

Abstract
The double helix is a ubiquitous feature of RNA molecules and provides a target for nucleases involved in RNA maturation and decay. Escherichia coli ribonuclease III participates in maturation and decay pathways by site-specifically cleaving double-helical structures in cellular and viral RNAs. The site of cleavage can determine RNA functional activity and half-life and is specified in part by local tertiary structure elements such as internal loops. The involvement of base pair sequence in determining cleavage sites is unclear, because RNase III can efficiently degrade polymeric double-stranded RNAs of low sequence complexity. An alignment of RNase III substrates revealed an exclusion of specific Watson–Crick bp sequences at defined positions relative to the cleavage site. Inclusion of these “disfavored” sequences in a model substrate strongly inhibited cleavage in vitro by interfering with RNase III binding. Substrate cleavage also was inhibited by a 3-bp sequence from the selenocysteine-accepting tRNASec, which acts as an antideterminant of EF-Tu binding to tRNASec. The inhibitory bp sequences, together with local tertiary structure, can confer site specificity to cleavage of cellular and viral substrates without constraining the degradative action of RNase III on polymeric double-stranded RNA. Base pair antideterminants also may protect double-helical elements in other RNA molecules with essential functions.
RNA maturation and decay reactions involve site-specific cleavages carried out by a diverse ensemble of cellular ribonucleases. The site of cleavage can determine RNA half-life and functional activity and is established by specific sequence and structural features in RNA (1). Double-helical regions provide a target for enzymatic cleavage and can be formed through long-range or short-range intramolecular base pairing. Double-stranded (ds) RNA structures also are created by antisense RNA binding to complementary RNA sequences, or by symmetric transcription of cellular or viral DNA. Moreover, many viral chromosomes are dsRNA molecules. Thus, dsRNA cleavage reactions are not only necessary for RNA maturation, function, and decay, but also may provide an antiviral strategy (2, 3).
Ribonuclease III of Escherichia coli [EC 3.1.24] is a dsRNA-specific nuclease with important functions in cellular and viral RNA metabolism (2, 4, 5). RNase III cleaves the primary transcript of the ribosomal RNA operons to provide the immediate precursors to the mature 16S and 23S ribosomal RNAs. RNase III also participates in the maturation of cellular and phage mRNAs and can control mRNA translation and half-life (2, 5). RNase III is a homodimeric phosphodiesterase, cleaving substrate to create 5′-phosphate, 3′-hydroxyl termini. The only required cofactor is a divalent metal ion, preferably Mg2+. Substrate cleavage in vitro at physiologically relevant salt concentrations accurately reflects the in vivo cleavage pattern (2, 5, 7, 8). Nucleases similar to RNase III are present in all cells and perform similar functions, including ribosomal RNA maturation (2, 5, 6, 9).
RNase III recognition and cleavage of its diverse substrates presents a puzzle. On the one hand, RNase III can efficiently degrade polymeric dsRNA of low sequence complexity, yielding short (12–15 bp) duplexes (7, 8). On the other hand, cellular and viral substrates are precisely cleaved at one or two sites. Although most of these substrates display a ≈20-bp double-helical structure within which cleavage occurs, the scissile bond(s) are not determined by the distance from one end of the double helix (10). The role of RNA sequence in determining cleavage sites has been controversial. Although an earlier study noted weakly conserved sequences symmetric to the cleavage site (11), any proposed involvement of sequence must be reconciled with the apparent sequence-nonspecific action of RNase III. We show in this report that Watson-Crick (W-C) bp sequence plays a critical role in the reactivity of RNase III substrates, but in an unanticipated manner.
MATERIALS AND METHODS
Materials.
RNAs were synthesized in vitro by using T7 RNA polymerase and DNA oligonucleotide templates and purified by gel electrophoresis as described (10). Internally 32P-labeled RNAs (≈5,000 dpm/pmol) were prepared by including [α-32P]UTP in transcription reactions, and 5′-32P-labeled RNAs (≈7 × 106 dpm/pmol) were prepared by treating dephosphorylated RNA with T4 kinase and [γ-32P]ATP. RNase III and the [Glu117Lys] RNase III mutant were purified as described (12).
RNA Cleavage Assays.
Cleavage assays were performed as described (13). Briefly, 32P-labeled RNA and RNase III were combined in potassium glutamate buffer, and reactions (37°) were initiated by adding 10 mM MgCl2. So that initial cleavage rates would reflect changes in Km or kcat, enzyme and substrate concentrations (≈10 nM and ≈50 nM, respectively) were below the Km for R1.1[WC] RNA cleavage, which was ≈1 μM (data not shown). Reactions were analyzed by gel electrophoresis, and initial velocities were determined by radioanalytic imaging (AMBIS).
RNA Binding Assays.
RNase III binding to substrate was measured by a gel shift assay as described (10, 12). Briefly, 5′-32P-labeled RNA (≈104 dpm) was combined with the [Glu117Lys] RNase III mutant in binding buffer containing MgCl2 (12), then electrophoresed in a 6% polyacrylamide gel containing Tris⋅borate⋅EDTA buffer supplemented with 10 mM MgCl2. K′D values were determined as described (12). Because some dissociation of the RNA-protein complex occurred during electrophoresis, the amount of free (unbound) RNA was measured, which was used to calculate the bound fraction.
RESULTS
To assess the involvement of RNA sequence in substrate reactivity we aligned 10 well-characterized RNase III substrates (see Fig. 1 legend). Fig. 1A provides the frequency of each W-C bp at positions +1 to −12 relative to the cleavage site. The alignment reveals deviations from random sequence at positions −4, −5, and −6 (hereafter termed the “proximal box”), and at −11 and −12 (“distal box”). In particular, the proximal and distal boxes are distinguished by the near or complete absence of specific sequences. Thus, the proximal box lacks GC/CG at position −5, with GC underrepresented at positions −4 and −6, whereas the distal box lacks UA and CG at positions −11 and −12, respectively. The position-dependent exclusion of sequence is statistically significant (P < 0.05). Fig. 1B displays the “disfavored” bp in a dsRNA structure, while Fig. 1C demonstrates the degeneracy of the “favored” bp consensus.
Figure 1.

Position-specific exclusion of sequence in RNase III substrates. (A) Substrate alignment analysis. Ten substrates were aligned whose cleavage sites were accurately determined, either by direct RNA sequence analysis or by primer extension of RNA cleaved in vitro by purified RNase III: 16S rRNA precursor (14), 23S rRNA precursor (15), T7 R0.3 (16), R0.5 (16), R1.1 (16) and R1.3 (16) substrates; lambda N leader (17), lambda sib (18) lambda cIII (19), and the RNase III operon transcript 5′-leader (20, 21). Each substrate provided two sequences for alignment, reflecting a two-fold symmetry about the cleavage site (11, 12). The cleavage site is indicated by vertical arrowhead between −1 and +1. The data represent the number of times (n = 20) each W-C bp occurs at positions +1 to −12. Position −12 represented the duplex limit for most substrates. P values from a Chi squared analysis also are provided. The significance of the disfavored A and U at positions −1 and −2, respectively, and the preferred C at −2 are not known, but may reflect specific sequence requirements for a structured asymmetric internal loop in a number of the substrates (22). The preference for CG at −6 is not known. (B) The “disfavored” bp, displayed in a dsRNA structure. The proximal box (PB) and distal box (DB) are included within an 11-bp helix. S = C or G, with S′ complementary to S. N, N′ indicate complementary nucleotides; while n, n′ indicate less strict complementarity. Arrowheads indicate the (blocked) cleavage sites. (C) Absence of conservation (degeneracy) of RNase III substrate sequence. H = A, G, U, with D′ (A, C, U) complementary to H; B = C, G, U, with V′ (G, C, U) complementary to B; W, W′ = A, U. (D) Secondary structure of the T7 R1.1 RNase III substrate, showing the proximal and distal boxes and the single cleavage site (arrowhead).
The position-specific exclusion of specific W-C bp prompted the question of whether the presence of these sequences can affect cleavage of substrate. We examined the effects of bp substitution on the cleavage of a variant of the T7 phage R1.1 RNase III substrate (Fig. 1D). The R1.1 substrate occurs between T7 genes 1 and 1.1 and is precisely cleaved within the internal loop, thereby separating the two coding sequences and providing a hairpin at the 3′ end of the gene 1 mRNA (16). To avoid potential complications imposed by the internal loop in interpreting cleavage rates we used R1.1[WC] RNA (Fig. 2A), which possesses a fully base-paired structure, and which is efficiently cleaved in vitro at the two indicated sites. The first set of R1.1[WC] RNA variants contained proximal and/or distal boxes fully substituted with the disfavored bp. All of the variants exhibit reduced cleavage rates, ranging from 3-fold (var-5) to >1,000-fold (var-6) lower than that for R1.1[WC] RNA (Fig. 2). In contrast, substitution of CG/GC bp for the four AU/UA bp spanning the cleavage sites does not inhibit cleavage (Fig. 2, var-7).
Figure 2.
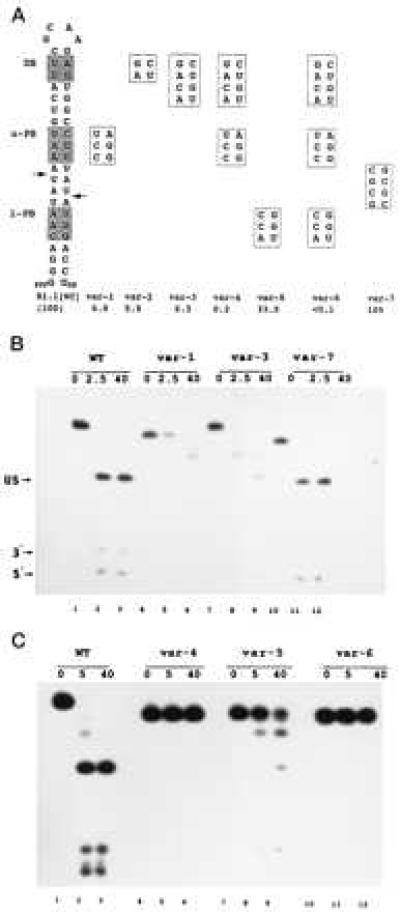
Bp-sequence-dependent inhibition of substrate cleavage. (A) Structure of R1.1[WC] RNA and the relevant sequences of seven variants. Cleavage of R1.1[WC] RNA occurs at the two indicated sites (arrows), determined by oligonucleotide sequence analysis of cleavage products (not shown). DB, distal box; u-PB, upper proximal box; l-PB, lower proximal box. Numbers given below the R1.1[WC] variants are cleavage rates relative to the R1.1[WC] RNA rate (100%, or ≈150 fmol product/min). The reported values are the average of at least three experiments, with standard deviations ≤13%. (B) Cleavage time course pattern for internally 32P-labeled R1.1[WC] RNA (WT, lanes 1–3) and variant 1 (lanes 4–6), variant 3 (lanes 7–9), and variant 7 (lanes 10–12). Time points (minutes) are indicated. (C) Same as experiment in B, but examining variant 4 (lanes 4–6), variant 5 (lanes 7–9), and variant 6 (lanes 10–12). Lanes 1–3 are for R1.1[WC] RNA. The cleavage products are the upper stem (US, 28 nucleotides); a 5′-end-containing fragment (5′, 10 nucleotides), and a 3′-end-containing fragment (3′, 8 nucleotides). The slighter slower rate of cleavage for R1.1[WC] RNA in the experiment in the lower panel reflects typical variation from experiment to experiment. The differing gel mobilities of the uncleaved RNAs reflect conformational differences in 7M urea, also seen elsewhere (23). For several of the variants, the two species with mobilities between the substrate and the 28-nt upper stem product represent products of single-site cleavage.
The inhibitory effects of box substitution are cumulative, as R1.1[WC] RNA variants with a single substituted box (Fig. 2, var-1, -2, -3, and -5) are more reactive than the variant with two substituted boxes (var-4), which in turn is more reactive than the triply substituted RNA (Fig. 2, var-6). The two variants containing either a single substituted distal box or an upper proximal box (var-2 and var-5) exhibit equal loss of reactivity (Fig. 2). However, the greater inhibitory effect of a substituted upper proximal box than a substituted lower proximal box (Fig. 2, compare variants 1 and 5) indicates an asymmetric interaction of RNase III with substrate, which also was noted in a deletion analysis of R1.1 RNA (10). The bp substitutions do not shift the cleavage site, as autoradiographic overexposure reveals comigration of the variant RNA 5′-end-containing and 3′-end-containing products with the corresponding cleavage products of R1.1[WC] RNA (data not shown).
The inhibition of cleavage could reflect interference with RNase III binding. Alternatively, binding may occur, but the cleavage step is inhibited. To distinguish between these possibilities we carried out gel shift assays on the R1.1[WC] RNA variants, by using the [Glu117Lys] RNase III mutant, which can bind substrate in the same manner as wild-type enzyme but cannot catalyze cleavage (5, 12). Under conditions where R1.1[WC] RNA efficiently binds the RNase III mutant, the poorly cleaved substrates exhibit greatly weakened or undetectable binding (Fig. 3A). There is a strong correlation (r = 0.985) between cleavage rates (Vi) and binding affinities (K′A), wherein the cleavage rate extrapolates to zero as all binding affinity is lost (Fig. 3B). Thus, the disfavored W-C bp weaken RNase III binding rather than inhibiting the cleavage step.
Figure 3.
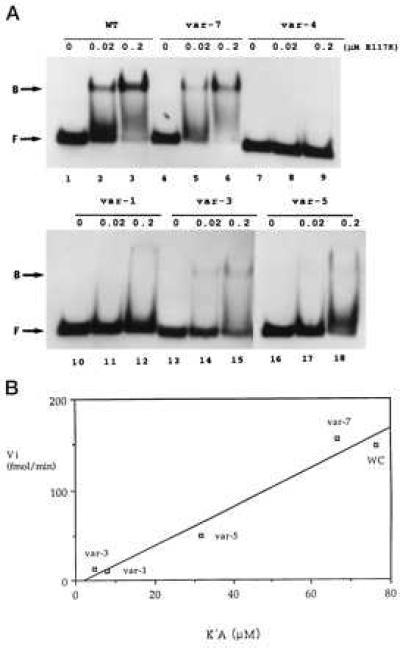
W-C bp substitution inhibits RNase III binding. (A) Gel shift assay of R1.1[WC] RNA variant binding to RNase III. 5′-32P-labeled RNA (104 dpm; 1.4 fmol) was combined with 0, 0.02, or 0.2 μM of the [E117K] RNase III mutant, then electrophoresed in a nondenaturing gel. (Upper) Lanes 1–3, R1.1[WC] RNA (WT); lanes 2–4, variant 7; and lanes 7–9, variant 4. (Lower) Lanes 1–3, variant 1; lanes 4–6, variant 3; and lanes 7–9, variant 5. (B) Correlation between initial cleavage rate (Vi) and binding affinity (1/K′D). WC refers to R1.1[WC] RNA. The Vi values are expressed as fmol product formed per minute (37°). The measured K′D values (nM ± SD) are the average of at least three experiments, and are: R1.1[WC] RNA, 13.1 ± 9.3; var-7, 15.0 ± 4.0; var-5, 31.5 ± 16.5; var-1, 126 ± 30; and var-3, 213 ± 117. Given the similarity in cleavage rates (Fig. 2A), the binding affinity of variant 2 was assumed to be similar to that for variant 3. The weak binding affinities of variants 4 and 6 prevented K′D determination.
Is complete W-C bp substitution within the proximal or distal box required for maximal inhibition of cleavage? Within the distal box, the two single bp substitutions are less inhibitory than double substitution (Fig. 4A, compare variants 8 and 9 with variant 2). In the proximal box, however, substitution of CG in the middle position (variant 10) inhibits comparable to substitution of all three bp (variant 1). Although double substitutions in the proximal box are more inhibitory in general than single bp substitutions (Fig. 4A, compare variants 14–16 with variants 10–13), there is no clear correlation of sequence with reactivity, suggesting the inhibitory effects are sequence context-dependent (see also below).
Figure 4.
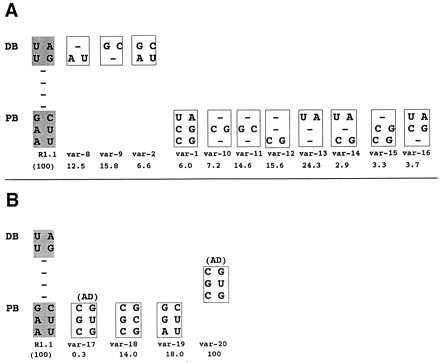
Inhibition of cleavage by single or double bp substitutions and by the tRNASec AD. (A) Relative cleavage rates of internally 32P-labeled R1.1[WC] RNA variants exhibiting single or double W-C bp substitution in the distal or proximal box. The proximal box (PB) and distal box (DB) sequences of R1.1[WC] RNA are shown on the left. (B) Inhibition by the tRNASec AD and sequence variants. The var-1 RNA value is from Fig. 2. (A and B) The numbers below the variant RNAs are cleavage rates, relative to that of R1.1[WC] RNA (100%), and represent the average of three experiments, with SDs averaging ≤22% of the values.
The inhibitory action of the disfavored W-C bp may be because of alteration of R1.1[WC] RNA secondary structure. To examine this possibility, 5′-32P-labeled R1.1[WC] RNA and variants were probed with RNase T2, which cuts within unstructured regions, and with RNase V1, which cuts within helical regions (24). No differences were observed between the cleavage patterns of R1.1[WC] RNA and its variants, all of which display RNase T2-sensitive tetraloops and RNase V1-sensitive stems (data not shown). We conclude that the W-C bp substitutions do not cause significant changes in the hairpin structure of R1.1[WC] RNA.
Transfer RNAs contain antideterminant (AD) sequences that block recognition by noncognate aminoacyl-tRNA synthetases or elongation factors (25, 26). We noted a similarity of the disfavored sequence in the proximal box (CSC/GS′G, with S = G or C; and S′ complementary to S) (Fig. 1B) with the selenocysteine (Sec)-tRNASec AD sequence (CGC/GUG), which blocks elongation factor Tu (EF-Tu) binding (26). The tRNASec AD also strongly inhibits RNase III action, as an R1.1[WC] RNA variant that contains the AD sequence in the upper proximal box (Fig. 4B, var-17) exhibits a 330-fold lower cleavage rate compared with the parent RNA. This inhibition is substantially greater than that imposed by any other proximal box substitution. Similar to the other disfavored bp substitutions, and similar to its action on EF-Tu, the transplanted tRNASec AD inhibits RNase III binding (data not shown). Importantly, an R1.1[WC] RNA variant with the AD positioned between the proximal and distal box is as reactive as the parent substrate (Fig. 4B, var-20). Thus, tRNASec AD inhibition (and by inference the other disfavored bp) reflects a localized interference with RNase III binding rather than a global perturbation of R1.1 RNA structure, which would be AD position-independent. The G⋅U pair is a primary inhibitory feature of the AD, as the variant with a CGC/GCG sequence (with the underlined C representing the change) exhibits only a ≈6-fold lower cleavage rate than the parent substrate (Fig. 4B, var-18). However, the G⋅U inhibition is most pronounced within the context of the AD, as single substitution of G⋅U in the proximal box middle position (AGG/CUU; var-19) causes only a ≈5-fold decrease in cleavage rate. There are no clear similarities between the primary sequences of RNase III and EF-Tu that would suggest a common mechanism of inhibition by the tRNASec AD.
DISCUSSION
We have shown that specific W-C bp sequences at defined positions within a model RNase III substrate can inhibit enzymatic cleavage in vitro. The control of dsRNA cleavage by bp sequence is unprecedented among known ribonuclease-substrate recognition mechanisms (27) and provides a mechanism by which RNase III can cleave cellular and viral substrates in a site-specific manner. We propose that RNase III recognizes its substrates by directly binding the proximal and distal box helices. These interactions are indicated by (i) a significant drop in cleavage reactivity upon deletion of the R1.1 RNA distal box (10); (ii) proximal and distal box-localized phosphodiesters whose ethylation interferes with RNase III binding, and ribose residues that are protected by RNase III from hydroxyl radicals (12); and (iii) the inhibition of cleavage by mutations that disrupt W-C base pairing in the proximal box (28, 29). Furthermore, this report has shown that RNase III binding is inhibited by the presence in the proximal or distal box of disfavored W-C bp, as identified through sequence alignment. The unequal inhibitory effects of the GC and CG bp in the middle position of the proximal box suggests (30) incompatible RNA-protein interactions localized to the major groove, which apparently are absent with either AU or UA in the same position. However, it is also possible that RNase III does not interact with functional groups in either groove, but is sensitive to subtle structural changes in the sugar-phosphate backbone caused by one or more disfavored bp.†
We propose that the inhibitory W-C bp represent RNase III antideterminants, and that these elements play a key role in selecting the cleavage site. Thus, the presence of antideterminants at specific sites in a substrate can block cleavage of otherwise reactive phosphodiesters, with the scissile bonds selected (by default) through the absence of antideterminants within the corresponding proximal and/or distal box positions. The additional imposition of an internal loop can constrain cleavage to one phosphodiester (2, 8). Examination of other possible cleavage sites in R1.1[WC] RNA reveal one or more antideterminant bp within the positions corresponding to the proximal or distal box (Fig. 2A)‡. Although this report has described only the inhibition of substrate cleavage in vitro, antideterminant bp apparently inhibit RNase III action in vivo: whereas a transcript containing the T7 R1.1 sequence is efficiently cleaved at the canonical site in vivo, there is no detectable cleavage of a transcript containing the R1.1 sequence substituted with antideterminant bp (W. Liao and A.W.N., unpublished data). The control of RNase III action by W-C bp sequence does not constrain RNase III cleavage of polymeric dsRNA: the lack of conserved sequences within (as well as outside of) the proximal and distal boxes correlates with the efficient cleavage of dsRNA of low sequence complexity, and the occurrence of multiple cleavage sites within antisense RNA-target RNA duplexes (e.g., see ref. 11). The “relaxed” specificity toward these substrates may reflect a key role of RNase III in dsRNA turnover, which originally may have involved the degradation of viral RNA replicative intermediates and restricting genetic exchange at the RNA level (2, 4, 5).
Bulges, loops, and mismatches can block RNase III cleavage of substrate (e.g., see ref. 34). However, these motifs perturb double-helical structure, which may be required for proper function of the RNA. W-C bp antideterminants can confer protection from RNase III without disrupting secondary structure. For example, the E. coli 4.5S RNA, the T7 RNA polymerase transcription terminator (Tφ), and the E. coli rrnB operon T1 transcription terminator contain W-C bp stems 15–20 bp in length, which are functionally vital. However, these structures are not cleaved by RNase III in vivo (16, 35, 36). Examination of the dsRNA sequences reveal potential cleavage sites that are masked by antideterminant bp. Bacterial RNA processing antideterminants can be expanded to include 5′- and 3′-end secondary structures (e.g., see refs. 37 and 38), which together with internal sequences can control cleavage by cellular ribonucleases, many of which exhibit substantial nonspecificity in isolated form (39). W-C bp antideterminants may control the action of other dsRNA-binding or dsRNA-modifying proteins, including the eukaryotic dsRNA adenosine deaminase, which exhibits little apparent specificity with long dsRNAs (40), but edits cellular and viral RNAs with high precision (41).
Acknowledgments
We thank Hugh Robertson for RNA fingerprinting analyses, insight on the manuscript, and his long-standing interest in RNase III; Irina Calin for analysis of var-20 RNA; Elizabeth Ries for DNA syntheses; Mary Murray for comments on the manuscript; and Phil Cunningham for help with statistical analysis and comments on the manuscript. This project was supported by National Institutes of Health Grants GM41283 and GM56457.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: AD, antideterminant; ds, double-stranded; W-C, Watson-Crick; Sec, selenocysteine.
A previous analysis (11) revealed a modest conservation of W-C bp sequence at specific positions relative to the cleavage site. However, more subtle features may have been missed because (i) the conservation may reflect genetic duplication of RNase III substrates within a given genome (e.g., T7 phage), and (ii) the aligned substrates included ones whose cleavage sites were not accurately mapped. In this regard, RNAs processed in vivo often exhibit 3′-end heterogeneity because of 3′-exoribonuclease and/or poly(A) polymerase action (31, 32), and mapping by nuclease protection can be imprecise because of fraying of RNA-DNA hybrid termini (19).
It was reported previously (23) that specific W-C bp substitutions within the upper proximal box of R1.1 RNA do not significantly inhibit cleavage in vitro. We attribute the sustained reactivity of the R1.1 RNA variants in part to (i) the use of lower salt (≤160 mM KCl) in the reactions, which can promote cleavage of less reactive substrates (33), and (ii) the existence in R1.1 RNA of a longer double-helical element, with two proximal+distal box sets (Fig. 1D), which partially would compensate for mutations in a single proximal box.
References
- 1.Belasco J G, Brawerman G, editors. Control of Messenger RNA Stability. San Diego: Academic; 1993. [Google Scholar]
- 2.Nicholson A W. Prog Nucleic Acids Res Mol Biol. 1996;52:1–65. doi: 10.1016/s0079-6603(08)60963-0. [DOI] [PubMed] [Google Scholar]
- 3.Meegan J M, Marcus P I. Science. 1989;244:1089–1091. doi: 10.1126/science.2471268. [DOI] [PubMed] [Google Scholar]
- 4.Robertson H D, Webster R E, Zinder N D. J Biol Chem. 1968;243:82–91. [PubMed] [Google Scholar]
- 5.Court D. In: Control of Messenger RNA Stability. Belasco J G, Brawerman G, editors. San Diego: Academic; 1993. pp. 71–116. [Google Scholar]
- 6.Abou-Elela S, Igel H, Ares M. Cell. 1996;85:115–124. doi: 10.1016/s0092-8674(00)81087-9. [DOI] [PubMed] [Google Scholar]
- 7.Dunn J J. In: The Enzymes. Boyer P, editor. Vol. 15. San Diego: Academic; 1982. pp. 485–499. [Google Scholar]
- 8.Robertson H D. Cell. 1982;30:669–672. doi: 10.1016/0092-8674(82)90270-7. [DOI] [PubMed] [Google Scholar]
- 9.Rotondo G, Frendewey D. Nucleic Acids Res. 1996;24:2377–2386. doi: 10.1093/nar/24.12.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chelladurai B S, Li H, Zhang K, Nicholson A W. Biochemistry. 1993;32:7549–7558. doi: 10.1021/bi00080a029. [DOI] [PubMed] [Google Scholar]
- 11.Krinke L, Wulff D L. Nucleic Acids Res. 1990;18:4809–4815. doi: 10.1093/nar/18.16.4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Nicholson A W. EMBO J. 1996;15:101–113. [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Chelladurai B S, Zhang K, Nicholson A W. Nucleic Acids Res. 1993;21:1919–1925. doi: 10.1093/nar/21.8.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young R A, Steitz J A. Proc Natl Acad Sci USA. 1978;75:3593–3597. doi: 10.1073/pnas.75.8.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bram R J, Young R A, Steitz J A. Cell. 1980;19:393–401. doi: 10.1016/0092-8674(80)90513-9. [DOI] [PubMed] [Google Scholar]
- 16.Dunn J J, Studier F W. J Mol Biol. 1983;166:477–535. doi: 10.1016/s0022-2836(83)80282-4. [DOI] [PubMed] [Google Scholar]
- 17.Steege D A, Cone K C, Rosenberg M. J Biol Chem. 1987;262:17651–17658. [PubMed] [Google Scholar]
- 18.Schmeissner U, McKenney K, Rosenberg M, Court D. J Mol Biol. 1984;176:39–53. doi: 10.1016/0022-2836(84)90381-4. [DOI] [PubMed] [Google Scholar]
- 19.Altuvia S, Kornitzer D, Kobi S, Oppenheim A B. J Mol Biol. 1991;218:723–733. doi: 10.1016/0022-2836(91)90261-4. [DOI] [PubMed] [Google Scholar]
- 20.Bardwell J C A, Régnier P, Chen S-M, Nakamura Y, Grunberg-Manago M, Court D L. EMBO J. 1989;8:3401–3407. doi: 10.1002/j.1460-2075.1989.tb08504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsunaga J, Simons E L, Simons R W. RNA. 1996;2:1228–1240. [PMC free article] [PubMed] [Google Scholar]
- 22.Schweisguth D C, Chelladurai B S, Nicholson A W, Moore P B. Nucleic Acids Res. 1994;22:604–612. doi: 10.1093/nar/22.4.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chelladurai B S, Li H, Nicholson A W. Nucleic Acids Res. 1991;19:1759–1766. doi: 10.1093/nar/19.8.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehresmann C, Baudin F, Mougel M, Romby P, Ebel J-P, Ehresmann B. Nucleic Acids Res. 1987;15:9109–9128. doi: 10.1093/nar/15.22.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Putz J, Puglisi J D, Florentz C, Giegé R. EMBO J. 1993;12:2949–2957. doi: 10.1002/j.1460-2075.1993.tb05957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudinger J, Hillenbrandt R, Sprinzl M, Giegé R. EMBO J. 1996;15:650–657. [PMC free article] [PubMed] [Google Scholar]
- 27.D’Alessio G, Riordan J F, editors. Ribonucleases: Structures and Functions. San Diego: Academic; 1997. [Google Scholar]
- 28.Saito H, Richardson C C. Cell. 1981;27:533–542. doi: 10.1016/0092-8674(81)90395-0. [DOI] [PubMed] [Google Scholar]
- 29.Montanez C, Bueno J, Schmeissner U, Court D L, Guarneros G. J Mol Biol. 1986;191:29–37. doi: 10.1016/0022-2836(86)90420-1. [DOI] [PubMed] [Google Scholar]
- 30.Seeman N, Rosenberg J M, Rich A. Proc Natl Acad Sci USA. 1976;73:804–808. doi: 10.1073/pnas.73.3.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coburn G A, Mackie G A. J Biol Chem. 1996;271:15776–15781. doi: 10.1074/jbc.271.26.15776. [DOI] [PubMed] [Google Scholar]
- 32.Haugel-Nielsen J, Hajnsdorf E, Régnier P. EMBO J. 1996;15:3144–3152. [PMC free article] [PubMed] [Google Scholar]
- 33.Dunn J J. J Biol Chem. 1976;251:3807–3814. [PubMed] [Google Scholar]
- 34.Hjalt T A H, Wagner E G H. Nucleic Acids Res. 1995;23:571–579. doi: 10.1093/nar/23.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wood H, Luirink J, Tollervey D. Nucleic Acids Res. 1992;20:5919–5925. doi: 10.1093/nar/20.22.5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christiansen J. Nucleic Acids Res. 1988;16:7457–7476. doi: 10.1093/nar/16.15.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu F, Cohen S N. Nature (London) 1995;374:180–183. doi: 10.1038/374180a0. [DOI] [PubMed] [Google Scholar]
- 38.Bouvet P, Belasco J G. Nature (London) 1992;360:488–491. doi: 10.1038/360488a0. [DOI] [PubMed] [Google Scholar]
- 39.Deutscher M P. In: Nucleases. Linn S M, Lloyd R S, Roberts R J, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1993. pp. 377–406. [Google Scholar]
- 40.Polson A G, Bass B L. EMBO J. 1994;13:5701–5711. doi: 10.1002/j.1460-2075.1994.tb06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Polson A G, Bass B L, Casey J L. Nature (London) 1996;380:454–456. doi: 10.1038/380454a0. [DOI] [PubMed] [Google Scholar]