamine conformation and aggregation are relatively recent.7; 12; 32; 33; 34; 35; 36; 37. Thakur et al. assessed the effect of the Nt17 segment on aggregation rates for a range of polyglutamine containing peptides. These rates were measured by quantifying the rate of loss of soluble material. They compared these rates to those for constructs of the type K2-QN-K2 and noted that they were significantly higher with the Nt17 segment present. While K2-QN-K2 constructs did not form detectable intermediates, constructs with the Nt17 segment formed distinct oligomeric intermediates that are spherical in nature and were observed in electron micrographs taken at early time points. Application of a thermodynamic nucleus model38 to analyze aggregation kinetics yielded an apparent nucleus size of -1 suggesting that the aggregation of Nt17-containing constructs shows marked deviation from the homogenous nucleation process assumed to be valid for the aggregation of K2-QN-K2 constructs.24 Combined with mutational analysis, Thakur et al. interpreted their results to implicate direct interactions between Nt17 segments as the reason for the enhancement of aggregation rates in constructs with this segment.
Information regarding comparative rates for the loss of soluble material provides an important constraint for understanding how the Nt17 segment modulates the aggregation properties of polyglutamine expansions in mutant huntingtin. However, such information is inherently incomplete because it does not provide precise structural information regarding the modulation of polyglutamine conformations and interactions by the Nt17 segment. In this work, we report the use of atomistic simulations as the main tool to answer several questions regarding the effects of the Nt17 segment on intrinsic properties of constructs of the type Ac-Nt17-QN-Nme, where Ac denotes the acetyl group, and Nme denotes the N-methylamide group. The questions of interest are as follows: How are secondary structural preferences and other conformational characteristics of Nt17 and polyglutamine domains modulated as a function of polyglutamine length in Ac-Nt17-QN-Nme constructs? What is the nature of intramolecular interfaces that form between the Nt17 and polyglutamine domains? How does dimerization for Ac-Nt17-QN-Nme constructs differ from that of Ac-QN-Nme and Ac-K2-QN-K2-Nme constructs, respectively? Finally, are intermolecular associations in Ac-Nt17-QN-Nme constructs driven mainly by contacts between the hydrophobic groups of the Nt17 segment, by the polyglutamine domain alone, or both? In answering these questions, we compare the interpretations from our analysis to those presented by Thakur et al.37 We conclude with a discussion that provides a mechanism for the transitions that might connect our simulation results on monomer-dimer equilibria to fibril formation.
Results
Secondary structure propensities of Nt17 and polyglutamine as a function of polyglutamine length
Recent experiments and simulations suggest that the Nt17 peptide might form transient helical segments in aqueous solutions.37; 39 However, secondary structure propensities of the polyglutamine domain have yet to be determined. We investigated the dependence of α-helical propensities on polyglutamine lengths in Ac-Nt17-QN-Nme constructs. Panel A of Figure 1 shows simulation results for the temperature dependence of helical propensities within Nt17 segments for a series of polyglutamine lengths. For constructs without polyglutamine attachments, the highest helical content of 34% is achieved at the lowest simulated temperature (298 K). The intrinsic helical content of the Nt17 segment decreases with increasing temperature and at the highest simulated temperature (385 K) it drops below 1%. In the presence of polyglutamine expansions, it decreases as N increases. Panel B shows that for N=5 there is an increase in the α-helical content within the polyglutamine stretch. However, this induced helical content decreases with increasing N and drops below 5% for N ≥ 25 for all simulated temperatures. Therefore, for large enough N, entire Ac-Nt17-QN-Nme constructs encompassing the Nt17 and polyglutamine segments become disordered as assessed in terms of helical content and this disorder prevails even at low temperatures. The two domains are clearly coupled and the coupling ensures that the intrinsic preferences of the polyglutamine domain prevail over the entire peptide construct as N increases.
Figure 1. Fractional α-helical contents for different Nt17 constructs.

Helical contents were computed by counting the fraction of conformations that had at least seven consecutive residues (two helical turns) with (ϕ,ψ)-angles in the helical basin of a Ramachandran map. Panel A: Shows the temperature dependent fractional helical contents within the Nt17 domain in Ac-Nt17-QN-Nme constructs. Panel B: Shows the temperature dependent fractional helical contents within the polyglutamine domains in Ac-Nt17-QN-Nme constructs. In the interest of clarity, we left out the error bars from panels A and B. These plots, with error bars, have been reproduced in Figure S1 to establish the statistical significance of the results.
The loss of helical content is not a rigorous indicator of increasing disorder. Instead, it could signal an “order-to-order” transition. To interrogate this possibility, we computed the propensity for populating β-strand segments within the Nt17 and polyglutamine domains as a function of temperature. These results are shown in panels A and B of Figure S2 of the supplementary material. Neither panel shows significant β-content or discernible trends with polyglutamine length. The results shown in Figure S1 are consistent with published data,26; 31; 40 which demonstrate that monomeric polyglutamine domains have low probabilities of forming structures with high β-content in aqueous solutions at physiologically relevant temperatures. The Nt17 segment does not alter this intrinsic behavior thereby contradicting recent speculation that there might be an increase in β-content as polyglutamine lengths increase in Nt17-QN constructs.39
The results presented above provide support for the hypothesis that Ac-Nt17-QN-Nme peptides become disordered as polyglutamine lengths increase. Before moving on to further analysis of conformational characteristics and intermolecular associations of these constructs, we present an experimental assessment of α-helical content. We performed UV-CD measurements on the Nt17 peptide and compared the estimates obtained from these experiments to those presented in panel A of Figure 1. Panel A of Figure 2 shows UV-CD spectra for the Nt17 peptide in 0%, 5% and 20% (v/v) trifluoroethanol (TFE), respectively. Characteristic helical spectra are obtained as the TFE content is increased. This suggests that the intrinsic helicity in the absence of cosolutes of Nt17 can be unmasked through appropriate titration of cosolutes that increase or decrease the measurable helical content. We assumed a two-state model and used TFE / urea titrations to obtain the baseline molar ellipticities at 222 nm corresponding to 100% and 0% helical contents, respectively. Following the approach of Scholtz et al.,41 we assumed the helical content to be proportional to the molar ellipticity at 222 nm ([θ]222). The baseline ellipticity for 100% helix was taken to be the value of [θ]222 in 50% (v/v) TFE based on the titration shown in panel B of Figure 2. Similarly, the baseline value for [θ]222 corresponding to 0% helix was taken to be the one measured in 6 M urea (see panel B of Figure 2). These values are −3.3×104 deg-cm2/dmol for and 2.0×103 deg-cm2/dmol for , respectively. In the absence of cosolutes, the value of [θ]222 is −9.8×103 deg-cm2/dmol. The intrinsic helical content was estimated as to yield a value of approximately 34% at 298 K. As an additional test, we estimated the value of for a peptide of finite chain length using the value for an infinitely long helix and the formula41 where N denotes the number of peptide residues. Setting N=17, we obtain the estimated value for to be −3.41×104 deg-cm2/dmol, which is similar to the value obtained in the presence of 50% (v/v) of TFE. This justifies the use of the TFE titration to estimate . We conclude that 34% is a reasonable estimate of the helical content for the Nt17 peptide in aqueous solution at 298K. This analysis demonstrates that the criterion (see caption to Figure 1) used to analyze helical content from simulation results is reasonable and yields quantitative agreement with estimates obtained from analysis of experimental data.
Figure 2. UV-CD data for the Nt17 peptide.

Panel A: UV-CD CD data for Nt17 in two different amounts of TFE. Panel B: The molar ellipticity at [θ]222 25°C plotted against % TFE (filled circles) and urea concentration (filled squares). The dashed red line corresponds to the estimate for the helical content of Nt17 at 25°C in native buffer. The value of approximately 34% is identified from the intercept with the ordinate on the right.
Polymeric properties of Ac-Nt17-QN-Nme constructs
Previous studies showed that monomeric forms of polyglutamine molecules form collapsed, disordered globules in aqueous solutions at room temperature.10; 28 For the ABSINTH implicit solvation model used in this work, it was shown that Ac-QN-Nme constructs undergo well defined globule-to-coil transitions as temperature increases.31 Panel A of Figure 3 shows comparative analysis of the normalized average radius of gyration for the peptides Ac-QN-Nme and Ac-Nt17-QN-Nme plotted against increasing temperature for different polyglutamine lengths. A general comparison of simulation results shows that peptides with and without the Nt17 segment show similar responses to changes in temperature, with all peptides showing a convergent theta temperature of ca. 385 K. The theta temperature refers to the temperature at which chain-chain and chain-solvent interactions are counterbalanced and can be detected by the presence of a point along the coil-to-globule transition curve where the normalized 〈Rg〉 values coincide for different chain lengths. It appears that the rate of change of 〈Rg〉 with increasing shows stronger dependence on chain length than the precise nature of the polypeptide sequence. However, it is also true that results for Ac-Nt17-Q35-Nme and Ac-Q35-Nme are indistinguishable indicating that past a certain value of N, the effects of overall peptide length and polyglutamine length are not independent of one another.
Figure 3. Polymeric properties of the Nt17 segment analyzed using different measures.

Panel A: Coil-to-globule transitions of Ac-Nt17-QN-Nme constructs compared to similar profiles for Ac-QN-Nme constructs. The plot shows normalized ensemble-averaged radii of gyration (along the ordinate) where and the abscissa denotes simulation temperature. Transitions are generally sharper and more pronounced for the Ac-Nt17-QN-Nme constructs because they are longer than the homopolymeric constructs. Panel B: Ensemble averaged radius of gyration (〈Rg〉) of the Nt17 segment within Ac-Nt17-QN-Nme constructs plotted against the length of the polyglutamine domain. The dashed line represents the radius of gyration of the Nt17 segment in a regular, uninterrupted, ideal a-helix conformation (8.83 Å); 〈Rg〉 values are shown for Nt17-QN constructs at two different temperatures, 305 K (black diamonds) and 385K (gray squares). Note that at the high temperature the Nt17 segment is maximally expanded for all polyglutamine lengths because the Ac-Nt17-QN-Nme constructs sample canonical random coil ensembles at this temperature.
Data based on Förster resonance energy transfer experiments from Thakur et al.42 showed an N-dependent expansion of Nt17. In panel B of Figure 3 we plot the average radius of gyration (〈Rg〉) of the Nt17 segment at 298 K and 385 K against N, the length of the polyglutamine domain. At the lower temperature, 〈Rg〉 increases systematically from 7.5Å to 9.5 Å for values of N between 5 and 25. This translates to a 50% decrease in the average density of the Nt17 segment. For polyglutamine lengths of N ≥ 25, the value of 〈Rg〉 for the Nt17 segment converges, within error, to a value of ≈ 9.3 Å. For small N, the 〈Rg〉 values are below those obtained for a straight α-helix for the Ac-Nt17-Nme peptide. The helix-content observed in Figure 1 does not result from the Nt17 segment forming a straight α-helix; rather it results from a population of transient, short helical segments, longer segments with disordered ends, and occasionally, bent helices.39 Expansion of the Nt17 segment with increased polyglutamine length correlates with the loss of α-helix content seen in panel A of Figure 1, where it is shown that the helical content within the NT17 and polyglutamine domains fall below 5% for all N ≥ 25. In fact, both 〈Rg〉 of the Nt17 segment and helical contents for the entire Ac-Nt17-QN-Nme constructs cease to show significant changes as N increases (for T=298 K). The observation of Nt17 expansion as a function of increasing N – for N ≤ 25 – is the result of an intramolecular, inter-domain interface that forms between the Nt17 and polyglutamine domains and this is illustrated next.
Figure 4 shows the results of cluster analysis, where each structure represents the central structure taken from the most populated cluster for each peptide construct at 305K. Statistics such as the number of clusters and the fraction of clusters required to account for 95% of the population are summarized in Table S2 of the supplementary material. For Ac-Nt17-Q5-Nme, Figure 4 depicts the collapse of the Nt17 segment onto itself, which allows the hydrophobic residues to form a semi-accessible core and an interface with the glutamine residues. The primary sequence constrains the hydrophobic residues to remain spatially proximal to the charged residues. As the length of the polyglutamine domain increases, the representative structures illustrate the expansion of the Nt17 segment and the partial sequestration of hydrophobic groups in an inter-domain interface.
Figure 4. Central structures of the most populated cluster for Ac-Nt17-QN-Nme monomers.
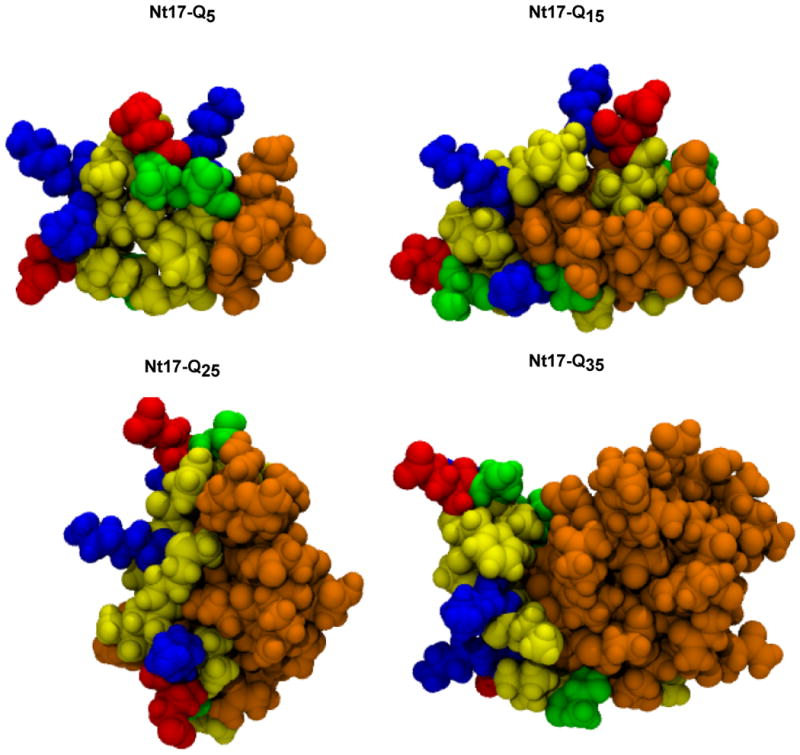
Graphics were generated using VMD.58 Atoms are drawn in space-filling representation and colored according to residue type; positively charged (blue), negatively charged (red), polar excluding glutamine (green), hydrophobic (yellow), and glutamine (orange).
Characteristics of the intramolecular, inter-domain interfaces in Ac-Nt17-QN-Nme peptides
In this section, we place the qualitative observations of Figure 4 on a quantitative footing. Panel A in Figure 5 shows the enhancement / depletion of different types of inter-residue intramolecular contacts for different polyglutamine lengths in Ac-Nt17-QN-Nme constructs at 305K. As discussed in Methods, we first computed the probabilities of realizing different types of inter-residue contacts and then calibrated them vis-à-vis a random mixture model that accounts for the combinatorial contributions to the observed contacts that arise from the nature of the sequence constructs. If the values along the ordinate of panel A of Figure 5 are positive, then these contacts are preferentially enhanced within the conformational ensemble and, conversely, negative values indicate depletion. Based on these results, we identify three defining features of intramolecular interactions within Ac-Nt17-QN-Nme peptides: (i) Contacts within the Nt17 segment become depleted as N increases, with only the hydrophobic groups showing persistent, albeit weak clustering for large N. This is consistent with the expansion of the Nt17 segment quantified above. (ii) The inter-domain interface causes a clear spatial separation between the glutamine residues and the charged / polar groups within the Nt17 segment. This spatial separation becomes pronounced as N increases. (iii) Contacts between glutamine residues are enhanced as polyglutamine lengths increase.
Figure 5. Quantitative analysis of intramolecular interfaces in Ac-Nt17-QN-Nme constructs.

Panel A shows the length dependence of the enhancement / depletion in the probability of observing intramolecular inter-residue contacts between specific residues classes in monomers of Ac-Nt17-QN-Nme constructs. Results are shown for 305 K. The definition of a contact and the calculation of a random prior model are described in Methods. Panels B and C show the volumetric, C1(T), and surface energy contributions C2(T), for polyglutamine domains in Ac-Nt17-QN-Nme and homopolymeric constructs, respectively. The energetic terms were obtained from fits to the linear regression equation shown in the text. Fits were obtained using results for chain lengths N=(15, 20, 25, 30, 35, 45). The quality of the fits is assessed through the plotted errors bars, which result from a propagation of error though the fitting procedure. Only chains of length N≥15 are used, as shorter chains are not likely to have a volumetric contribution to the potential energy.
Homopolymeric polyglutamine forms collapsed, disordered globules in aqueous solutions.10; 28; 31 Collapse achieves the dual effect of minimizing the unfavorable surface energy with the surrounding solvent while maximizing favorable volumetric intramolecular inter-glutamine contacts. The analysis of contact probabilities does not reveal the sequestration of glutamine residues within an inter-domain interface, which should cause a reduction of the unfavorable interface of the polyglutamine domain with the surrounding solvent. We can ask if energetic contributions to the effective “surface tension” and volumetric interactions are influenced by the presence of the Nt17 segment, especially when compared to homopolymeric constructs. We fit the temperature dependence of the ensemble-averaged potential energies using: . For a given conformation of Ac-Nt17-QN-Nme, Utotal denotes the total potential energy of the system including the milieu of counter- and co-ions; conversely, UNt17 is the N-independent total potential energy for Ac-Nt17-Nme at the same temperature and solution conditions. A similar decomposition was computed for the Ac-QN-Nme peptides; note that for this system UNt17 is ignored. Regression yields the volumetric term C1(T) and the surface energy term C2(T). The results are shown in panels B and C of Figure 5. The presence of the Nt17 segment causes a loss of favorable intra-polyglutamine domain self-interactions worth ∼5 kcal/mol with respect to the homopolymeric constructs (panel B). This provides quantitative evidence that the globules of polyglutamine domains in Ac-Nt17-QN-Nme peptides have reduced self-interactions compared to homopolymeric Ac-QN-Nme constructs. In Figure 3 we showed that the driving force for collapse depends only weakly on the details of the peptide sequence. This means that the reduction in the favorability of inter-glutamine contacts does not compromise the ability of Ac-Nt17-QN-Nme peptides to form collapsed structures. This is explained by results in panel C of Figure 5 that show significant diminution in “surface tension” for the polyglutamine domain in Ac-Nt17-QN-Nme peptides for T < 360. This diminution arises from the sequestration of glutamine residues in inter-domain interfaces. Evidence for this assertion comes from the temperature dependence of the C2(T) term. As T increases, the difference between the C2(T) term for polyglutamine domains in the two constructs decreases. This is because the inter-domain interface loosens with increasing T and the polyglutamine domain in Ac-Nt17-QN-Nme peptides behaves like it would in the homopolymeric constructs.
For temperatures below 350 K, peptides prefer to form collapsed structures indicating a preference for self-interactions to interactions with the surrounding solvent. Previous work showed that for these temperatures the homopolymeric constructs also form stable dimers because dimerization leads to further self-interactions and minimization of unfavorable interactions with the solvent.31; 40 Based on the preceding paragraph, we expect that the formation of favorable intramolecular, inter-domain interfaces cause a diminution in the spontaneity of dimer formation for Ac-Nt17-QN-Nme peptides vis-à-vis their homopolymeric counterparts. We test this hypothesis next.
Comparative analysis of dimer formation
We quantified the thermodynamics of intermolecular associations for three sequence constructs, viz. Ac-QN-Nme, Ac-Nt17-QN-Nme, and Ac-K2-QN-K2-Nme, respectively. We performed simulations of dimer formation by confining the peptides to a spherical droplet of radius 200Å. For each peptide construct and simulation temperature we quantified the probability of association, which refers to the probability of finding a pair of molecules within a volume that is less than 0.02% of the total available simulation volume. These probabilities were obtained from analysis of cumulative distribution functions for intermolecular separations of the type shown in Figure S3. The higher this value, the stronger the intermolecular associations. The results from our calculations are shown in Figure 6.
Figure 6. Temperature dependent probabilities of intermolecular associations.

Results were computed as discussed in the results section using the CDFs shown in Figure S3. Panels A, B, and C show simulation results for Ac-QN-Nme, Ac-Nt17-QN-Nme, and Ac-K2-QN-K2-Nme constructs, respectively.
Panel A of Figure 6 shows the temperature dependence of the probability of association for the homopolymeric constructs, Ac-QN-Nme. At the lowest temperature (298 K), peptides of length N ≥ 15 are strongly associative. For peptides of this length, there are numerous ways of realizing complementary intermolecular interfaces. The size of the interface and the number of ways of realizing these contacts depends on chain length. This is reflected in the temperature dependence of the probability of association shown in panel A of Figure 6. As temperature increases, the longer chains remain more associative when compared to the shorter globule-forming chains (N ≥ 15). Short chains such as Ac-Q5-Nme have negligibly small probabilities of association across the entire temperature range.
Panel B of Figure 6 shows the same results for the Ac-Nt17-QN-Nme constructs. Two features stand out when comparing these results to those in panel A. First, even at the lowest temperature, there is a more pronounced length dependence whereby longer chains are always more associative than shorter chains. This trend is preserved in the temperature dependence of the probability of association. Clearly, longer polyglutamine domains are needed to promote favorable intermolecular associations suggesting that intermolecular interfaces are promoted primarily by contacts that involve the polyglutamine domains. Secondly, we note that for a given polyglutamine length and temperature (as long as N ≥ 15), the probability of association is always higher for the homopolymeric constructs (panel A of Figure 6) when compared to the Ac-Nt17-QN-Nme constructs. This suggests that the Nt17 segment causes a diminution in the probability of intermolecular associations. We explain this finding by analyzing the types of dimer structures that are realized for the Ac-Nt17-QN-Nme constructs.
Figure 7 shows representative structures extracted from the dominant cluster obtained for each Ac-Nt17-QN-Nme construct at 298 K. The polar, albeit solvophobic28 polyglutamine domains partition to the interior of what we refer to as incipient micellar structures. The bottom row in Figure 7 shows the other face of Nt17-QN dimers, which is comprised of surface exposed charge groups. While the charged groups are exposed to the surface, the hydrophobic groups are largely sequestered from the solvent and form both intra- and intermolecular interfaces with the polyglutamine domain. This suggests that formation of higher order “polar inside” micellar oligomers, the likelihood of which increases with increasing polyglutamine length, has the additional advantage of leading to increased sequestration of a majority of the hydrophobic groups of the Nt17 segment. Figure 8 shows quantification of the signed percent change in solvent accessible volume of all of the hydrophobic residues of the Nt17 segment in dimers of Ac-Nt17-QN-Nme constructs at 305 K. The reference state for these calculations is the isolated Nt17 segment at 305 K. The analysis shows that a majority of the hydrophobic residues are sequestered from solvent when compared to the reference state and that the degree of burial increases with increasing N.
Figure 7. Central structures of the most populated cluster for associated Ac-Nt17-QN-Nme dimers.
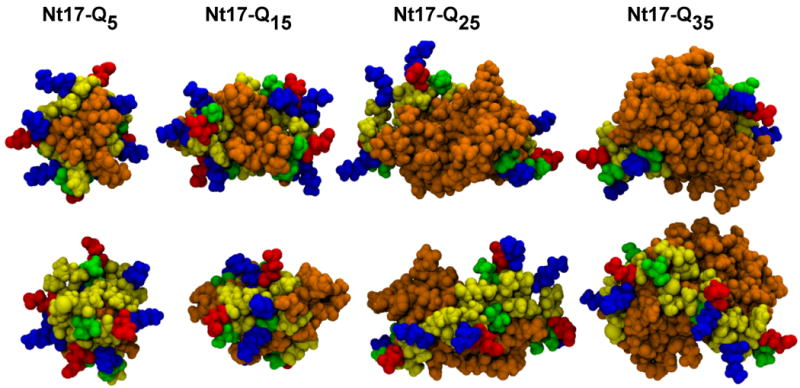
Graphics were generated using VMD58. The bottom row is the same structure as the top row, rotated by 180° around a vertical axis. Atoms are drawn identically to Figure 4.
Figure 8. Sequestration of the hydrophobic groups of Nt17 in dimers of Nt17-QN constructs.

The changes in residue-level solvent accessible volume (SAV) for each of the hydrophobic groups in Nt17 in dimers of Ac-Nt17-QN-Nme constructs was computed using the conformational ensemble of monomeric Ac-Nt17-Nme as the reference. For each of the residues shown along the abscissa, the ordinate plots the quantity 100(〈rSAVDimer〉 − 〈rSAVNt17-monomer〉), where 〈rSAVX〉 refers to the residue's average solvent accessible volume in the appropriate construct designated by the subscript, X. Analysis was carried out for T=305K. SAV values were computed using the formalism of Vitalis and Pappu. 45
We compared the probabilities of association shown in panels A and B of Figure 6 to those for the Ac-K2-QN-K2-Nme constructs. Experiments that study the kinetics of polyglutamine aggregation use similar peptide constructs as proxies for homopolymeric polyglutamine.8; 24; 25; 26; 30 The hope is that the lysine residues do not have a perturbing effect on polyglutamine conformational preferences, aggregation mechanisms, and aggregate morphologies that are intrinsic to polyglutamine.8; 24; 25; 30; 43; 44 Recently, Thakur et al. showed that constructs with the Nt17 segment aggregated more rapidly than constructs with pairs of lysine residues at the N- and C-termini. This was interpreted to mean that the Nt17 segment promotes enhanced intermolecular interactions vis-à-vis homopolymeric polyglutamine. Panel C of Figure 6 shows the probabilities of association that we obtained for Ac-K2-QN-K2-Nme constructs. Although the likelihood of association is generally higher for longer polyglutamine domains, probabilities of association are typically negligible compared to either of the constructs for which results are shown in panels A and B. The results for Ac-K2-Q35-K2-Nme obtained at the lowest temperatures seem to imply that the effects of lysine residues may be overcome at the right environmental conditions and large enough N. However, this result may also be fraught with some of the sampling challenges that we inevitably encounter for longer chain lengths. Hence, the main result is that N- and C-terminal lysine residues inhibit non-specific intermolecular associations of glutamine-rich peptides, vis-à-vis the true homopolypeptides. We propose that this is partly because the number of poses that promote intermolecular interactions is diminished to avoid the effects of unfavorable electrostatic repulsions. Our findings agree with Thakur et al. who suggested that Nt17-containing constructs aggregate more readily when compared to constructs with N- and C-terminal lysine residues. However, our interpretation for the mode of associations for Nt17-QN constructs is different because we were able to assess the associativities relative to true homopolymeric constructs.
Finally, Figure 9 shows the results of our analysis of the enhanced / depleted intermolecular inter-residue contacts in Ac-Nt17-QN-Nme constructs vis-à-vis a random contact model. Akin to the results shown in panel A of Figure 5, we find that dimer formation is the result of enhanced intermolecular contacts between glutamine residues and a depletion of all other types of contacts. This emphasizes the point made illustratively in Figure 7, which suggests that oligomers of Ac-Nt17-QN-Nme constructs are best described as being micellar with the charges on the surface, the polyglutamine domains buried within the interior, and the hydrophobic groups sequestered from solvent in inter-domain interfaces. It also counters the scheme that emerged from the analysis of Thakur et al., which, as our results suggest, might be due to the use of the lysinated polyglutamine constructs as the reference state for homopolymeric polyglutamine.
Figure 9. Enhancement / depletion of different types of contacts in dimers of Ac-Nt17QN-Nme constructs.

The figure shows the length dependence of the enhancement / depletion in the probability of observing intermolecular, inter-residue contacts between specific residue classes in dimers of Ac-Nt17-QN-Nme constructs. Analysis was carried out for T=305K.
Discussion
Atomistic, Metropolis Monte Carlo simulations utilizing the ABSINTH implicit solvation model45 allowed us to investigate the effect of the Nt17 segment on polyglutamine conformations and intermolecular associations. In accord with published data37; 39, our simulations and UV-CD spectroscopy uncovered an intrinsic preference for helical segments within the Nt17 peptide in aqueous solution at 25°C. The hydrophobic groups form clusters leading to increased compaction of the Nt17 peptides compared to an uninterrupted α-helix. The Nt17 segment goes through an expansion as N increases in Ac-Nt17-QN-Nme constructs. This expansion leads to increased sequestration of a majority of the Nt17 segment's hydrophobic groups and their partitioning in an inter-domain interface (Figure 4). For a given temperature, both the Nt17 and polyglutamine domains become more disordered as the polyglutamine length increases in the Ac-Nt17-QN-Nme constructs. Intramolecular contacts are dominated by glutamine-glutamine interactions for large N (panel A of Figure 5).
For specific values of N and temperature, the probability that a pair of molecules forms dimers is lower for the Ac-Nt17-QN-Nme constructs when compared to Ac-QN-Nme constructs. This suggests that the Nt17 segment leads to partial suppression of the intrinsic associativity because it reduces the interaction surface by (a) partial sequestration of the polyglutamine domain in an intramolecular, inter-domain interface and (b) intramolecular spatial separation of the polyglutamine domain from the charged groups, which causes some fraction of intermolecular poses to be unfavorable because of electrostatic repulsions. Intermolecular associations are dominated by interactions between the polyglutamine domains (Figure 9).
Thakur et al. suggested that the Nt17 segment enhances aggregation vis-à-vis polyglutamine, specifically K2-QN-K2 constructs. We were able to recapitulate their findings when we compared the probability of associations for the Ac-Nt17-QN-Nme constructs to those of the Ac-K2-QN-K2-Nme constructs. Both intra- and intermolecular repulsions between the N- and C-terminal lysine residues in the latter influence the polyglutamine conformations and significantly diminish intermolecular associations. Indeed, intramolecular electrostatic repulsions modulate the intrinsic preference of polyglutamine chains for collapsed conformations,10; 46 especially when N is short (N ≤ 15). Singh and Lapidus44 used Trp/Cys quenching in their studies of monomeric forms of K2-W-QN-C-K2 peptides for N ≤ 14 and concluded that these molecules prefer extended and stiff conformations on average. Recently, Walters and Murphy47 used a combination of spectroscopic tools to quantify the apparent discrepancy between the proposals of Singh and Lapidus and those that have emerged from simulations,9; 10; 29; 31; 35 theory,48 small ensemble fluorescence correlation spectroscopy measurements,28 and recent single molecule atomic force microscopy experiments,46 all of which show or argue that monomeric polyglutamine forms collapsed structures in water. Walters and Murphy47 studied peptide constructs of the form Ac-K2-W-QN-K2-X-NH2 for N=8, 12, 16, 20, and 24; here, X is either alanine or a dansylated lysine. They found that these peptide constructs formed compact structures for N ≥ 16 and that peptide compaction was necessary for aggregation – a finding that appears to be consistent with previous simulation results.31 Elevation of the pH beyond the pKa of lysine caused significant compaction and aggregation of the longer constructs, thereby demonstrating the modulating effect of net charge of the peptide context on the intrinsic conformations and aggregation propensities of polyglutamine.
Our results have two implications for ongoing studies on polyglutamine aggregation: First, they point to the advantages of using simulations as a complement to the experiments of Thakur et al. because the two approaches have access to different types of details and a clearer picture emerges when results from both approaches are combined to generate a mechanistic picture. Second, they highlight the importance of choosing appropriate constructs as reference states for the intrinsic conformational properties and association propensities of polyglutamine.
Figure 10 summarizes our proposal for the mechanism by which Ac-Nt17-QN-Nme constructs might assemble to form amyloid fibrils. This proposal represents an extrapolation from the observations presented here and accommodates the structural observations of Thakur et al. Incipient micellar structures could grow through monomer / oligomer addition via interactions between polyglutamine domains to yield metastable higher-order oligomers. These oligomers could either be spherical or cylindrical. The number of molecules per oligomer will, in all likelihood, depend on the length of the polyglutamine domain. The central tenet of our proposal is that β-sheet formation is likely to occur within spherical or cylindrical micellar oligomers, and this might be a pre-requisite for fibril formation and elongation via monomer / oligomer addition. There is precedent for the sphere-to-rod transitions49 depicted in Figure 10. Such transitions have characteristic concentration dependencies and growth laws that make our proposal amenable to experimental scrutiny. Our proposal in Figure 10 is congruent with the model of Krishnan and Lindquist50 for the aggregation of Sup35 and that of Marina et al.51 for the aggregation of Aβ alloforms.
Figure 10. Proposed scheme for the assembly of Ac-Nt17-QN-Nme constructs into amyloid fibrils.

The color scheme is identical to that of Figures 4 and 7. Details of the proposal are described in the discussion section.
Kim et al. 52 recently published a report on the crystal structure of a fusion protein of maltose-binding protein (MBP) and the exon 1 stretch of huntingtin that had seventeen residues in the polymorphic polyglutamine region. They referred to this construct as MBP-Htt17Q-EX1. The MBP domain facilitates crystallization and the construct crystallized as a trimer. The structure was solved using molecular replacement based on the known structure of MBP. Two salient features stand out from the structural analysis of Kim et al. First, The Nt17 fragment and the first five residues of the polyglutamine domain reliably form α-helical structures. Second, Kim et al. found that the polyglutamine domain adopts multiple conformations that includes α-helices, “random coil”, and other extended conformations. Kim et al. proposed that disorder (referred to as random coils) within the polyglutamine domain is likely to increase with increasing polyglutamine length. They also proposed that this increased disorder provides part of the driving force for increased intermolecular interactions. This hypothesis is borne out in the quantitative analysis reported in this work. The surface exposure of the Nt17 segment in oligomers and aggregates may allow it to engage in productive interactions in vivo. Our results might explain why such interactions may not be altered even when Nt17-QN constructs form soluble oligomers.53 The observed coupling between the Nt17 and polyglutamine domains suggests the need for further studies on the effects of other flanking sequences on polyglutamine conformations and associations.
Materials and Methods
Circular dichroism
Trevor Creamer provided us with chemically synthesized Nt17 peptide in purified form. CD spectra were collected using a Jasco J715 spectropolarimeter with a 1-mm path length quartz cuvette. Nt17 was disaggregated by dissolving 1 mg in a mixture of 1 mL of trifluoroacetic acid (TFA) and 1 mL of hexafluoroisopropanol (HFIP). This solution was evaporated under nitrogen stream followed by lyophilization for 1 hour to remove any residual TFA/HFIP. The peptide was resuspended to a concentration of 1 mg/mL in one of freshly prepared 6 M urea in native buffer, 50% TFE in native buffer, or native buffer (100 mM phosphate buffer at pH 7.2). These stock solutions were diluted to a final working concentration of 0.1 mg/mL. Results shown in panels A and B of Figure 2 represent an average of 10 scans taken at 50 nm per minute in steps of 0.1 nm at a temperature of 25°C.
System setup and conformational sampling
The following peptide constructs were used: Ac-Nt17-QN-Nme, Ac-QN-Nme, and Ac-K2-QN-K2-Nme. Table S3 provides a detailed inventory of peptide sequences used in each of the simulations, which were carried out using the ABSINTH implicit solvation model45 and molecular mechanics potential functions. For each peptide investigated, Table S3 lists the number of independent sets of replicates that were used to obtain the data presented in the main text. A single replicate is defined as a single thermal replica exchange run encompassing ten different temperatures. For each replica we carried out 15×107 production steps after 107 steps of equilibration. These settings were needed to obtain quantitatively realiable results for the longest peptides studied. Partial charges and parameters for torsions that maintain planarity of peptide units were taken from the OPLS-AA/L forcefield54. Parameters for Lennard-Jones potentials were taken from previous work.45 For all simulations, the quality of sampling was enhanced using thermal replica exchange.55 Thermal replicas were set using the following schedule: T = 298K, 305K, 315K, 325K, 335K, 345K, 355K, 365K, 375K, 385K. The temperature schedule was chosen based on results for coil-to-globule transitions shown in panel A for Figure 3 and criteria described in previous work.40
Markov chain Metropolis Monte Carlo (MC) simulations were performed in the canonical ensemble using the CAMPARI software package (http://lima.wustl.edu/software.php). Details of the move sets are provided in Table S4. The peptides were enclosed in a spherical droplet whose boundary was modeled as a stiff, one-sided harmonic potential. Random cluster moves attempt to perturb the rigid-body coordinates of two molecules simultaneously: two molecules are chosen at random, translated by a common vector, and rotated around their mutual center of mass. By preserving their relative orientation, tightly associated molecules can be sampled much more effectively. To be able to probe multiple length scales simultaneously, Monte Carlo moves in CAMPARI either fully randomize a given degree of freedom, or perform a stepwise perturbation that has a maximum size. The frequencies for different moves are chosen to reflect the relevance of the various degrees of freedom to both the conformational equilibria and the association of these peptides. Additionally, these choices reflect the associated computational cost. As an example, we sampled ω-angles relatively infrequently as these values are expected to remain close to the ideal trans-conformation. Note that there were a small number of moves for each simulation, which were used as swap attempts for replica exchange. Details on the individual moves are as follows: Rigid-body moves simultaneously change rotational and translational degrees of freedom of the whole molecule. The first value listed in parentheses of row 1, column 2 of Table S4 is the fraction of moves assigned to finite perturbations whereas the remaining attempts fully randomize the respective degrees of freedom. The second and third values are the maximum translational and rotational step-sizes associated with the finite perturbations. For perturbations of ω angles there are N+1 ω-angles for a chain length of N due to the acetyl and N-methylamide capping groups. The two sets of values in parentheses of row 2, column2 of Table S4 are the fraction of ω-moves, which attempt a stepwise perturbation along with the maximum step-size. Sidechain moves perturb the χ-angles of a given sidechain in the peptide. In each attempt to alter sidechain degrees of the freedom, a random number of χ-angles are given random orientations. Sidechain moves are inexpensive and therefore several sidechains are sampled during each “move” (first value in parentheses of row 3, column2 of Table S4). The remaining two values in parentheses again give the fraction of χ-moves with a finite perturbation and the maximum value of that perturbation. Lastly, the most important moves are those that perturb both the ϕ- and the ψ-angle of a given residue. The values in parentheses are interpreted the same way as for ω-moves. It is important to remind the reader that appropriate design of Metropolis Monte Carlo (MMC) move sets allow us to simultaneously probe multiple, disparate length scales rather efficiently, taking advantage of the low overall density. This situation is unlike molecular dynamics sampling which is quite inefficient for sampling large-scale conformational changes as well as intermolecular associations / dissociations. The latter is hindered by slow diffusion and will require adaptive approaches and indeed MMC sampling may be viewed as a variant of such an adaptive approach.
At pH 7, the Ac-Nt17-QN-Nme constructs are expected to carry a net charge of +1e. We added explicit chloride counterions plus excess Na+ and Cl- ions to mimic a salt concentration of 2.5 mM. Identical conditions were used in simulations of Ac-K2-QN-K2-Nme peptides. For simulations of dimerization, enclosing two peptides in a droplet of radius 200 Å mimics an effective concentration of 100 μM. In all simulations, snapshots were saved every 5000 steps, and intermolecular distances and polymeric properties were recorded every 500 steps. In general, errors were quantified by running simulations as multiple replicates, where at least three independent replica exchange simulations were carried out for each system.
Analysis of contacts
We computed the probabilities of realizing different types of intra- and intermolecular inter-residue contacts for Ac-Nt17-QN-Nme peptides. Raw values for these contact probabilities can be misleading since the peptide sequence influences the computed probabilities. We formulated a prior using a random mixture model to assess the contacts that are enhanced or depleted vis-à-vis random expectations. Residues in Ac-Nt17-QN-Nme peptides are classified as hydrophobic (h=M, L, F, A), glutamine (q) or other (o=S, T, E, K). A contact is defined if any two atoms of two residues have a distance of separation of less than 3.5Å. Nearest neighbors in the linear sequence are always in contact; hence, these are excluded in our enumeration of contacts and in setting up the random mixture model. For the latter we have 17+N total residues that can form (N+17)(N+16)/2 unique non-identical, intramolecular contact pairs. We excluded N+16 unique, nearest neighbor contacts, and the total number of unique possible contacts is: CN = (N+16)(N+15)/2. For Ac-Nt17-QN-Nme peptides, there are N glutamine residues, nine hydrophobic residues, and eight other residues. The N+16 excluded nearest neighbor contacts are composed of ten h-o contacts, three h-h contacts, three o-o contacts, one h-q contact, and N-1 q-q contacts. According to the random mixture model, the probabilities of realizing different types of contacts are: phh=(33/CN); pho=(62/CN); phq = (9N - 1)/CN; poo = (25/CN); poq = (8N/CN); and pqq =(1/CN)(N-1)(N-2)/2. The prior probabilities from the random mixture model for intermolecular contacts are simpler because there are no nearest neighbors to be excluded. Here, both molecules contribute nine hydrophobic residues, N glutamine residues, and eight other residues. Therefore the total contact number is CN2 = (N+17)(N+17) and the individual probabilities are: phh2=(81/CN2); pho2=(144/CN2); phq2=(18N/CN2); poo2=(64/CN2); poq2=(16N/CN2); pqq2 = (N2/CN2). Note that for combinatorial reasons, contacts between different groups have an intrinsically higher weight (factor of two). For a pair of residue types x and y, we estimated the enhancement / depletion of probabilities for contacts computed from analysis of simulated ensembles (psimxy) by evaluating the difference psimxy − pxy. For positive values, this quantity reports an enhancement of contacts vis-à-vis the random mixture model and for negative values, this quantity reports depletions relative to expectations based on the random mixture model.
Cluster analysis
To identify clusters of structures within ensembles for one or two peptides of the type Ac-Nt17-QN-Nme, we used a bottom-up clustering algorithm56 implemented in the GROMACS 4.0 utility g_cluster.57 A matrix of pairwise all-atom root mean square deviation (RMSD) values was computed for all structures within a trajectory at 305K (a minimum of 10,300 structures). The number of neighbors of each structure, within a cutoff value of 3.0 Å, was counted and the structure with the largest number of neighbors was removed from the pool along with all neighbors. This was repeated on the remaining members of the pool until every structure was assigned to a cluster. The number of clusters was not explicitly defined prior to the analysis. Statistics for the fraction of the ensemble represented by the dominant cluster and the number of clusters required to represent 95% of the ensemble are shown in Table S2 of the supplementary material.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Williams AJ, Paulson HL. Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci. 2008;31:521–8. doi: 10.1016/j.tins.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker FO. Huntington's disease. The Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 3.Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, Strand A, Torcassi C, Savage J, Hurlburt A, Cha GH, Ukani L, Chepanoske CL, Zhen Y, Sahasrabudhe S, Olson J, Kurschner C, Ellerby LM, Peltier JM, Botas J, Hughes RE. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007;3:e82. doi: 10.1371/journal.pgen.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rockabrand E, Slepko N, Pantalone A, Nukala VN, Kazantsev A, Marsh JL, Sullivan PG, Steffan JS, Sensi SL, Thompson LM. The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum Mol Genet. 2007;16:61–77. doi: 10.1093/hmg/ddl440. [DOI] [PubMed] [Google Scholar]
- 5.Orr AL, Li S, Wang CE, Li H, Wang J, Rong J, Xu X, Mastroberardino PG, Greenamyre JT, Li XJ. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci. 2008;28:2783–92. doi: 10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL. SUMO modification of Huntingtin and Huntington's disease pathology. Science. 2004;304:100–4. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 7.de Chiara C, Menon RP, Dal Piaz F, Calder L, Pastore A. Polyglutamine is not all: The functional role of the AXH domain in the ataxin-1 protein. Journal Of Molecular Biology. 2005;354:883–893. doi: 10.1016/j.jmb.2005.09.083. [DOI] [PubMed] [Google Scholar]
- 8.Chen S, Berthelier V, Yang W, Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. Journal Of Molecular Biology. 2001;311:173–182. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 9.Wang XL, Vitalis A, Wyczalkowski MA, Pappu RV. Characterizing the conformational ensemble of monomeric polyglutamine. Proteins-Structure Function And Bioinformatics. 2006;63:297–311. doi: 10.1002/prot.20761. [DOI] [PubMed] [Google Scholar]
- 10.Vitalis A, Wang X, Pappu RV. Quantitative characterization of intrinsic disorder in polyglutamine: insights from analysis based on polymer theories. Biophys J. 2007;93:1923–37. doi: 10.1529/biophysj.107.110080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ignatova Z, Gierasch LM. Extended polyglutamine tracts cause aggregation and structural perturbation of an adjacent beta barrel protein. Journal Of Biological Chemistry. 2006;281:12959–12967. doi: 10.1074/jbc.M511523200. [DOI] [PubMed] [Google Scholar]
- 12.Bulone D, Masino L, Thomas DJ, San Biagio PL, Pastore A. The interplay between polyQ and protein context delays aggregation by forming a reservoir of protofibrils. PLoS ONE. 2006;1:e111. doi: 10.1371/journal.pone.0000111. art. no. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 14.Bader R, Seeliger MA, Kelly SE, Ilag LL, Meersman F, Limones A, Luisi BF, Dobson CM, Itzhaki LS. Folding and fibril formation of the cell cycle protein Cks1. Journal Of Biological Chemistry. 2006;281:18816–18824. doi: 10.1074/jbc.M603628200. [DOI] [PubMed] [Google Scholar]
- 15.Berke SJS, Paulson HL. Protein aggregation and the ubiquitin proteasome pathway: gaining the UPPer hand on neurodegeneration. Current Opinion in Genetics & Development. 2003;13:253–261. doi: 10.1016/s0959-437x(03)00053-4. [DOI] [PubMed] [Google Scholar]
- 16.Holmberg CI, Staniszewski KE, Mensah KN, Matouschek A, Morimoto RI. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO Journal. 2004;23:4307–4318. doi: 10.1038/sj.emboj.7600426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Molecular Cell. 2004;14:95–104. doi: 10.1016/s1097-2765(04)00151-0. [DOI] [PubMed] [Google Scholar]
- 18.Ratovitski T, Gucek M, Jiang H, Chighladze E, Waldron E, D'Ambola J, Hou Z, Liang Y, Poirer MA, Hirschhorn RR, Graham R, Hayden MR, Cole RN, Ross CA. Mutant Huntingtin N-terminal fragments of specific size mediate aggregation and toxicity in neuronal cells. J Biol Chem. 2009;284:10855–67. doi: 10.1074/jbc.M804813200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ratovitski T, Nakamura M, D'Ambola J, Chighladze E, Liang Y, Wang W, Graham R, Hayden MR, Borchelt DR, Hirschhorn RR, Ross CA. N-terminal proteolysis of full-length mutant huntingtin in an inducible PC12 cell model of Huntington's disease. Cell Cycle. 2007;6:2970–81. doi: 10.4161/cc.6.23.4992. [DOI] [PubMed] [Google Scholar]
- 20.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine Repeats As Polar Zippers - Their Possible Role In Inherited Neurodegenerative Diseases. Proceedings Of The National Academy Of Sciences Of The United States Of America. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perutz MF, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2002;99:5591–5595. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altschuler EL, Hud NV, Mazrimas JA, Rupp B. Random coil conformation for extended polyglutamine stretches in aqueous soluble monomeric peptides. Journal Of Peptide Research. 1997;50:73–75. doi: 10.1111/j.1399-3011.1997.tb00622.x. [DOI] [PubMed] [Google Scholar]
- 23.Sharma D, Sharma S, Pasha S, Brahmachari SK. Peptide models for inherited neurodegenerative disorders: conformation and aggregation properties of long polyglutamine peptides with and without interruptions. FEBS Letters. 1999;456:181–185. doi: 10.1016/s0014-5793(99)00933-3. [DOI] [PubMed] [Google Scholar]
- 24.Chen S, Ferrone FA, Wetzel R. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci U S A. 2002;99:11884–9. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen SM, Berthelier V, Hamilton JB, O'Nuallain B, Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 2002;41:7391–7399. doi: 10.1021/bi011772q. [DOI] [PubMed] [Google Scholar]
- 26.Bhattacharyya AM, Thakur AK, Wetzel R. polyglutamine aggregation nucleation: thermodynamics of a highly unfavorable protein folding reaction. Proc Natl Acad Sci U S A. 2005;102:15400–5. doi: 10.1073/pnas.0501651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khare SD, Ding F, Gwanmesia KN, Dokholyan NV. Molecular origin of polyglutamine aggregation in neurodegenerative diseases. Plos Computational Biology. 2005;1:230–235. doi: 10.1371/journal.pcbi.0010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crick SL, Jayaraman M, Frieden C, Wetzel R, Pappu RV. Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2006;103:16764–16769. doi: 10.1073/pnas.0608175103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marchut AJ, Hall CK. Side-chain interactions determine amyloid formation by model polyglutamine peptides in molecular dynamics simulations. Biophysical Journal. 2006;90:4574–4584. doi: 10.1529/biophysj.105.079269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee CC, Walters RH, Murphy RM. Reconsidering the mechanism of polyglutamine peptide aggregation. Biochemistry. 2007;46:12810–20. doi: 10.1021/bi700806c. [DOI] [PubMed] [Google Scholar]
- 31.Vitalis A, Wang X, Pappu RV. Atomistic simulations of the effects of polyglutamine chain length and solvent quality on conformational equilibria and spontaneous homodimerization. J Mol Biol. 2008;384:279–97. doi: 10.1016/j.jmb.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masino L, Musi V, Menon RP, Fusi P, Kelly G, Frenkiel TA, Trottier Y, Pastore A. Domain architecture of the polyglutamine protein ataxin-3: a globular domain followed by a flexible tail. FEBS Letters. 2003;549:21–25. doi: 10.1016/s0014-5793(03)00748-8. [DOI] [PubMed] [Google Scholar]
- 33.Bhattacharyya A, Thakur AK, Chellgren VM, Thiagarajan G, Williams AD, Chellgren BW, Creamer TP, Wetzel R. Oligoproline effects on polyglutamine conformation and aggregation. Journal Of Molecular Biology. 2006;355:524–535. doi: 10.1016/j.jmb.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 34.Darnell G, Orgel JPRO, Pahl R, Meredith SC. Flanking Polyproline Sequences Inhibit ?-Sheet Structure in Polyglutamine Segments by Inducing PPII-like Helix Structure. Journal of Molecular Biology. 2007;374:688. doi: 10.1016/j.jmb.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 35.Barton S, Jacak R, Khare SD, Ding F, Dokholyan NV. The length dependence of the polyQ-mediated protein aggregation. Journal of Biological Chemistry. 2007;282:25487–25492. doi: 10.1074/jbc.M701600200. [DOI] [PubMed] [Google Scholar]
- 36.Ignatova Z, Thakur AK, Wetzel R, Gierasch LM. In-cell aggregation of a polyglutamine-containing chimera is a multistep process initiated by the flanking sequence. Journal of Biological Chemistry. 2007;282:36736–36743. doi: 10.1074/jbc.M703682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, L Byeon IJ, Anjum DH, Kodali R, Creamer TP, Conway JF, Gronenborn AM, Wetzel R. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol. 2009;16:380–9. doi: 10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferrone F. Analysis of protein aggregation kinetics. Methods in Enzymology. 1999;309:256–274. doi: 10.1016/s0076-6879(99)09019-9. [DOI] [PubMed] [Google Scholar]
- 39.Kelley NW, Huang X, Tam S, Spiess C, Frydman J, Pande VS. The predicted structure of the headpiece of the Huntingtin protein and its implications on Huntingtin aggregation. J Mol Biol. 2009;388:919–27. doi: 10.1016/j.jmb.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vitalis A, Lyle N, Pappu RV. Thermodynamics of beta-Sheet Formation in Polyglutamine. 2009;97:303–311. doi: 10.1016/j.bpj.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scholtz JM, Qian H, York EJ, Stewart JM, Baldwin RL. Parameters of helix-coil transition theory for alanine-based peptides of varying chain lengths in water. Biopolymers. 1991;31:1463–70. doi: 10.1002/bip.360311304. [DOI] [PubMed] [Google Scholar]
- 42.Thakur AK, Wetzel R. Mutational analysis of the structural organization of polyglutamine aggregates. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2002;99:17014–17019. doi: 10.1073/pnas.252523899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma D, Shinchuk LM, Inouye H, Wetzel R, Kirschner DA. Polyglutamine homopolymers having 8-45 residues form slablike beta-crystallite assemblies. Proteins-Structure Function And Bioinformatics. 2005;61:398–411. doi: 10.1002/prot.20602. [DOI] [PubMed] [Google Scholar]
- 44.Singh VR, Lapidus LJ. The intrinsic stiffness of polyglutamine peptides. Journal of Physical Chemistry B. 2008;112:13172. doi: 10.1021/jp805636p. [DOI] [PubMed] [Google Scholar]
- 45.Vitalis A, Pappu RV. ABSINTH: a new continuum solvation model for simulations of polypeptides in aqueous solutions. J Comput Chem. 2009;30:673–99. doi: 10.1002/jcc.21005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dougan L, Li J, Badilla CL, Berne BJ, Fernandez JM. Single homopolypeptide chains collapse into mechanically rigid conformations. Proceedings of the National Academy of Sciences. 2009 doi: 10.1073/pnas.0900678106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walters RH, Murphy RM. Examining Polyglutamine Peptide Length: A Connection between Collapsed Conformations and Increased Aggregation. Journal Of Molecular Biology. 2009;393:978–992. doi: 10.1016/j.jmb.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pappu RV, Wang X, Vitalis A, Crick SL. A polymer physics perspective on driving forces and mechanisms for protein aggregation. Archives of Biochemistry and Biophysics. 2008;469:132–141. doi: 10.1016/j.abb.2007.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.May S, Ben-Shaul A. Molecular theory of the sphere-to-rod transition and the second CMC in aqueous micellar solutions. Journal Of Physical Chemistry B. 2001;105:630–640. [Google Scholar]
- 50.Krishnan R, Lindquist SL. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature. 2005;435:765–772. doi: 10.1038/nature03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marina GB, Kirkitadze D, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid beta-protein assembly: Abeta40 and Abeta42 oligomerize through distinct pathways. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I. Secondary Structure of Huntingtin Amino-Terminal Region. Structure. 2009;17:1205–1212. doi: 10.1016/j.str.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum Mol Genet. 2008;17:345–56. doi: 10.1093/hmg/ddm311. [DOI] [PubMed] [Google Scholar]
- 54.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. Journal Of Physical Chemistry B. 2001;105:6474–6487. [Google Scholar]
- 55.Mitsutake A, Sugita Y, Okamoto Y. Generalized-ensemble algorithms for molecular simulations of biopolymers. Biopolymers. 2001;60:96–123. doi: 10.1002/1097-0282(2001)60:2<96::AID-BIP1007>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 56.Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, Mark AE. Peptide folding: When simulation meets experiment. ANGEW CHEM INT EDIT. 1999;38:236–240. [Google Scholar]
- 57.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J CHEM THEORY COMPUT. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 58.Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. Journal of Molecular Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.