

Abstract
The visual pigment rhodopsin is a prototypical G protein-coupled receptor. These receptors have seven transmembrane helices and are activated by specific receptor–ligand interactions. Rhodopsin is unusual in that its retinal prosthetic group serves as an antagonist in the dark in the 11-cis conformation but is rapidly converted to an agonist on photochemical cis to trans isomerization. Receptor–ligand interactions in rhodopsin were studied in the light and dark by regenerating site-directed opsin mutants with synthetic retinal analogues. A progressive decrease in light-dependent transducin activity was observed when a mutant opsin with a replacement of Gly121 was regenerated with 11-cis-retinal analogues bearing progressively larger R groups (methyl, ethyl, propyl) at the C9 position of the polyene chain. A progressive decrease in light activity was also observed as a function of increasing size of the residue at position 121 for both the 11-cis-9-ethyl- and the 11-cis-9-propylretinal pigments. In contrast, a striking increase of receptor activity in the dark—i.e., without chromophore isomerization—was observed when the molecular volume at either position 121 of opsin or C9 of retinal was increased. The ability of bulky replacements at either position to hinder ligand incorporation and to activate rhodopsin in the dark suggests a direct interaction between these two sites. A molecular model of the retinal-binding site of rhodopsin is proposed that illustrates the specific interaction between Gly121 and the C9 methyl group of 11-cis-retinal. Steric interactions in this region of rhodopsin are consistent with the proposal that movement of transmembrane helices 3 and 6 is concomitant with receptor activation.
G protein-coupled receptors (GPCRs) comprise a large family of integral membrane proteins involved in signal transduction across cell membranes (1). These receptors share a common architecture consisting of seven transmembrane (TM) helices. The cytoplasmic surface of the protein, which provides the binding site for heterotrimeric G proteins, is formed by the C terminus and the three loops connecting the transmembrane helices. The cavity formed within the helix bundle, and in some cases the extracellular surface of the protein, provide ligand-binding sites. The range of different GPCRs and their associated ligands is staggering. For example, both olfaction and visual transduction are mediated by multiple GPCRs. Sequence differences provide selectivity in binding different ligands and generate functional differences between receptor subclasses. In the case of olfaction, there are at least 1,000 distinct genes for putative GPCRs that may recognize the many structurally diverse odorants (2).
In contrast, all visual receptors, from those of insects to humans, have 11-cis-retinal or its derivative as their photoreactive chromophore. Differences in the primary structures of these receptors give rise to their characteristic functional differences. For example, the absorption maxima (λmax) of cone pigments, which are responsible for trichromatic color vision in humans, range from about 430 to 565 nm because of specific amino acid differences (3, 4).
In rhodopsin, the retinal is attached to Lys296 through a protonated Schiff base bond. The retinal-binding site favors the bent 11-cis geometry, and when bound, the 11-cis-retinal suppresses a small but measurable basal activity of the apoprotein opsin at low pH (5, 6). Linear all-trans- and 13-cis-retinals are not able to bind efficiently and/or stably in the dark (7–9). Because the visual receptors all bind 11-cis-retinal, key residues that are responsible for specific ligand recognition are expected to be conserved in this subclass of GPCR.
The retinal-binding pocket is largely defined by conserved residues on TM helices 3, 5, and 6. Based in part on NMR constraints, we recently proposed a detailed model for how the retinal binds in the interior of rhodopsin (10, 11). The counterion to the protonated Schiff base is Glu113, which is located at the extracellular end of TM helix 3 (12–14). This residue is conserved in the vertebrate visual receptors (15). A second glutamic acid (Glu122) is found on TM helix 3 only in the rhodopsin receptors and is pairwise conserved with His211 on TM helix 5 (11, 15, 16). There are several conserved aromatic residues on helix 6 that contribute to the retinal-binding pocket. Crosslinking studies with retinal analogues have shown that Trp265 is near the β-ionone ring of the retinal (17, 18). Trp265 is strongly conserved in the visual pigments, as well as in the GPCR family in general. The environment of Trp265 changes in the formation of metarhodopsin II (19). Tyr268 encloses the retinal-binding pocket on the extracellular side of the receptor and is strictly conserved in the visual pigments (11, 15, 16).
We recently identified Gly121 to be a crucial residue in the retinal-binding site. Gly121 is strictly conserved in the visual pigments (20) but not in other GPCRs. Replacement of Gly121 by a larger residue causes increased steric interactions with the chromophore that affect spectral properties of the mutant pigments (20, 21). In addition, Gly121 mutants regenerated with 11-cis-retinal are able to catalyze GTP uptake by transducin in the dark (29). An increase in the packing volume at the position 121 side chain is associated with larger levels of dark activity. We concluded that bulky residues introduced in place of Gly121 make van der Waals contacts with the retinal chromophore, most likely at the C9 position (11, 20).‖ This conclusion is consistent with the observation of Ganter et al. (22) that regeneration of opsin with the 9-demethylretinal analogue resulted in reduced ability of the artificial pigment to activate transducin.
In this paper, we combine Gly121 mutants with various retinal analogues to demonstrate the proposed interaction between Gly121 and the C9 methyl group of the retinal chromophore. The hypothesis is that an increase in the packing volume caused by changing the moiety at the C9 position should augment dark activity and mimic the effect of replacement of Gly121. Retinals bearing ethyl and propyl substituents at position C9 of the retinal (11-cis-9-ethylretinal and 11-cis-9-propylretinal, respectively) were synthesized and reconstituted with opsin and seven opsin mutants with amino acid replacements at position 121. We show that bulky replacements at either position 121 of the opsin or at C9 of the retinal hinder ligand incorporation and activate rhodopsin in the dark. In addition, we compare the ability of different retinal isomers, β-ionone, and its related compounds (Fig. 1) to activate the G121L mutant opsin. These studies identify the specific region of the retinal chromophore that is in steric contact with TM helix 3 in the vicinity of Gly121.
Figure 1.
Structures of retinal isomers and synthetic retinal analogues used in the present study. Individual carbons are numbered for 11-cis-retinal. For simplicity, common names are given for analogues altered at the C9 position. For example, 11-cis-9-ethylretinal would formally be 11-cis-19-methylretinal. All compounds are shown in the 6-s-cis conformation.
MATERIALS AND METHODS
Synthesis of C9-Ethyl- and C9-Propyl-11-cis-Retinals.
All reagents were from Aldrich and used without further purification. β-Cyclocitral and N-methoxy-N-methyl-3-(2,6,6-trimethyl-1-cyclohexenyl)-2-propenamide were prepared as previously described (23). The C9-ethyl- and C9-propylretinals were prepared from the propenamide by reaction with the appropriate Grignard reagent, CH3CH2MgCl or CH3CH2CH2MgCl, respectively. The ketone products of the Grignard reaction were converted into the corresponding retinals by using standard polyene elongation methods (23). The required 11-cis isomers were obtained by irradiation in acetonitrile followed by HPLC separation. The structure and configuration of the retinals were confirmed by 1H NMR and high resolution mass spectrometry.
Expression of Opsins, Regeneration with Retinal Analogues, and Measurement of Transducin Activity.
The procedures for mutant construction, expression, and characterization of the opsin Gly121 replacement mutants have been described previously (20). COS cell membrane suspensions containing wild-type or mutant opsins (30 μl) were incubated with 1 μl of either native retinal or retinal analogues (5 mM ethanolic solution) in the dark at room temperature for 30 min before assaying for transducin activity. The transducin activation assays of COS cell membrane samples were carried out as previously described (20).
RESULTS AND DISCUSSION
Activation of Opsin by 11-cis-9-Ethyl- and 11-cis-9-Propylretinal in the Dark.
We have previously shown that an increase in the volume of the residue side chain at position 121 roughly correlated with an increase in transducin activity in the dark on the addition of all-trans-retinal, the natural agonist for rhodopsin activation (20). The size of the residue at position 121 also correlated with the degree of blue shift in the λmax of the pigment and its reactivity with hydroxylamine in the dark. These changes suggested that a specific steric interaction exists between the retinal chromophore and Gly121. The interaction between Gly121 and retinal appears to be specific, because similar bulky replacements at Gly114 and Ala117, two and one helical turns away on TM helix 3, respectively, show no such effects (20).
Recently, we have shown that 11-cis-retinal, normally an inverse agonist of rhodopsin activation, can serve as a partial agonist for the Gly121 mutant opsins (24). Gly121 mutants regenerated with 11-cis-retinal are able to catalyze GTP uptake by transducin in the dark (Fig. 2 and Table 1). Furthermore, the relative dark activity correlates with the size of the side chain at position 121. The first question we posed in this study was whether an increase in the molecular volume of the side chain at the C9 position of retinal would also lead to similar transducin activity in the dark. To address this question, retinal analogues with an ethyl or propyl substituent at C9 were synthesized. The structures of 11-cis-retinal, 11-cis-9-ethylretinal, and 11-cis-9-propylretinal analogues are shown in Fig. 1. The volume occupied by the methyl group at position 9 increases from ≈25 Å3 to ≈50 and 75 Å3 in the ethyl- and propylretinal analogues, respectively (25). These three retinals were incubated with COS cell membranes containing wild-type opsins to generate opsin–retinal complexes. Receptor activation was determined by the ability of opsin in the presence of each retinal analogue to catalyze guanine nucleotide exchange by transducin with a filter-binding assay (20).
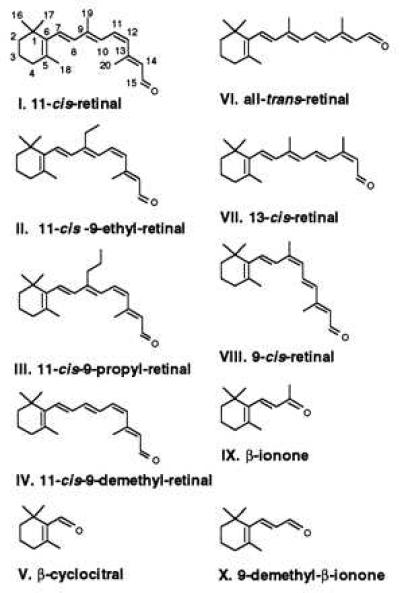
Figure 2.
Activation of transducin by opsin–retinal complexes in COS cell membranes in the dark. Numerical values are given in Table 1. (A) Dark activity of G121A, G121S, G121T, G121V, G121L, G121I, and G121W mutant opsins incubated with 11-cis-retinal. (B) Dark activity of wild-type opsin incubated with 11-cis-retinal, 11-cis-9-ethylretinal, and 11-cis-9-propylretinal.
Table 1.
Transducin activation by mutant opsins in the presence of C9-retinal analogues measured in COS cell membranes
| Mutant | 11-cis-Retinal, Dark* | 11-cis-9-Ethylretinal
|
11-cis-9-Propylretinal
|
||
|---|---|---|---|---|---|
| Dark* | Light† | Dark* | Light† | ||
| Wild type | 0.8 ± 0.2 | 5.4 ± 1.2 | 80.2 ± 19.5 | 11.2 ± 5.3 | 55.9 ± 10.5 |
| G121A | 2.3 ± 1.3 | 4.6 ± 1.2 | 86.1 ± 6.3 | 7.6 ± 0.6 | 78.6 ± 7.2 |
| G121S | 5.0 ± 2.6 | 7.7 ± 2.3 | 62.4 ± 15.4 | 7.2 ± 3.2 | 53.9 ± 11.1 |
| G121T | 5.6 ± 1.8 | 10.4 ± 3.5 | 41.2 ± 15.0 | 17.5 ± 6.8 | 16.4 ± 7.9 |
| G121V | 7.8 ± 3.7 | 8.8 ± 0.9 | 47.0 ± 16.4 | 4.4 ± 1.2 | 19.8 ± 9.4 |
| G121L | 14.6 ± 2.5 | 32.1 ± 4.8 | 49.7 ± 16.4 | 65.1 ± 6.8 | 16.1 ± 6.3 |
| G121I | 21.1 ± 4.8 | 47.7 ± 12.5 | 32.2 ± 10.8 | 78.9 ± 6.2 | 14.5 ± 4.6 |
| G121W | 30.0 ± 6.2 | 29.9 ± 4.0 | 15.4 ± 4.7 | 41.4 ± 15.3 | 10.2 ± 4.0 |
All of the values are presented as the mean ± SE from at least three measurements using at least two independent membrane preparations.
The dark activities are given as a percentage of transducin guanosine 5′-[γ-thio]triphosphate (GTP[γS]) activity measured in the dark normalized to the GTP[γS] uptake of the sample measured under continuous illumination.
The light activities of the mutant opsins in the presence of retinal analogues are percentages of the transducin activation by the opsin–retinal analogue complex in the light normalized to the transducin activation by the same opsin regenerated with retinal.
Fig. 2 shows that extension of the C9 methyl group to ethyl or propyl leads to increased receptor activation in the dark—i.e., without chromophore isomerization—similar to that observed in Gly121 mutants regenerated with 11-cis-retinal. Increasing the size of the side chain at position 121 of opsin by mutagenesis or increasing the size of the group at position C9 of 11-cis-retinal by analogue synthesis results in transducin activation in the dark. These observations are consistent with the hypothesis that there is a direct steric interaction between Gly121 and the C9 methyl group of retinal in rhodopsin.
Synergistic Activation of Opsin by Increasing the Molecular Volume at Both Gly121 and Retinal C9.
Twenty-four opsin–retinal complexes were prepared by combining COS cell membranes containing either wild-type opsin or one of the seven Gly121 mutant opsins (G121A, V, S, T, L, I, and W) with 11-cis-retinal, 11-cis-9-ethylretinal, or 11-cis-9-propylretinal. Each complex was tested for its ability to activate transducin in the presence (light activity) and the absence (dark activity) of light. The ability of the 24 opsin–retinal complexes to activate transducin in the light is shown in Fig. 3. All of the activities are normalized to the light activity of wild-type rhodopsin. The light activity reflects both the degree of binding of the retinal analogue with each opsin and the intrinsic ability of the analogue to activate the receptor after isomerization. The relative contributions of these two effects are difficult to quantitate because the complexes between 9-ethyl or 9-propylretinal and the Gly121 opsins are typically not stable enough to be purified in dodecyl maltoside detergent. Detergent-solubilized pigment is needed to assess the level of regeneration through measurements of the spectral ratio (20).
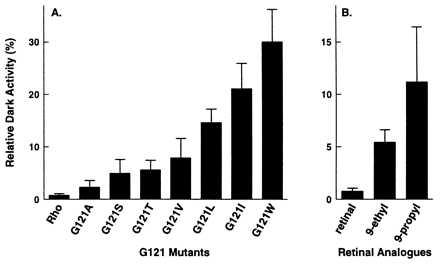
Figure 3.
Three-dimensional plot of light-dependent transducin activation by 24 opsin–retinal analogue complexes in COS cell membranes. The level of light-dependent transducin activation is plotted as a function of the amino acid at position 121 in the recombinant opsin and the chromophore used for regeneration in the membranes. In the case of wild-type rhodopsin, glycine is located at position 121 and the chromophore is 11-cis-retinal. The size of the amino acid side chain at position 121 is varied in combination with the size of the aliphatic group on the C9 position of the retinal polyene chain. Transducin activities are all normalized to the light activity of wild-type rhodopsin and are directly comparable. Numerical values are presented in Table 1.
The Gly121 mutants regenerated with 11-cis-retinal in most cases can fully activate transducin (Fig. 3). This observation suggests that a bulky side chain at position 121 can be tolerated. The kinetics of the retinal regeneration, however, are greatly reduced for G121L opsin compared with wild-type opsin (20). In contrast to the results with 11-cis-retinal, there was a decrease in the ability of the Gly121 opsins to activate transducin when regenerated with 11-cis-9-ethylretinal (Fig. 3). This decrease was even more marked for the Gly121 opsins regenerated with 11-cis-9-propylretinal. Based on the observation that the 11-cis-9-ethyl- and 11-cis-9-propylretinal analogues are capable of significantly activating wild-type and G121A opsin in the light (Fig. 3), these retinals seem to be intrinsically capable of full light-induced activation. If, in general, the bound 11-cis-9-ethyl- and 11-cis-9-propylretinals can fully activate rhodopsin on illumination, then lowered ligand incorporation is most likely the primary cause for the decreased transducin activity observed in Fig. 3 and likely results from an increase in the size of the side chain at position 121 and/or in the size of the group at the C9 position of the retinal.
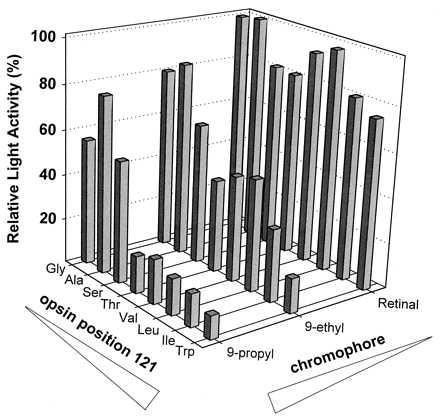
The transducin activities of the 24 opsin–retinal complexes in the dark are presented in Fig. 4. The dark activity of any given complex is lower than 30% of the light activity of rhodopsin. However, in a few cases (e.g., 11-cis-9-propylretinal–G121I) the dark activity is very close to the light activity of the same complex. By normalizing the dark activity to the light activity measured for the same opsin–retinal complex in COS cell membranes, differences in regeneration levels and the intrinsic ability of each analogue to activate the mutant opsins are mitigated. Normalization allows a reasonable comparison of the ability of each opsin–retinal complex to activate transducin in the dark. The relative dark activity is presented as a function of both the residue at position 121 and the retinal analogue in a three-dimensional plot in Fig. 4. A striking trend of increased dark activity is observed as a function of increasing molecular volume at both position 121 on TM helix 3 and at C9 of the retinal (the only exceptions being 9-ethylretinal–G121W and 9-propylretinal–G121W). The dark activity ranges from undetectable for native rhodopsin to 80% for the 9-propylretinal–G121I complex. The trend strongly suggests that the high levels of dark activity result from the increased steric interaction between the side chain at position 121 and the group at the C9 of retinal. These two factors appear to act synergistically, implying a direct interaction between the two positions. In contrast to 11-cis-9-ethyl and 11-cis-9-propylretinals, 11-cis-9-demethylretinal did not activate any of the Gly121 mutant opsins (unpublished results).
Figure 4.
Three-dimensional plot of activities of the 24 opsin–retinal analogue complexes in COS cell membranes in the dark. The level of transducin activation in the dark is plotted as a function of the amino acid at position 121 in the recombinant opsin and the chromophore used for regeneration in the membranes. In the case of wild-type rhodopsin, glycine is located at position 121, and the chromophore is 11-cis-retinal. The size of the amino acid side chain at position 121 is varied in combination with the size of the aliphatic group on the C9 position of the retinal polyene chain. The relative dark activity is defined as the percent activity measured in the dark vs. that measured in the light. Numerical values are presented in Table 1.
Activation of G121L Opsin by β-Ionone.
Retinal analogue studies have shown that β-ionone can competitively inhibit the binding of 11-cis-retinal (26). Moreover, the binding of β-ionone is faster than the formation of the Schiff base linkage to 11-cis-retinal when both ligands are co-incubated with opsin (26). These studies led to the suggestion of a high affinity β-ionone ring-binding site in rhodopsin. Additional support for this idea has come from studies with acyclic retinal analogues, which reveal a requirement for the cyclohexyl ring for binding of the chromophore and forming activated rhodopsin (27, 28). Moreover, Oprian and coworkers (29) have shown that the Schiff base linkage is not necessary for receptor activation. Together these studies argue that the hydrophobic β-ionone ring drives the binding of retinal to opsin.
We measured transducin activation by wild-type and G121L opsin in the dark in the presence of β-ionone and other related compounds, 9-demethyl-β-ionone and β-cyclocitral (Fig. 1). The aim of the experiment was to identify the region of 11-cis-retinal that is necessary for conferring dark activity on G121L opsin. β-Ionone, 9-demethyl-β-ionone, and β-cyclocitral do not significantly activate wild-type opsin (Fig. 5 and Table 2). This is consistent with the finding that β-ionone is capable of relieving the desensitization on bleaching of cone cells (30). In contrast, G121L opsin can be significantly activated by β-ionone. The extent of activity was similar to that induced by 11-cis-retinal. Therefore, β-ionone, like 11-cis-retinal, serves as a partial agonist for G121L mutant opsin. Because β-ionone has slightly higher agonist activity than 11-cis-retinal, the polyene segment from C10 to the Schiff base does not contribute to the partial agonism of 11-cis-retinal. The agonist activity is greatly reduced when the C9 methyl group of β-ionone is removed (9-demethyl-β-ionone). The C9 methyl group of β-ionone must play a role in the agonist activity of β-ionone. In contrast, the removal of the C9 methyl group from 11-cis-retinal (11-cis-9-demethylretinal) abolished the agonist activity of 11-cis-retinal. This observation, together with the β-ionone results, suggests that the polyene chain from C10 to the Schiff base of 11-cis-retinal may serve to antagonize the basal activity of the receptor. The agonist efficiency of β-cyclocitral, which can be viewed as β-ionone further shortened by removing C8 and C9 (Fig. 1), is again lower than that of 9-demethyl-β-ionone. The activity of G121L opsin in the presence of β-cyclocitral is only slightly higher than that of G121L opsin alone. In summary, the order of the efficiency of these compounds for activating G121L opsin in the dark is: β-ionone > 11-cis-retinal ≫ 9-demethyl-β-ionone > β-cyclocitral. The C9 methyl group appears extremely important for conferring partial agonist activity of both β-ionone and 11-cis-retinal in the Gly121 mutants.
Figure 5.
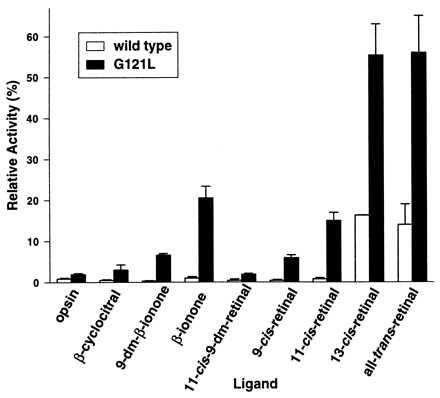
Transducin activation by G121L mutant opsin in the presence of different compounds in the dark. The relative dark activity is presented as the percent activity measured in the dark vs. that measured in the light for each opsin regenerated with 11-cis-retinal. Numerical values are presented in Table 2.
Table 2.
Transducin activation by wild-type or G121L mutant opsin in the presence of various retinal isomers and analogues
| Retinal analogue | Wild type | G121L |
|---|---|---|
| None (opsin alone) | 0.9 ± 0.2 | 1.9 ± 0.3 |
| β-Cyclocitral (V) | 0.5 ± 0.2 | 3.0 ± 1.2 |
| 9-Demethyl-β-ionone (X) | 0.3 ± 0.1 | 6.6 ± 0.4 |
| β-Ionone (IX) | 1.0 ± 0.4 | 20.5 ± 2.9 |
| 11-cis-9-Demethylretinal (IV) | 0.4 ± 0.4 | 1.9 ± 0.3 |
| 9-cis-Retinal (VIII) | 0.4 ± 0.2 | 5.9 ± 0.7 |
| 11-cis-Retinal (I) | 0.8 ± 0.3 | 14.6 ± 2.5 |
| 13-cis-Retinal (VII) | 16.3 ± 0.1 | 55.4 ± 7.6 |
| All-trans-retinal (VI) | 14.3 ± 4.8 | 56.2 ± 4.8 |
All of the values are presented as the mean ± SE from at least three measurements using at least two independent membrane preparations. The activities measured in the dark are given as a percentage of transducin guanosine 5′-[γ-thio]triphosphate (GTP[γS]) activity measured in the dark normalized to the GTP[γS] uptake of the sample measured under continuous illumination. Structures of the analogues are shown in Fig. 1.
Activation of G121L Opsin by Various Isomers of Retinal.
The retinal isomers 9-cis-, 11-cis-, 13-cis-, and all-trans-retinal were tested in parallel for their ability to activate wild-type and G121L opsins in the dark (Fig. 5). Interestingly, 13-cis-retinal activates wild-type opsin in COS cell membranes although it has been reported to be unable to form a stable pigment with opsin (31). The behavior of 13-cis-retinal is very similar to that of all-trans-retinal. No activation of opsin in the dark was observed for 11-cis- and 9-cis-retinal. Palczewski et al. (32) observed a similar correlation for transducin activation as well as for opsin phosphorylation by rhodopsin kinase. In contrast to the case of wild-type opsin, all of the retinal isomers above can activate G121L opsin and therefore can act as partial agonists. The order of the agonist efficiency of the retinal isomers is: all-trans ≈ 13-cis > 11-cis > 9-cis. The trend suggests that the more similar an isomeric structure is to the all-trans conformation, the higher its agonist efficiency. It is noteworthy that in every aspect tested, 13-cis-retinal and all-trans-retinal behave nearly identically. Although the all-trans conformation is the only chromophore normally formed after illumination of the 11-cis-retinal in the binding site of rhodopsin, wild-type opsin and G121L mutant opsin may not distinguish between the 13-cis and all-trans isomers when added exogenously.
Steric Interactions in the Retinal-Binding Site of Rhodopsin.
In this study, we identified a specific steric interaction between the C9 methyl group of retinal and Gly121 on TM helix 3 of opsin. This structural constraint can be incorporated into a molecular model proposed earlier (11), which was based in part on the position of the retinylidene Schiff base relative to its counterion (10) and the complementarity between Gly121 and Phe261 on TM helix 6 (20, 21). Fig. 6 shows the molecular model of rhodopsin illustrating the interaction between Gly121 (green) and the C9 methyl group (yellow) of the retinal chromophore. The β-ionone portion is thought to be tightly packed in the interior of the protein between TM helices 3, 5, and 6. Key residues surrounding the chromophore from the β-ionone ring to C9 are Gly121 and Glu122 on helix 3, His211 on helix 5, and Phe261, Trp265, and Tyr268 on helix 6 (11, 15, 16, 20, 21). Gly121 is strictly conserved in the visual receptors, Glu122 and His211 are pairwise conserved in the vertebrate rhodopsin-like receptors, and the three aromatic residues on helix 6 are largely conserved across the GPCR family. Residue 211 is a serine in insect rhodopsins, which interestingly may interact with a 3-OH group on the ionone ring in these pigments (33). In the β-adrenergic receptor, two serines that are essential for ligand binding and receptor activation are found at positions 204 and 207, also on TM helix 5 (34). These residues interact with the hydroxyl groups on the catechol ring, whereas a phenylalanine on TM helix 6 is thought to interact with the catechol ring itself through π-orbital interactions.
Figure 6.
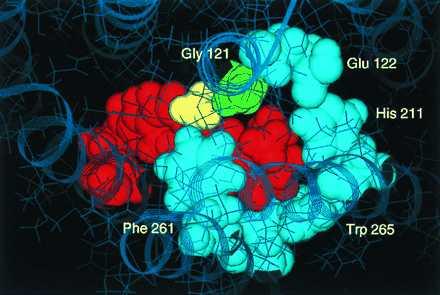
Molecular model of the 11-cis-retinal (red) binding site in rhodopsin centering on the β-ionone ring region and demonstrating the interaction of the retinal C9-methyl group (yellow) with Gly121 (green). The view is from above the plane of the membrane. Several of the residues forming the β-ionone-binding pocket (Glu122, His211, Trp265, Phe261) are shown in light blue.
Our model reveals that replacement of the Gly121 with leucine leads to overlap of the leucine side chain with the β-ionone portion of the retinal chromophore between C4 and C9. This interaction results from the fact that the retinal is tightly packed in the vicinity of position 121 and the Cα—Cβ bond is oriented toward the extracellular surface. Similarly, replacement of the C9 methyl group of the retinal with a propyl group leads to overlap of the propyl group carbons with TM helix 3. The overlap between TM helix 3 and retinal is even more exaggerated when there are bulky substitutions on both position 121 and C9 of retinal, such as in the 11-cis-9-propylretinal–G121I complex. Such molecular overlap in the model may have two possible consequences in the actual receptor that are not mutually exclusive. First, the ligand cannot fully occupy the overcrowded binding site. Second, if the ligand does bind, then there has to be outward movements of the relevant helices to relieve the increased steric interaction in the binding site. It is indeed observed that no pigment can be purified in detergent when wild-type, G121A, G121V, and G121L opsins are regenerated with 11-cis-9-ethyl- or 11-cis-9-propylretinal. Furthermore, light-independent transducin activation of Gly121 mutant opsins in the presence of 11-cis-9-ethylretinal in COS cell membranes decreases when the size of the side chain at position 121 increases (Fig. 3). This decrease in transducin activation is even more pronounced when 11-cis-9-propylretinal is used (Fig. 3). As discussed above, we believe that the decreased transducin activity primarily reflects the decreased level of ligand incorporation in these opsin–retinal complexes.
The increased steric interaction between opsin and retinal either by a leucine substitution at 121 or by the presence of a C9 propyl group on retinal can cause the outward movement of TM helix 3. Because we have shown that Phe261 on TM 6 and Gly121 on TM helix 3 interact with retinal in a complementary manner (21), it is also possible that the increased steric interaction between TM helix 3 and retinal can be partially relieved by the outward movement of TM helix 6. Site-directed spin labeling studies of rhodopsin demonstrate that outward rigid body movements of TM helices 3 and 6 are involved during receptor activation (35–37). The partial movements of TM helices 3 and/or 6 caused by bulky substitution on position 121 of opsin, or C9 of retinal, or both, are likely to be the mechanism for the partial receptor activation observed in the dark (Figs. 2 and 4). The role of TM helices 3 and 6 (relative to 1, 2, and 7) is consistent with the higher dark activity of β-ionone relative to 11-cis-retinal.
Finally, we propose that the specific interaction between the strictly conserved Gly121 in visual pigments and the retinal ligand is important for receptor activation. Because the increased steric interaction between the two positions can cause partial receptor activation in the dark, it is likely that they are both involved in the formation of R* (the active state of rhodopsin) during light activation. In several other GPCRs, the functional importance of the residue at the equivalent position of rhodopsin Gly121 has been demonstrated (38). Studies on the dopamine D2 receptor show that this site is water-accessible and forms part of the ligand-binding pocket (39, 40). Val118 in the NK-1 receptor confers species selectivity for several nonpeptide antagonists (41, 42), whereas Ser159 in the 5-HT2A receptor is involved in agonist binding (43). It is therefore conceivable that this position in the middle of TM helix 3 may be important for ligand binding and/or receptor activation of GPCRs in general.
Acknowledgments
M.H. is a Charles H. Revson Fellow in Biomedical Research. T.P.S. is an Associate Investigator of the Howard Hughes Medical Institute. This work was supported in part by the Allene Reuss Memorial Trust (to T.P.S.) and by National Institutes of Health Grant GM 41412 (to S.O.S.).
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: GPCR, G protein-coupled receptor; TM, transmembrane.
According to IUPAC-IUB nomenclature, the carbon of the methyl group on the C9 position is numbered C19, and 9-demethylretinal would be denoted 19-nor-retinal. Likewise, the common names 9-ethylretinal and 9-propylretinal would formally be 19-methylretinal and 19-ethylretinal, respectively. For clarity and consistency with much of the existing biochemical literature, we reluctantly use the common names in this report.
References
- 1.Strader C D, Fong T M, Tota M R, Underwood D. Annu Rev Biochem. 1994;63:101–132. doi: 10.1146/annurev.bi.63.070194.000533. [DOI] [PubMed] [Google Scholar]
- 2.Buck L, Axel R. Cell. 1991;65:175–187. doi: 10.1016/0092-8674(91)90418-x. [DOI] [PubMed] [Google Scholar]
- 3.Aseñjo A B, Rim J, Oprian D D. Neuron. 1994;12:1131–1138. doi: 10.1016/0896-6273(94)90320-4. [DOI] [PubMed] [Google Scholar]
- 4.Merbs S L, Nathans J. Nature (London) 1992;356:433–435. doi: 10.1038/356433a0. [DOI] [PubMed] [Google Scholar]
- 5.Cohen G B, Oprian D D, Robinson P R. Biochemistry. 1992;31:12592–12601. doi: 10.1021/bi00165a008. [DOI] [PubMed] [Google Scholar]
- 6.Robinson P R, Cohen G B, Zhukovsky E A, Oprian D D. Neuron. 1992;9:719–725. doi: 10.1016/0896-6273(92)90034-b. [DOI] [PubMed] [Google Scholar]
- 7.Hubbard R, Wald G. J Gen Physiol. 1952;31–32:268–313. doi: 10.1085/jgp.36.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsumoto H, Yoshizawa T. Vision Res. 1978;18:607–609. doi: 10.1016/0042-6989(78)90212-2. [DOI] [PubMed] [Google Scholar]
- 9.Liu R S H, Asato A E, Denny M, Mead D. J Am Chem Soc. 1984;106:8298–8300. [Google Scholar]
- 10.Han M, Smith S O. Biochemistry. 1995;34:1425–1432. doi: 10.1021/bi00004a037. [DOI] [PubMed] [Google Scholar]
- 11.Shieh T, Han M, Sakmar T P, Smith S O. J Mol Biol. 1997;269:373–384. doi: 10.1006/jmbi.1997.1035. [DOI] [PubMed] [Google Scholar]
- 12.Sakmar T P, Franke R R, Khorana H G. Proc Natl Acad Sci USA. 1989;86:8309–8313. doi: 10.1073/pnas.86.21.8309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhukovsky E A, Oprian D D. Science. 1989;246:928–930. doi: 10.1126/science.2573154. [DOI] [PubMed] [Google Scholar]
- 14.Nathans J. Biochemistry. 1990;29:937–942. doi: 10.1021/bi00456a013. [DOI] [PubMed] [Google Scholar]
- 15.Alkorta I, Du P. Protein Eng. 1994;7:1231–1238. doi: 10.1093/protein/7.10.1231. [DOI] [PubMed] [Google Scholar]
- 16.Pogozheva I D, Lomize A L, Mosberg H I. Biophys J. 1997;70:1963–1985. doi: 10.1016/S0006-3495(97)78842-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang H, Lerro K A, Yamamoto T, Lien T H, Sastry L, Gawinowicz M A, Nakanishi K. J Am Chem Soc. 1994;116:10165–10173. [Google Scholar]
- 18.Nakayama T A, Khorana H G. J Biol Chem. 1990;265:15762–15769. [PubMed] [Google Scholar]
- 19.Lin S W, Sakmar T P. Biochemistry. 1996;35:11149–11159. doi: 10.1021/bi960858u. [DOI] [PubMed] [Google Scholar]
- 20.Han M, Lin S W, Smith S O, Sakmar T P. J Biol Chem. 1996;271:32330–32336. doi: 10.1074/jbc.271.50.32330. [DOI] [PubMed] [Google Scholar]
- 21.Han M, Lin S W, Minkova M, Smith S O, Sakmar T P. J Biol Chem. 1996;271:32337–32342. doi: 10.1074/jbc.271.50.32337. [DOI] [PubMed] [Google Scholar]
- 22.Ganter U M, Schmid E D, Perez-Sala D, Rando R R, Siebert F. Biochemistry. 1989;28:5954–5962. doi: 10.1021/bi00440a036. [DOI] [PubMed] [Google Scholar]
- 23.Groesbeek M, Lugtenburg J. Photochem Photobiol. 1996;56:903–908. [Google Scholar]
- 24.Han M, Lou J, Nakanishi K, Sakmar T P, Smith S O. J Biol Chem. 1997;272:23091–23095. doi: 10.1074/jbc.272.37.23081. [DOI] [PubMed] [Google Scholar]
- 25.Chothia C. Nature (London) 1975;254:304–308. doi: 10.1038/254304a0. [DOI] [PubMed] [Google Scholar]
- 26.Matsumoto H, Yoshizawa T. Nature (London) 1975;258:523–526. doi: 10.1038/258523a0. [DOI] [PubMed] [Google Scholar]
- 27.Nakanishi K, Crouch R. Isr J Chem. 1995;35:253–272. [Google Scholar]
- 28.Jäger F, Jäger S, Krautle O, Friedman N, Sheves M, Hofmann K P, Siebert F. Biochemistry. 1994;33:7389–7397. doi: 10.1021/bi00189a045. [DOI] [PubMed] [Google Scholar]
- 29.Zhukovsky E A, Robinson P R, Oprian D D. Science. 1991;251:558–560. doi: 10.1126/science.1990431. [DOI] [PubMed] [Google Scholar]
- 30.Jin J, Crouch R K, Corson D W, Katz B M, MacNichol E F, Cornwall M C. Neuron. 1993;11:513–522. doi: 10.1016/0896-6273(93)90155-k. [DOI] [PubMed] [Google Scholar]
- 31.Liu R S H, Mirzadegan T. J Am Chem Soc. 1988;110:8617–8623. [Google Scholar]
- 32.Palczewski K, Jåger S, Buczylko J, Crouch R K, Bredberg D L, Hofmann K P, Asson-Batres M A, Saari J C. Biochemistry. 1994;33:13741–13750. doi: 10.1021/bi00250a027. [DOI] [PubMed] [Google Scholar]
- 33.Oprian D D. J Bioenerg Biomembr. 1992;24:211–217. doi: 10.1007/BF00762679. [DOI] [PubMed] [Google Scholar]
- 34.Strader C D, Candelore M R, Hill W S, Sigal I S, Dixon R A F. J Biol Chem. 1989;264:13572–13578. [PubMed] [Google Scholar]
- 35.Farahbakhsh Z T, Hideg K, Hubbell W L. Science. 1993;262:1416–1419. doi: 10.1126/science.8248781. [DOI] [PubMed] [Google Scholar]
- 36.Altenbach C, Yang K, Farrens D L, Farahbakhsh Z T, Khorana H G, Hubbell W L. Biochemistry. 1996;35:12470–12478. doi: 10.1021/bi960849l. [DOI] [PubMed] [Google Scholar]
- 37.Farrens D L, Altenbach C, Yang K, Hubbell W L, Khorana H G. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 38.van Rhee A M, Jacobson K A. Drug Dev Res. 1996;37:1–38. doi: 10.1002/(SICI)1098-2299(199601)37:1<1::AID-DDR1>3.0.CO;2-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Javitch J A, Li X, Kaback J, Karlin A. Proc Natl Acad Sci USA. 1994;91:10355–10359. doi: 10.1073/pnas.91.22.10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Javitch J A, Fu D, Chen J, Karlin A. Neuron. 1995;14:825–831. doi: 10.1016/0896-6273(95)90226-0. [DOI] [PubMed] [Google Scholar]
- 41.Fong T M, Yu H, Strader C D. J Biol Chem. 1992;267:25668–25671. [PubMed] [Google Scholar]
- 42.Jensen C J, Gerard N P, Schwartz T W, Gether U. Mol Pharmacol. 1994;45:294–299. [PubMed] [Google Scholar]
- 43.Almaula N, Ebersole B, Zhang D, Weinstein H, Sealfon S C. J Biol Chem. 1996;271:14672–14675. doi: 10.1074/jbc.271.25.14672. [DOI] [PubMed] [Google Scholar]