

Abstract
Sequence variants that may result in splicing alterations are a particular class of inherited variants for which consequences can be more readily assessed, using a combination of bioinformatic prediction methods, and in vitro assays. There is also a general agreement that a variant would invariably be considered pathogenic on the basis of convincing evidence that it results in transcript(s) carrying a premature stop codon or an in-frame deletion disrupting known functional domain(s). This commentary discusses current practices used to assess the clinical significance of this class of variants, provides suggestions to improve assessment, and highlights the issues involved in routine assessment of potential splicing aberrations. We conclude that classification of sequence variants that may alter splicing is greatly enhanced by supporting in vitro analysis. Additional studies that assess large numbers of variants for induction of splicing aberrations and exon skipping are needed to define the contribution of splicing/exon skipping to cancer and disease. These studies will also provide the impetus for development of algorithms that better predict splicing patterns. We call for the deposition of all laboratory data from splicing analyses in national and international databases in order to facilitate variant classification and development of more specific bioinformatic tools.
Keywords: unclassified variant, splicing, bioinformatic prediction, cancer, oncology
Introduction
While development of likelihood based prediction models integrating data from a variety of sources (see Goldgar et al, 2008) will be a very useful tool for the classification of rare sequence variants in “high-risk” disease genes, there are several limitations to the application of such models. In the first instance, classification may be hampered by the availability of data on co-occurrence with clearly deleterious mutations, segregation, and tumor pathology for rare variants identified in individual families. There is also a possibility that results could be confounded if a “neutral” variant occurs in cis with an undetected mutation. This problem would be exacerbated for genes such as the mismatch repair (MMR) genes, where immunohistochemical testing is often used to prioritize gene testing. Assays which assess the influence of specific missense alterations on protein function can assist with the clinical classification of individual variants. The utility of such data in the prediction process will be greatly increased when methods have been sufficiently developed for their inclusion in multifactorial models which use multiple lines of evidence, and there is already great promise for this aspect of multifactorial likelihood classification (see Couch et al, 2008, and Goldgar et al, 2008). However, it will take some time and effort to generate reference data across a range of mutation types and positions for any given functional assay or gene.
Sequence variants that may result in splicing alterations are a particular class of variants for which the functional and clinical consequences can be more readily assessed. In the first instance, there are a variety of prediction programs that can be used to predict the probability that a sequence change will disrupt the “normal” splicing pattern. Secondly, given the availability of suitable biological material from a variant carrier, including lymphocytes or lymphoblastoid cell lines (LCL), the predictions can be relatively easily tested in vitro by analysis of mRNA in normal tissue. Thirdly, if there is convincing evidence that the variant results in a transcript (or transcripts) carrying a premature stop codon, or an in-frame deletion disrupting known functional domain(s), the variant would invariably be considered pathogenic without need for additional data required for multifactorial likelihood analysis. The aim of this commentary paper is to discuss current practices used to assess the clinical significance of this class of variants, to provide suggestions on how to improve assessment, and to highlight the caveats, advantages, and obstructions to routine assessment of potential splicing aberrations.
Prediction and verification of potential splicing aberrations
A number of web-based programs may be used for bioinformatic analysis of potential splicing aberrations. Details and descriptions of commonly-used programs are shown in Table 1. Virtually all of these programs were originally developed for gene hunting, and primarily take into account the consensus sequence or other sequence features like reading frames. That is, the programs were not designed for prediction of the consequences of sequence alterations. Analyses are done in a rather cumbersome fashion by generating results for the wildtype sequence separately to the variant sequence, and there is no specific output or interpretation comparing wildtype and variant sequences directly. Also, it is generally more straightforward to assess loss of splice consensus sites than creation of de novo sites because of the nature of the data input methods.
Table 1.
Web-based programs commonly used for bioinformatic analysis of potential splicing aberrations.
| Prediction Program | Website | Original Paper Describing Method | General Comment | Ease of Use - Batching | Flexibility of Use - Thresholds, Scores |
|---|---|---|---|---|---|
| MaxEntScan | http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html | Yeo, G and Burge, CB (2004), J Comput Biol 11: 377–94. | MaxEntScan is based on the approach for modelling the sequences of short sequence motifs such as those involved in RNA splicing which simultaneously accounts for non-adjacent as well as adjacent dependencies between positions. This method is based on the ‘Maximum Entropy Principle’ and generalizes most previous probabilistic models of sequence motifs such as weight matrix models and inhomogeneous Markov models. | Upload or pasting of sequences, batching possible - sequences in FASTA format. | No threshold alteration possible but possibility to choose probabilistic model. Separate sequence submissions for calculation of scores for acceptor and donor sites. |
| GENSCAN | http://genes.mit.edu/GENSCAN | Burge C and Karlin S (1997), J Mol Biol 268: 78–94. Burge CB (1998). In Salzberg, S, Searles, D, and Kasif, S, eds, Computational Methods in Molecular Biology, Elsevier Science, Amsterdam, pp 127–163. Burge, CB (1998), Curr Opin Struct Biol 8: 346–354. | As a rule, internal exons are predicted more accurately than initial or terminal exons, and exons are predicted more accurately than polyadenylation or promoter signals | Single sequences only, upload, pasting or FASTA format. | Some alteration possible (suboptimal exon cutoff). Scores provided for exon prediction. |
| NNSPLICE | http://www.fruitfly.org/seq_tools/splice.html | Reese MG, Eeckman, FH, Kulp, D, Haussler, D (1997), J Comp Biol 4: 311–23. | No use of branch point sequence. Has wider coverage - possibly better prediction of donor sites. | Pasting of sequences, batching possible - sequences in FASTA format. | Threshold alteration possible. Scores provided for acceptor and donor sites. |
| SpliceSiteFinder | http://violin.genet.sickkids.on.ca/~ali/splicesitefinder.html | In-house algorithm to calculate scores of donoe and acceptor sequences is based on Shapiro and Senapathy (1987), Nucleic Acids Research 15: 7155–7174. The algorithm to calculate the score of branch sequence is similar to that of donor, based on the matrix compiled by Senapathy et al (1990), Methods Enzymol 183:252–78. | Uses and accounts for strength of upstream branch point - possibly better prediction of acceptor sites. | Single sequences only, upload, pasting and GCG, EMBL or FASTA format. | Threshold alteration possible. Scores provided for acceptor, donor, branch point sites. |
| NetGene2 | http://www.cbs.dtu.dk/services/NetGene2 | Hebsgaard, SM, Korning, PG, Tolstrup, N, Engelbrecht, J, Rouze, P, Brunak, S (1996), Nucleic Acids Research 24: 3439–3452. Brunak, S., Engelbrecht, J., and Knudsen, S (1991), Journal of Molecular Biology 220: 49–65. | In-house algorithm, and extent of coverage is not clear. Existence of ectopic splice sites not predicted. | Single sequences only, upload, pasting or FASTA format. | No threshold alteration possible. Scores provided for acceptor and donor sites. Also graphical output. |
| GeneSplicer | http://www.tigr.org/tdb/GeneSplicer/gene_spl.html | Pertea, M, Lin, X and Salzberg, SL (2001). Nucleic Acids Research 29: 1185–1190. | Combination of Markov modelling techniques, MDD (Maximal dependence decomposition) and features coding and non-coding sequences. | Single sequences only, upload, pasting or FASTA format. | Threshold alteration possible. Scores provided for acceptor and donor sites. |
| Human Splicing Finder (HSF)formerly Automated splicing mutation analysis | https://splice.uwo.ca | Rogan PK, Faux B, Schneider TD (1998) Hum Mutat. 12: 153–171. Nalla, VK and Rogan PK (2005) Hum Mutat 25: 334–342. | Analysis based on information theory. Uses a weight matrix derived from the nucleotide frequencies at each position of a splice site sequence database. Possibility to check effects of mutations based on HGVS format. Guest access limits use to 20 analyses, further analyses possible. | Single sequences only, pasting or easy retrieval of sequences using HUGO gene name or accession number from UCSC genome browser. | No threshold alteration possible. Score provided for donor, acceptor, branch and ESE sites. Effects mutations compared to reference sequence. Tabular and sequence walker output. |
| Splicing Sequences Finder | www.umd.be/SSF/ | This website was developed specifically to better understand intronic mutations and exonic mutations leading to splicing defects. It provides assess to new prediction algorithms developed to calculate the consensus values of potential splice sites and search for branch points. The tool has also integrated matrices from 6 other sources for regulatory sequence motifs (ESE, ESS, ISE and ISS), and new matrices to identify hnRNP A1, Tra2 and 9G8. | Use of Ensembl sequences and search options. Intron and exon sequences are marked. Possibility to check effects of mutations and SNPs. | Single sequences only, pasting or easy retrieval of sequences from Ensembl. | Many different options for regulatory sequences. Threshold alteration possible. Scores provided for regulatory sequences, acceptor, donor and branch point sites. Also graphical output. |
| ESEfinder | http://rulai.cshl.edu/tools/ESE | Liu, H-X, Zhang, M, and Krainer, AR. (1998). Genes Dev.12: 1998–2012. | Searches for ESEs only. Provides scores for the consensus binding motifs for the SR proteins SRp40, SRp55, SC35, and SF2. | Upload or pasting of sequences, batching possible - sequences in FASTA format. | Threshold alteration possible. Score provided - allows for subsequent filtering. Also graphical output. |
| RESCUE-ESE | http://genes.mit.edu/burgelab/rescue-ese/ | Fairbrother, WG, Yeh, RF, Sharp, PA, Burge, CB (2002). Science 297:1007–13. | Searches for ESEs only. Recognises a different panel of ESE motifs, predicted on the basis of exon-intron and splice site composition and in vitro tests of enhancer activity. Predictions also possible for mouse, zebrafish and pufferfish. | Pasting of sequences only, batching possible - sequences in FASTA format. | No threshold alteration possible. No score provided - post-analysis filtering not possible. |
| PESX | http://cubweb.biology.columbia.edu/pesx | Zhang XH, Chasin LA. (2004). Genes Dev 18:1241–50; Zhang XH, Kangsamaksin T, Chao MS, Banerjee JK, Chasin LA. (2005). Mol Cell Biol 25:7323–32. | Searches for ESE and ESS sites, using algorithm derived from computational search of exonic splicing signals. | Pasting of sequences only. Can submit 10K sequence in FASTA format. | Threshold alteration possible. No score provided - post-analysis filtering not possible. |
There are two broad classes of programs. The most clinically useful are those which can be used to analyze the effect of sequence alterations in consensus sequences of donor and acceptor splice sites which may disrupt or weaken canonical sites, or sequence alterations which may create cryptic splice sites in intronic or exonic regions. In addition, several programs have been designed to predict possible exonic or intronic splice enhancer (ESE/ISE) and exonic or intronic splice silencer (ESS/ISS) sites. It is important to note that these splice enhancer/silencer programs recognize multiple sequences that are potential binding sites of splicing factors, but cannot denote which sites are actually used under physiological conditions.
There is a general acceptance that there is fairly good prediction of the disruption of the canonical splice donor and splice acceptor sites, the intronic dinucleotides 5’GT and 3’AG that flank exons. Sequence changes in these highly conserved sites are well recognized to affect the splicing of the adjacent exon(s). Their evaluation in the clinical setting ranges from: classification as pathogenic on basis of sequence information alone; classification as pathogenic with the support of appropriate bioinformatic prediction; or classification as pathogenic with the support of experimental evidence as backup to bioinformatic prediction. Poorer (or no) recognition of normal splicing patterns for the wildtype sequence when analyzed using default settings of the relevant programs is likely to influence the decision to supplement bioinformatic predictions with experimental evidence. However, we know of at least one example where a variant affecting the invariant dinucleotide position at an intron extremity is apparently not associated with a splicing aberration (BRCA2 NM_000059.1:c.8331+1G>T), despite prediction of splice site loss by most bioinformatic programs tested (Radice, unpublished data). This suggests that even variants located in canonical splice donor and splice acceptor sites should not be unequivocally assumed to alter splicing.
It is recognized that the consequences of alterations in the exonic and intronic regions close to consensus acceptor and donor sites are more difficult to predict a priori. This is partly because, although there is some over-representation of specific nucleotides in these regions, their sequence shows substantial variability (Pettigrew and Brown, 2008). Furthermore, the location of the branch point can vary somewhat. Bioinformatic modeling can thus help assess the potential effect of such variants on splicing.
However, a major challenge to bioinformatic prediction is that use of cryptic splice sites (also known as ectopic splice sites) as a consequence of disruption of normal splicing is not very well predicted. This is often because the very nature of a cryptic site defines it as a site that is not recognized during normal splicing, or best recognized, in the event of low-abundance naturally-occurring splice variants. This may also be because the cryptic site is some distance from the original site and the sequence information may not be analyzed by bioinformatic prediction programs. Thus, while abrogation or creation of consensus or other splicing regions by a particular sequence variant may be relatively well predicted, the nature of the splice product(s) resulting from this sequence alteration cannot be accurately defined. Yet another challenge is the prediction of ectopic splice site use that results from an alteration that enhances use of the cryptic splice site, or creates a de novo splice site. Recent progress has been made in this field of research, indicating that size of exon likely to be skipped, splicing factor motif scores, availability of decoy splice sites and density of silencers are predictors of aberrant splicing events (Kralovicova and Vorechovsky, 2007). It also appears that cryptic 5’ donor splice sites are best predicted by computational algorithms that accommodate nucleotide dependencies, and take information about non-adjacent positions into account (Buratti, et al., 2007) and aberrant 3’ acceptor splice sites are characterized by higher purine content than authentic 3’ acceptor splice sites (Vorechovsky, 2006). However, this evidence has yet to be incorporated into modeling tools. Experimental evidence supporting bioinformatic predictions is thus preferable for clinical decision-making.
These statements are supported by a subset of published studies that have compared bioinformatic predictions with in vitro findings (Auclair, et al., 2006; Bonatti, et al., 2006; Buratti, et al., 2007; Claes, et al., 2002; Houdayer, et al., 2008; Lastella, et al., 2006; Sharp, et al., 2004; Tesoriero, et al., 2005; Tournier, et al., 2008), and by unpublished data of the authors on variants in the BRCA1 (MIM# 113705) and BRCA2 (MIM# 600185) (Sinilnikova et al, Radice et al), and MLH1 (MIM# 609310) and MSH2 (MIM# 609309) genes (Spurdle et al). Although few studies have comprehensively compared numerous bioinformatic programs to each other and to in vitro results, evidence to date suggests that use of any particular single program is not indicated (Houdayer, et al., 2008; Tournier, et al., 2008)(unpublished data of authors). In addition, no information is available on specific aspects of the analytical procedures, such as modification of default settings, or importance of the amount of input sequence, with respect to their influence the outcome of bioinformatic analyses. Thus, also taking into account the differences in baseline algorithms and sites assessed (Table 1), it should not be assumed that any single prediction program will be 100% efficient at predicting the existence of likely splicing aberration. Therefore, use of more than one program is preferable. In addition, in vitro analysis has an important role in confirming or establishing use of ectopic (cryptic or de novo) splice sites and defining what aberrant splice products are associated with a specific sequence variant.
The prediction of the effect of sequence alterations in possible enhancer and silencer splicing elements is even more challenging. Firstly, these sites are relatively poorly defined, so their accurate prediction is difficult. Moreover, their function in vivo in normal splicing is defined by other important variables, including distance from the splice site and splice site strength (Fairbrother, et al., 2004). Their activity is also likely to be influenced by context effects such as secondary structure or adjacent negative elements (Fairbrother, et al., 2004). So while there is evidence that exonic variants can alter ESEs at the nucleotide level to cause aberrant splicing (Aretz, et al., 2004; Cravo, et al., 2002; Liu, et al., 1998; Mazoyer, et al., 1998; McVety, et al., 2005; Sharp, et al., 2004; Zatkova, et al., 2004) there is also considerable evidence showing that splicing aberrations are rarely associated with sequence changes that alter the prediction scores for such sites, whether it be loss or gain of the site (Anczukow, et al., 2008; Lastella, et al., 2006)(Spurdle et al, Sinilnikova et al, and Radice et al, unpublished data). It has been suggested that identification of ESEs might be improved by increasing standard thresholds used by bioinformatic programs, considering distance from the intron-exon boundary, and assessing nucleotide evolutionary conservation of ESEs, although the latter is of value for assessing loss of (evolutionarily conserved) sites only (Pettigrew and Brown, 2008; Pettigrew, et al., 2007). However it is currently difficult to estimate the efficiency of such filters given the small number of studies which have comprehensively studied potential splicing aberrations associated with such sequence variants, and the relative paucity of variants known to alter splicing elements to provide positive controls for such filtering approaches. Accumulation of further data will thus be necessary to provide the stimulus and empirical information for further development of prediction programs. The introduction of large-scale array-based studies of alternative splice product expression (Carninci, et al., 2005), especially if coupled with high throughput next generation sequencing of transcripts from multiple samples, are likely to prove helpful in this regard. Additional research into the role of auxiliary splicing signals in the selection of aberrant splice sites in introns and exons will also provide a basis for further development of currently available prediction tools (Kralovicova and Vorechovsky, 2007). Assessing sequence variants based on loss or gain of these motifs is presently not pursued to any great extent outside the research setting, and this would appear to be an appropriate course of action in relation to use of health care funds.
Difficulties in interpretation of predicted and experimentally confirmed splicing aberrations
The general approach to prediction and verification of splicing aberrations associated with variants within or near the consensus splice sites is relatively straightforward. However, the interpretation of results may be more challenging. Whether confirmed in vitro or not, the clinical significance is not firmly established for in-frame single exon deletions which cover domains of unknown function, or for small in-frame insertions or deletions. For example, the BRCA2 NM_000059.1:c.8488-1G>A substitution resulting in skipping of the first 12 bases of exon 20 and an in-frame deletion of four amino acids is considered a variant of uncertain significance (Howlett, et al., 2002). This highlights the caution that should be taken with interpretation of variants that affect even the most conserved nucleotides of splice sites.
The relative amounts of alternative/aberrant splice products generated by a variant allele is also important for establishing clinical relevance (Claes, et al., 2002), particularly when there are several observed products but only one/some of these are clearly recognizable as transcripts with pathogenic consequences. Such a scenario would require additional in vitro quantitative expression studies. Since a number of naturally occurring splice products are known to be produced for most genes, and since the type and amount of alternative/aberrant splice product can differ depending on cell type and assay conditions (Lastella, et al., 2006; Speevak, et al., 2003), it is essential to include a large number of controls assessed under similar assay conditions to compare expression of “aberrant” splice products against the quantity of naturally occurring splice products. Ideally, samples from more than one variant carrier should also be tested. Even so, there would likely be some debate about the clinical significance of a variant which results in increased expression of a stable truncating transcript which occurs naturally at low abundance. It is thus important that experimental tests take into account a possible differential expression between wildtype and variant alleles which might be due to nonsense-mediated decay (NMD) of transcripts with a premature stop codon produced by variant allele, or lack of expression of the variant allele due to a defect in regulatory regions, etc. Useful information about the stability of transcripts carrying premature stop codons is provided when cell culture assays are conducted both with and without cycloheximide or other translation inhibitors, as a surrogate to assess NMD in vivo. Without cycloheximide treatment, certain mutant transcripts which are subject to NMD are almost undetectable due to their strong degradation, whereas the level of mutant transcripts is comparable to the level of normal transcripts in the presence of cycloheximide (Perrin-Vidoz, et al., 2002; Ware, et al., 2006). In addition, some sequence changes may be ‘leaky’, in that variant allele produces a full-length/normal mRNA in addition to abnormal transcripts, suggesting that the loss/inactivation of the constitutional wild-type allele at the somatic level would not completely abrogate the synthesis of a normal protein product (Bonnet, et al., 2008). How such a scenario translates to level of cancer risk is unknown. It has also been observed that some nucleotide variants increase the efficiency of physiological splice sites, leading to the loss of alternative transcripts normally produced by the wild-type allele (Radice, unpublished data). Since the role of alternative mRNA isoforms is generally still undefined, the effect of these changes in relation to cancer risk remains unclear.
Another less tractable issue is whether in vitro results generated from assays on cultured or even uncultured lymphocyte cells can be generalized to the target tissue in question (Claes, et al., 2002; Speevak, et al., 2003). This is particularly important for subtle variation in expression of an alternative splice product, which may be due in part to tissue-specific variation in splicing due to tissue-specific expression of splicing factors. Further information may be generated as more data from tissue microarray studies become available, but it is likely that a specific study to assess all possible naturally occurring splice variants in the target tissue would be required to address this issue. Lastly, the test itself is subject to experimental limitations, with the anticipated effect influencing the design of splicing assay components, including real-time PCR, fragment purification, sequencing, and use of coding polymorphisms or the variant itself to quantitate relative allele expression. Thus, some aberrant transcripts may be missed if they have not been predicted to exist, and the experimental design cannot accommodate their detection.
It is thus likely that a subset of variants predicted or confirmed to cause splicing aberrations will require further study using multifactorial methods (see Goldgar et al, 2008), including assessing segregation in family studies.
Application of in vitro assays in the clinical setting – feasibility and need
There is no doubt that further information regarding clinical significance of rare sequence variants would be obtained by introducing routine collection of fresh blood from such patients for clinical RNA-based assays of altered splicing. While this is technically possible for many clinical laboratories, such tests would introduce logistical issues from several aspects. Blood for DNA tests can be used successfully if 1–5 days old, but blood samples for RNA analysis would either require fresh collection for immediate processing or collection into specialized collection tubes to limit degradation and/or the induction of artefactual abnormal splice production resulting from environmental stress in aged blood (Speevak, et al., 2003). Yet another possibility is to establish a lymphoblastoid cell line, although that is time-consuming and would probably be considered excessive use of resources for a routine diagnostic test. There would also be a need to design and conduct variant-specific tests, requiring dedicated attention from molecular geneticists and technicians.
Another alternative for diagnostic testing could be the use of mini-gene assays, which obviate the need for clinical blood collection. A genomic fragment encompassing the exon of interest, and surrounding introns and immediate upstream and downstream exons, is cloned into a standard mammalian expression vector, which is then introduced into cells in culture (Anczukow, et al., 2008; Bonnet, et al., 2008; Zatkova, et al., 2004). The influence of variants in this exon of interest on splicing can then be assessed by in vitro RNA analysis. Theoretically this would allow for preparation of constructs to assay variants in the majority of exonic regions, but is subject to a number of limitations. The system is very artificial in that the entire gene sequence is not included in the assay, and that cell type may influence production of natural alternative/aberrant splice products. Moreover, there are technical challenges of creating constructs for very large exons, and the need to account for transfection efficiency. Use of these approaches is thus a less attractive alternative to blood-based assays which are already implemented to a degree in some laboratories, and is unlikely to be feasible without centralization of such tests.
The need and prioritization of splicing assays would be influenced by location of the variant relative to (predicted) splicing sequences, the results from bioinformatic prediction of aberrant splicing, and the probability of pathogenicity as currently determined from other data mining approaches (Easton, et al., 2007, Tavtigian et al, 2008). Suggested interpretation of bioinformatic splicing prediction results is shown in Table 2, including suggestions for classification of sequence variants according to the system proposed by Plon et al (2008). We specifically chose to use BRCA1 and BRCA2 as examples, given the availability of probabilities from data mining approaches for these genes, so that we can show how other available data modulate the interpretation of predicted splicing aberrations. Similar interpretation/ prioritization tables could be derived for any disease gene, including relevant data for that gene if it is available.
Table 2.
Suggested interpretation of bioinformatic prediction of aberrant splicing for variant classification and prioritization of in vitro splicing assays, using BRCA1 and BRCA2 as examples.
| Location of variant | Current predictions of posterior probability | Bioinformatic prediction of splicing aberration* | Suggested variant class** | Recommendations for in vitro splicing analysis | Examples of classification based on in vitro results*** |
|---|---|---|---|---|---|
| Conserved splice donor and splice acceptor sites† | 100% (95%CI 91%–100%) as per Easton et al (2007) | Splicing aberration predicted OR no aberration predicted. | 4 – likely pathogenic | Clinical testing | Class 5 if the variant allele produces major transcript(s) carrying a premature stop codon or an in-frame deletion disrupting known functional domain(s) |
| Class 4 if the variant allele produces minor transcript(s) carrying a premature stop codon or an in-frame deletion disrupting known functional domain(s), at a higher level than similar transcripts corresponding to physiological alternative splicing, or absent in wildtype controls. Multifactorial likelihood analysis recommended. | |||||
| Class 3 if the variant allele produces major or minor transcript(s) carrying an in-frame deletion not disrupting known functional domain(s), at a higher level than similar transcripts corresponding to physiological alternative splicing. Multifactorial likelihood analysis recommended. | |||||
| Class 2 if the variant allele produces the same transcript(s) as the wild-type allele. Multifactorial likelihood analysis recommended. | |||||
| Exonic variants resulting in a missense substitution near intron/exon boundary # | Missense variants inside the BRCA1 RING, BRCA1 BRCT or BRCA2 DBD domain, predictions as per Tavtigian et al (2008), according to A-GVGD category: C0 = 1% (0%–6%) C15, C25 = 29% (9%–56%) C35, C45, C55 = 66% (34% – 93%) C65 = 81% (61%–95%) | Splicing aberration predicted | 3- uncertain for all AGVGD categories, C0 to C65 | Clinical testing | As above for splice donor and splice acceptor sites if aberrant splicing is observed; |
| A-GVGD probability takes precedence if no splicing aberration is confirmed: Class 2 for C0 = 1% (0%–6%); Class 3 for C15, C25, C35, C45, C55 and C65 = 29% (9%–56%) to 81% (61%–95%). Multifactorial likelihood analysis recommended. | |||||
| Exonic variants resulting in a missense substitution near intron/exon boundary # | Missense variants outside the BRCA1 RING, BRCA1 BRCT or BRCA2 DBD domain: 1% (0%–4%) for all A-GVGD categories, with prior probabilities for individual categories yet to be determined - Tavtigian et al, 2008 | Splicing aberration predicted | 3 - uncertain | Clinical testing | As above for splice donor and splice acceptor sites if aberrant splicing is observed; |
| A-GVGD probability takes precedence if no splicing aberration is confirmed – suggest Class 2 for C0 and Class 3 for other AGVGD categories. Multifactorial likelihood analysis recommended. | |||||
| Exonic variants resulting in a missense substitution near intron/exon boundary # | Predictions according to A-GVGD class: 1% (0%–4%) to 81% (61%–95%) - Tavtigian et al, 2008. | No aberration predicted | 3 – uncertain for A-GVGD categories C0 to C65 | Clinical testing | As above for splice donor and splice acceptor sites if aberrant splicing is observed; |
| As above for exonic variants predicted to cause a splicing aberration, A-GVGD probability takes precedence if no splicing aberration is confirmed – Class 2 to Class 3. | |||||
| Silent substitutions and intronic variants near donor and acceptor splice sites$ | Not assessed | Any predicted splicing aberration OR no aberration predicted. | 3 - uncertain | Clinical testing | As above for splice donor and splice acceptor sites if aberrant splicing is observed; |
| Class 2 if no splicing aberration is observed. Multifactorial likelihood analysis recommended. | |||||
| Other exonic (silent/missense) or intronic variants | Predictions as per Tavtigian et al, 2008 (exonic missense), or not assessed (silent and intronic) | Creation of a splice site. | 3 - uncertain | Clinical testing | As above for splice donor and splice acceptor sites if aberrant splicing is observed; |
| Class 3 for exonic C15 to C65 missense substitutions, and Class 2 for all other alterations, if no splicing aberration is observed. Multifactorial likelihood analysis recommended. | |||||
| Other exonic (silent/missense) or intronic variants | Predictions as per Tavtigian et al, 2008 (exonic missense), or not assessed (silent and intronic) | Any predicted splicing aberration – excluding creation of splice site, or no aberration predicted | 3 - uncertain for exonic C15 to C65 missense substitutions, or 2 - likely not pathogenic for other alterations | Research testing only | Clinical testing and multifactorial analysis recommended if any splicing aberration is observed. |
IVS+/−1 and IVS+/−2
First 2 nucleotides and last 3 nucleotides of exon resulting in a missense substitution.
First 2 nucleotides and last 3 nucleotides of exon resulting in a silent substitution; IVS+/−3 to IVS+6 and IVS-20.
Using three different bioinformatic programs. Possible aberration considered if loss or creation of splice site is predicted by any of the three programs using default settings, or if loss or creation of the same ESE/ISE is predicted by at least two of three programs.
As per Plon et al, 2008: 5 - pathogenic, probability >99%; 4 – likely pathogenic, probability 95%–99%; 3 – uncertain, probability 5%- 94.9%; 2 - likely not pathogenic or of little clinical significance, probability 0.1%–4.9%; 1 – not pathogenic or of little clinical significance, probability <0.1%.
Assumes assays on fresh blood or LCLs with at least three reference controls.
Current data indicate that sequence changes in the highly conserved intronic dinucleotides flanking exons will almost invariably result in splicing aberrations. For the donor site these are GT, and for the acceptor site AG. A very strong association of such sequence alterations with pathogenicity is supported for the BRCA1 and BRCA2 genes at least, where 100% of splice site variants (95% CI 91%–100%) were estimated to be deleterious from estimates derived largely from family history analysis of a large BRCA1 and BRCA2 dataset from Myriad Genetics (Easton, et al., 2007). The confidence limits for this estimate are sufficiently wide that such variants might best be considered class 4 variants (likely pathogenic, probability 95–99%) according to the classification system in the accompanying article by Plon et al (Plon et al, 2008). Indeed, as noted above, evidence suggests that some variants within consensus splice sites may not actually alter splicing. Bearing in mind the limitations of bioinformatic analyses, particularly the difficulty in predicting the usage of cryptic splice sites, there is nevertheless benefit to determining the expected gene products for such splice site variants by exhaustive bioinformatic analysis. Using a range of programs to assess likelihood of altered splicing and/or prediction of alternative products has value for the design of experiments to assess the existence of any splicing aberrations. Moreover, if resources for further study are limited, it would allow prioritization of a subset of variants for in vitro assays. For example, variants predicted to generate products such as in-frame single exon deletions or naturally-occurring splice products would be considered of more equivocal clinical significance if confirmed. In vitro results for such variants would have implications for clinical management, and would also identify variants for further assessment using multifactorial likelihood analysis approaches.
Regarding variants near but not within the consensus splice donor and splice acceptor dinucleotides, routine clinical in vitro assays would most certainly improve understanding of the consequences of such variation given the poorer performance of prediction tools for these less conserved regions. However, the number of variants to be assayed would be considerable, unless preselection for analysis can be guided by other information such as cosegregation with disease or tumor characteristics. By example, a crude search of the BIC website (http://research.nhgri.nih.gov/projects/bic) for intervening sequence variants of uncertain clinical significance in BRCA1 and BRCA2 listed 150 BRCA1 variants (434 entries) and 121 BRCA2 variants (266 entries) in this category. It is also possible that the number identified will be much larger in routine testing, although a further complication is that variants in this category may not necessarily be reported outside the laboratory e.g. if more deeply intronic. Access to such information is likely to facilitate variant classification itself, as discussed in a separate paper by Greenblatt et al (2008). The role of bioinformatic analysis in this instance would be to provide matching bioinformatic and in vitro data to encourage and facilitate the further development of bioinformatic tools. Also, until argument can be made to increase funding to at least a subset of clinical laboratories to undertake such widespread testing as part of a routine service to patients, bioinformatic analysis would provide a means to prioritize some variants for in vitro assays, and for alternative methods of assessing variant pathogenicity such as multifactorial likelihood approaches. As shown in Table 2, in the absence of further data from splicing assays or multifactorial likelihood approaches, such variants could be considered class 3 variants (uncertain, probability 5%–94.9%) unless existing predictions e.g. those based on sequence alignment, suggest that a variant falls in class 4. As for variants within the conserved splice donor and splice acceptor sites, it would not be considered appropriate to assign class based on possible aberrant products resulting from a sequence change, given the relatively poor bioinformatic prediction of cryptic splice site use in particular.
While the situation is likely to change as prediction methods develop over time, current comparisons of bioinformatic predictions and in vitro data indicate poor prediction of the existence and/or alteration of exonic and intronic splicing elements. This suggests that in vitro screening of variants predicted to create or destroy such elements should currently be limited to the research setting. This is especially true for exonic variants, since current data from analysis of exonic sequence variants using protein sequence alignment-based methods (Tavtigian et al, 2008) indicate that the posterior probability of pathogenicity is only 1% (95% CI 0%–4%) overall for missense variants outside the BRCA1 Ring or BRCT domains, and outside the BRCA2 DBD domain. This suggests that such variants might best be considered class 2 variants unless splicing software predicts creation of a splice site and promotion to class 3. However, since prior probabilities for the individual A-GVGD categories C0 to C65 have yet to be determined for these regions of BRCA1 and BRCA2, particularly for several small but well conserved motifs, a conservative approach would be to rather consider any missense alterations in the categories C15 to C65 as class 3 at present. Risk associated with exonic silent or intronic sequence variants at a distance from the intron-exon boundary has not been specifically assessed for BRCA1 and BRCA2, but it is likely that the large majority of these are not associated with a high risk of cancer given the results of pooled analysis of difference classes of unclassified variants by Easton et al (Easton, et al., 2007). They should thus be considered class 2 variants, unless, as rationalized above, they are exonic C15 to C65 missense substitutions.
Although current evidence suggests a minor role of high-risk cancer-associated splicing aberrations due to alterations in ESEs and ISEs, there is some argument for routine RNA-based assays of all rare sequence variants of unknown clinical significance in the research setting, irrespective of bioinformatic predictions. There are examples of splicing alterations associated with apparent missense variants that are not convincingly predicted using bioinformatic methods (Farrugia, et al., 2008). Moreover, assay methods can be designed to identify variants associated with unstable mRNA expression, in addition to splicing aberrations. The conduct of larger numbers of research studies would also establish the overall clinical benefit of conducting in vitro assays on such variants,
The introduction of routine clinical splicing assays for even a subset of sequence variants would place considerable strain on resources at various levels, and provides a sufficiently strong argument to argue for additional health care spending to prevent overload on clinics and laboratories. However, despite the mentioned theoretical and technical problems, the analyses described have the potential to provide information of immediate use for the clinical management of at-risk individuals. It is important to note there is considerable evidence that mutations affecting mRNA splicing can be common molecular defects for inherited disorders in humans, and that a high proportion of these splicing mutations can occur at less stringently conserved splicing recognition sites (Ars, et al., 2000; Teraoka, et al., 1999).
An overview of the suggested approaches to be applied is shown in Figure 1. Overall, we suggest that additional studies assessing large numbers of variants for induction of splicing aberrations and exon skipping are carried out to define the contribution of splicing/exon skipping to disease. These studies will also provide the impetus for development of algorithms that better predict these effects. In addition, we call for the deposition of laboratory data from splicing analyses of any gene undergoing clinical testing in national and international databases in order to facilitate variant classification and development of more specific bioinformatic tools, and suggest that data deposition be a requisite of additional funding that would be required to support the increased workload to clinical laboratories.
Figure 1.
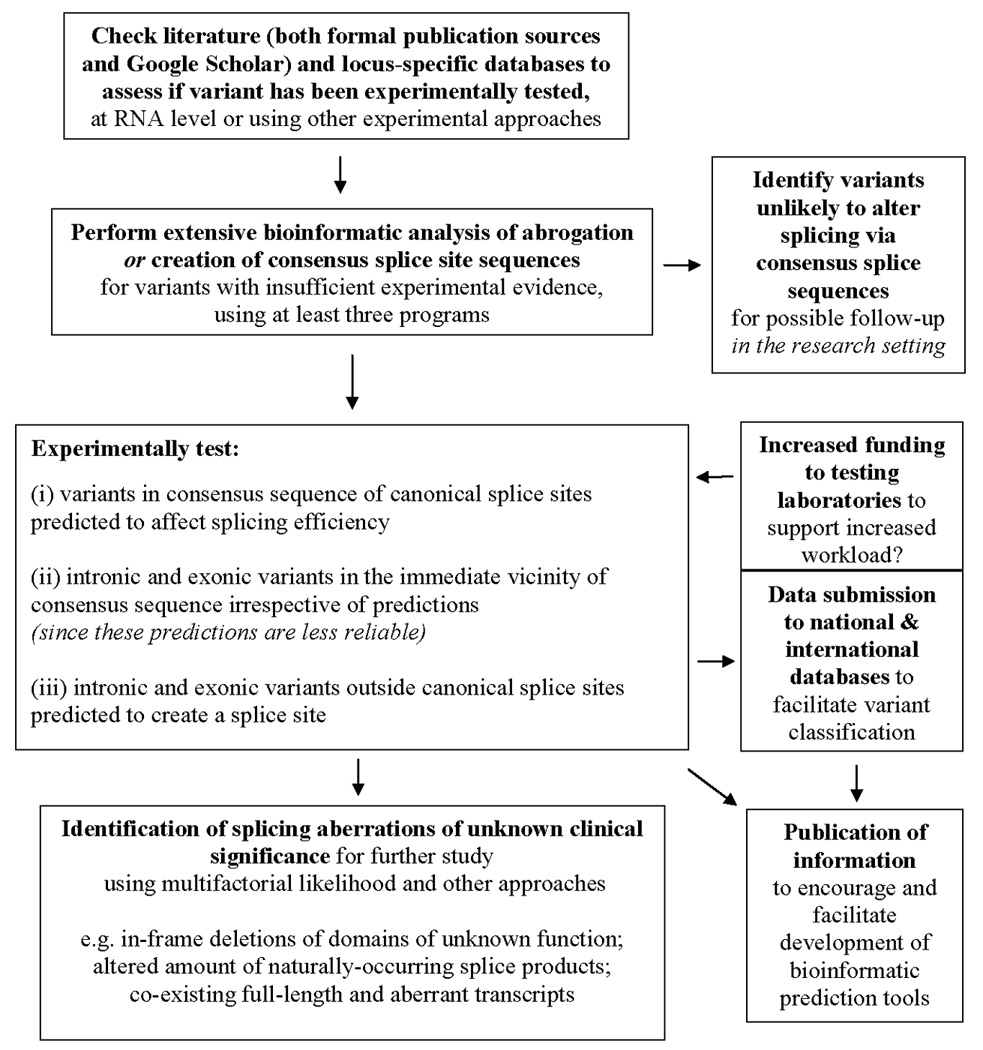
Schematic representation of proposed approaches to assessing splicing aberrations in the clinical setting.
Summary
Classification of sequence variants that may alter splicing would be greatly enhanced by supporting in vitro analysis. We suggest that use of multiple bioinformatic prediction programs is indicated for use in the clinical setting to assess the likely clinical significance of variants in or near consensus splice regions. The optimal situation for clinical reporting is that all variants in consensus splice site sequences predicted by bioinformatic tools to affect splicing should be experimentally verified. At a minimum, the subset of consensus splice region variants predicted to result in products of equivocal clinical significance, and those variants near consensus splice regions, should be prioritized for in vitro assays. Due to their clinical relevance of the information they can provide, it would be preferable that such assays are undertaken in the clinical setting as an initiative to evaluate variants for counseling purposes. However, this would necessitate provision of additional funding to clinical testing laboratories to support the resultant increased workload. It would be beneficial if a condition of such funding is that the results of such assays should be made available to the international clinical and research community through publication, and submission of information to appropriate databases, to further specific and general knowledge about the role of such variants in disease. The evaluation of splicing aberrations as a result of predicted alteration of exonic and intronic splicing elements should be considered an important area of research study to stimulate development of the relevant prediction tools. We recognize that the clinical significance of some variants with aberrant splicing products detected in vitro will remain uncertain, and further study using other approaches will be required to resolve their clinical significance.
Acknowledgments
We would like to thank Stephen Arnold for his assistance in collating information on bioinformatic tools. We thank the reviewers’ for helpful comments on the manuscript. The authors would like to acknowledge support from the Australian National Health and Medical Research Council (ABS), the NIH Breast Cancer Specialized Program in Research Excellence grant P50 CA116201 (FJC), NIH grant CA116167 (FJC), and American Cancer Society award RSG-04-220-01-CCE (FJC), and the Italian Association and Foundation for Cancer Research, AIRC/FIRC (PR).
Appendix
Members of the IARC Working Group on Unclassified Genetic Variants
Paolo Boffetta, IARC, France; Fergus Couch, Mayo Clinic, USA; Niels de Wind, Leiden University, the Netherlands; Douglas Easton, Cambridge University, UK; Diana Eccles, University of Southampton, UK; William Foulkes, McGill University, Canada; Maurizio Genuardi, University of Florence, Italy; David Goldgar, University of Utah, USA; Marc Greenblatt, University of Vermont, USA; Robert Hofstra, University Medical Center Groningen, the Netherlands; Frans Hogervorst, Netherlands Cancer Institute, the Netherlands; Nicoline Hoogerbrugge, University Medical Center Neimejen, the Netherlands; Sharon Plon, Baylor University, USA; Paolo Radice, Istituto Nazionale Tumori, Italy; Lene Rasmussen, Roskilde University, Denmark; Olga Sinilnikova, Hospices Civils de Lyon, France; Amanda Spurdle, Queensland Institute of Medical Research, Australia; Sean Tavtigian, IARC, France.
References
- Anczukow O, Buisson M, Salles MJ, Triboulet S, Longy M, Lidereau R, Sinilnikova OM, Mazoyer S. Unclassified variants identified in BRCA1 exon 11: Consequences on splicing. Genes Chromosomes Cancer. 2008 doi: 10.1002/gcc.20546. [DOI] [PubMed] [Google Scholar]
- Aretz S, Uhlhaas S, Sun Y, Pagenstecher C, Mangold E, Caspari R, Moslein G, Schulmann K, Propping P, Friedl W. Familial adenomatous polyposis: aberrant splicing due to missense or silent mutations in the APC gene. Hum Mutat. 2004;24(5):370–380. doi: 10.1002/humu.20087. [DOI] [PubMed] [Google Scholar]
- Ars E, Serra E, Garcia J, Kruyer H, Gaona A, Lazaro C, Estivill X. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet. 2000;9(2):237–247. doi: 10.1093/hmg/9.2.237. [DOI] [PubMed] [Google Scholar]
- Auclair J, Busine MP, Navarro C, Ruano E, Montmain G, Desseigne F, Saurin JC, Lasset C, Bonadona V, Giraud S others. Systematic mRNA analysis for the effect of MLH1 and MSH2 missense and silent mutations on aberrant splicing. Hum Mutat. 2006;27(2):145–154. doi: 10.1002/humu.20280. [DOI] [PubMed] [Google Scholar]
- Bonatti F, Pepe C, Tancredi M, Lombardi G, Aretini P, Sensi E, Falaschi E, Cipollini G, Bevilacqua G, Caligo MA. RNA-based analysis of BRCA1 and BRCA2 gene alterations. Cancer Genet Cytogenet. 2006;170(2):93–101. doi: 10.1016/j.cancergencyto.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Bonnet C, Krieger S, Vezain M, Rousselin A, Tournier I, Martins A, Berthet P, Chevrier A, Dugast C, Layet V others. Screening BRCA1 and BRCA2 unclassified variants for splicing mutations using reverse transcription PCR on patient RNA and an ex vivo assay based on a splicing reporter minigene. J Med Genet. 2008;45(7):438–446. doi: 10.1136/jmg.2007.056895. [DOI] [PubMed] [Google Scholar]
- Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991;220(1):49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- Buratti E, Chivers M, Kralovicova J, Romano M, Baralle M, Krainer AR, Vorechovsky I. Aberrant 5′ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2007;35(13):4250–4263. doi: 10.1093/nar/gkm402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268(1):78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Burge CB. Modeling dependencies in pre-mRNA splicing signals. In: Salzburg S, Searles D, Kasif S, editors. Computational Methods in Molecular Biology. Amsterdam: Elsevier Science; 1998. pp. 127–163. [Google Scholar]
- Burge CB, Karlin S. Finding the genes in genomic DNA. Curr Opin Struct Biol. 1998;8(3):346–354. doi: 10.1016/s0959-440x(98)80069-9. [DOI] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C others. The transcriptional landscape of the mammalian genome. Science. 2005;309(5740):1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- Claes K, Vandesompele J, Poppe B, Dahan K, Coene I, De Paepe A, Messiaen L. Pathological splice mutations outside the invariant AG/GT splice sites of BRCA1 exon 5 increase alternative transcript levels in the 5′ end of the BRCA1 gene. Oncogene. 2002;21(26):4171–4175. doi: 10.1038/sj.onc.1205520. [DOI] [PubMed] [Google Scholar]
- Cravo M, Afonso AJ, Lage P, Albuquerque C, Maia L, Lacerda C, Fidalgo P, Chaves P, Cruz C, Nobre-Leitao C. Pathogenicity of missense and splice site mutations in hMSH2 and hMLH1 mismatch repair genes: implications for genetic testing. Gut. 2002;50(3):405–412. doi: 10.1136/gut.50.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FJ, Rasmussen L, Hofstra R, Monteiro AANM, Greenblatt MS, de Wind N IARC Unclassified Genetic Variants Working Group. Assessment of Functional Effects of Unclassified Genetic Variants. Hum Mutat. 2008;29 [Google Scholar]
- Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, Tavtigian SV, Monteiro AN, Iversen ES, Couch FJ others. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81(5):873–883. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297(5583):1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- Fairbrother WG, Yeo GW, Yeh R, Goldstein P, Mawson M, Sharp PA, Burge CB. RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res. 2004;32(Web Server issue):W187–W190. doi: 10.1093/nar/gkh393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia DJ, Agarwal MK, Deffenbaugh AM, Wenstrup RL, Pruss D, Frye C, Wadum L, Johnson K, Mentlick J, Tavtigian SV others. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer Research. in press doi: 10.1158/0008-5472.CAN-07-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia DJ, Agarwal MK, Pankratz VS, Deffenbaugh AM, Pruss D, Frye C, Wadum L, Johnson K, Mentlick J, Tavtigian SV others. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer Res. 2008;68(9):3523–3531. doi: 10.1158/0008-5472.CAN-07-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgar DE, Easton DF, Byrnes GB, Spurdle AB, Iversen ES, Greenblatt MS IARC Unclassified Genetic Variants Working Group. Integration of various data sources for classifying uncertain variants into a single model. Hum Mutat. 2008;29 doi: 10.1002/humu.20897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt MS, Brody LC, Foulkes W, Genuardi M, Hofstra R, Plon S, Sijmons RH, Sinilnikova OM, Spurdle AB IARC Unclassified Genetic Variants Working Group. Locus-Specific Databases (LSDBs) and the Classification of Variants in Cancer Susceptibility Genes. Hum Mutat. 2008;29 [Google Scholar]
- Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouze P, Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996;24(17):3439–3452. doi: 10.1093/nar/24.17.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houdayer C, Dehainault C, Mattler C, Michaux D, Caux-Moncoutier V, Pages-Berhouet S, d'Enghien CD, Lauge A, Castera L, Gauthier-Villars M others. Evaluation of in silico splice tools for decision-making in molecular diagnosis. Hum Mutat. 2008;29(7):975–982. doi: 10.1002/humu.20765. [DOI] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G others. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297(5581):606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- Kralovicova J, Vorechovsky I. Global control of aberrant splice-site activation by auxiliary splicing sequences: evidence for a gradient in exon and intron definition. Nucleic Acids Res. 2007;35(19):6399–6413. doi: 10.1093/nar/gkm680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lastella P, Surdo NC, Resta N, Guanti G, Stella A. In silico and in vivo splicing analysis of MLH1 and MSH2 missense mutations shows exon- and tissue-specific effects. BMC Genomics. 2006;7:243. doi: 10.1186/1471-2164-7-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HX, Zhang M, Krainer AR. Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev. 1998;12(13):1998–2012. doi: 10.1101/gad.12.13.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazoyer S, Puget N, Perrin-Vidoz L, Lynch HT, Serova-Sinilnikova OM, Lenoir GM. A BRCA1 nonsense mutation causes exon skipping. Am J Hum Genet. 1998;62(3):713–715. doi: 10.1086/301768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVety S, Li L, Gordon PH, Chong G, Foulkes WD. Disruption of an exon splicing enhancer in exon 3 of MLH1 is the cause of HNPCC in a Quebec family. J Med Genet. 2005 doi: 10.1136/jmg.2005.031997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalla VK, Rogan PK. Automated splicing mutation analysis by information theory. Hum Mutat. 2005;25(4):334–342. doi: 10.1002/humu.20151. [DOI] [PubMed] [Google Scholar]
- Perrin-Vidoz L, Sinilnikova OM, Stoppa-Lyonnet D, Lenoir GM, Mazoyer S. The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum Mol Genet. 2002;11(23):2805–2814. doi: 10.1093/hmg/11.23.2805. [DOI] [PubMed] [Google Scholar]
- Pertea M, Lin X, Salzberg SL. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 2001;29(5):1185–1190. doi: 10.1093/nar/29.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettigrew CA, Brown MA. Pre-mRNA splicing aberrations and cancer. Front Biosci. 2008;13:1090–1105. doi: 10.2741/2747. [DOI] [PubMed] [Google Scholar]
- Pettigrew CA, Wayte N, Wronski A, Lovelock PK, Spurdle AB, Brown MA. Colocalisation of predicted exonic splicing enhancers in BRCA2 with reported sequence variants. Breast Cancer Res Treat. 2007 doi: 10.1007/s10549-007-9714-5. [DOI] [PubMed] [Google Scholar]
- Plon SE, Eccles DM, Easton DF, Foulkes W, Genuardi M, Greenblatt MS, Hogervorst FBL, Hoogerbrugge N, Spurdle AB, Tavtigian S IARC Unclassified Genetic Variants Working Group. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29 doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4(3):311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- Rogan PK, Faux BM, Schneider TD. Information analysis of human splice site mutations. Hum Mutat. 1998;12(3):153–171. doi: 10.1002/(SICI)1098-1004(1998)12:3<153::AID-HUMU3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Senapathy P, Shapiro MB, Harris NL. Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods Enzymol. 1990;183:252–278. doi: 10.1016/0076-6879(90)83018-5. [DOI] [PubMed] [Google Scholar]
- Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987;15(17):7155–7174. doi: 10.1093/nar/15.17.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp A, Pichert G, Lucassen A, Eccles D. RNA analysis reveals splicing mutations and loss of expression defects in MLH1 and BRCA1. Hum Mutat. 2004;24(3):272. doi: 10.1002/humu.9267. [DOI] [PubMed] [Google Scholar]
- Speevak MD, Young SS, Feilotter H, Ainsworth P. Alternatively spliced, truncated human BRCA2 isoforms contain a novel coding exon. Eur J Hum Genet. 2003;11(12):951–954. doi: 10.1038/sj.ejhg.5201063. [DOI] [PubMed] [Google Scholar]
- Tavtigian S, Byrnes GB, Goldgar DE, Thomas A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum Mutat. 2008;29 doi: 10.1002/humu.20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teraoka SN, Telatar M, Becker-Catania S, Liang T, Onengut S, Tolun A, Chessa L, Sanal O, Bernatowska E, Gatti RA others. Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet. 1999;64(6):1617–1631. doi: 10.1086/302418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesoriero AA, Wong EM, Jenkins MA, Hopper JL, Brown MA, Chenevix-Trench G, Spurdle AB, Southey MC. Molecular characterization and cancer risk associated with BRCA1 and BRCA2 splice site variants identified in multiple-case breast cancer families. Hum Mutat. 2005;26(5):495. doi: 10.1002/humu.9379. [DOI] [PubMed] [Google Scholar]
- Tournier I, Vezain M, Martins A, Charbonnier F, Baert-Desurmont S, Olschwang S, Wang Q, Buisine MP, Soret J, Tazi J others. A large fraction of unclassified variants of the mismatch repair genes MLH1 and MSH2 is associated with splicing defects. Hum Mutat. 2008 doi: 10.1002/humu.20796. [DOI] [PubMed] [Google Scholar]
- Vorechovsky I. Aberrant 3′ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2006;34(16):4630–4641. doi: 10.1093/nar/gkl535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware MD, DeSilva D, Sinilnikova OM, Stoppa-Lyonnet D, Tavtigian SV, Mazoyer S. Does nonsense-mediated mRNA decay explain the ovarian cancer cluster region of the BRCA2 gene? Oncogene. 2006;25(2):323–328. doi: 10.1038/sj.onc.1209033. [DOI] [PubMed] [Google Scholar]
- Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11(2–3):377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- Zatkova A, Messiaen L, Vandenbroucke I, Wieser R, Fonatsch C, Krainer AR, Wimmer K. Disruption of exonic splicing enhancer elements is the principal cause of exon skipping associated with seven nonsense or missense alleles of NF1. Hum Mutat. 2004;24(6):491–501. doi: 10.1002/humu.20103. [DOI] [PubMed] [Google Scholar]
- Zhang XH, Chasin LA. Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev. 2004;18(11):1241–1250. doi: 10.1101/gad.1195304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XH, Kangsamaksin T, Chao MS, Banerjee JK, Chasin LA. Exon inclusion is dependent on predictable exonic splicing enhancers. Mol Cell Biol. 2005;25(16):7323–7332. doi: 10.1128/MCB.25.16.7323-7332.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]