

Summary
dTOR (target of rapamycin) and dFoxo respond to changes in the nutritional environment to induce a broad range of responses in multiple tissue types. Both dTOR and dFoxo have been demonstrated to control the rate of age-related decline in cardiac function. Here, we show that the Eif4e-binding protein (d4eBP) is sufficient to protect long-term cardiac function against age-related decline and that up-regulation of dEif4e is sufficient to recapitulate the effects of high dTOR or insulin signaling. We also provide evidence that d4eBP acts tissue-autonomously and downstream of dTOR and dFoxo in the myocardium, where it enhances cardiac stress resistance and maintains normal heart rate and myogenic rhythm. Another effector of dTOR and insulin signaling, dS6K, may influence cardiac aging non-autonomously through its activity in the insulin-producing cells (IPCs), possibly by regulating dilp2 expression. Thus, elevating d4eBP activity in cardiac tissue represents an effective organ-specific means for slowing or reversing cardiac functional changes brought about by normal aging.
Keywords: cardiac senescence, heart failure, arrhythmia, Foxo, 4E-BP, eif4E, TSC, S6K, TOR
Introduction
Single-gene mutations have been demonstrated to extend lifespan in multiple organisms (Kim, 2007; Tatar et al., 2003). Tissue-specific targeted knockdown or overexpression of such genes can, in some cases, act locally to extend lifespan of the entire organism (Giannakou et al., 2004; Helfand and Rogina, 2003; Hwangbo et al., 2004b; Libina et al., 2003), presumably by regulating long-range signaling molecules. Less is known about how target tissues respond to such signals to slow age-related physiological changes. By understanding tissue-specific regulation of aging physiology, it may become possible to dramatically reduce the negative consequences that aging has on the function of critical organs.
Cardiac functional changes during aging have been described in Drosophila (Luong et al., 2006; Ocorr et al., 2007a-c; Paternostro et al., 2001; Taghli-Lamallem et al., 2008; Wessells and Bodmer, 2004; Wessells et al., 2004). Multiple mutations have been demonstrated to extend cardiac functional parameters in conjunction with extending lifespan (Luong et al., 2006; Wessells et al., 2004) Significantly, it has also been demonstrated that cardiac functional aging can be attenuated by virtue of tissue-specific genetic mutations, and that these effects are separable from changes in lifespan (Luong et al., 2006; Ocorr et al., 2007a; Wessells et al., 2004).
Two signaling pathways have been shown to regulate cardiac functional aging in a tissue-autonomous manner, the insulin signaling pathway (Wessells et al., 2004) and the TOR kinase signaling pathway (Luong et al., 2006). Although both pathways are interlinked (see diagrams in Oldham and Hafen, 2003; Junger et al., 2003), it has not been clear how they are coordinated to regulate long-term cardiac function. As the effects of insulin and TOR signaling on cardiac aging are remarkably similar, we decided to examine potential common factors under the control of both that may account for the cardiac aging phenotype.
A potential area of commonality between TOR and insulin signaling is that of translational control. Both TOR and the insulin receptor are involved in the control of translation (Oldham and Hafen, 2003). One important mechanism by which these pathways control the rate of protein synthesis is by regulating the expression or activity of 4eBP (Hay and Sonenberg, 2004; Junger et al., 2003; Teleman et al., 2006; Puig et al., 2003), a protein which impedes translation by binding and sequestering an essential component of the translation initiation complex (Hay and Sonenberg, 2004; Tee and Blenis, 2005). In C. elegans, lifespan extension by the Foxo homolog daf-16 is partially dependent on reduction in translation (Hansen et al., 2007), and activation of translation in rat cardiomyocytes is dependent on insulin and TOR signaling. (Wang et al., 2000).
Here, we examine whether the role of 4eBP downstream of both insulin and TOR signaling is sufficient to mimic the role of insulin and TOR signaling in controlling cardiac functional aging. We find that d4eBP acts autonomously in the heart and in a downstream fashion of both dTOR and dFoxo to modulate cardiac aging. This role is specific to d4eBP, as other targets of dFoxo and dTOR do not have similar effects. dMyc has no discernible effect on the rate of age-related cardiac decline, while dS6K appears to modulate cardiac aging by virtue of its activity in the insulin-producing cells (IPCs).
Results
dFoxo and dS6k loss-of-function have differential effects on cardiac aging
Overexpression of dFoxo in adipose tissue of the whole body (Giannakou et al., 2004) or just in the head (Hwangbo et al., 2004) has been shown to extend lifespan. Conversely, dFoxo null mutants have reduced stress resistance (Junger et al., 2003; Puig et al., 2003) and can block lifespan–extending effects of JNK pathway activation (Wang et al., 2005). We have previously established that external electrical pacing of the fly’s heart causes acute cardiac dysfunction (arrest or fibrillation, collectively termed ‘failure’) at a rate that is highly age-dependent and thus serves as a useful marker for cardiac functional aging (Wessells and Bodmer, 2004; Wessells et al., 2004). Cardiac-specific overexpression of dFoxo is known to prevent age-related decline of cardiac performance (Wessells and Bodmer, 2004; Wessells et al., 2004), but the effects of systemic dFoxo loss-of-function on cardiac performance have not been measured. dFoxo21/25 flies, which are null for dFoxo function (Junger et al., 2003), display cardiac stress resistance characteristic of wild-type flies (Fig. 1).
Figure 1. dS6K mutants have improved cardiac stress resistance.
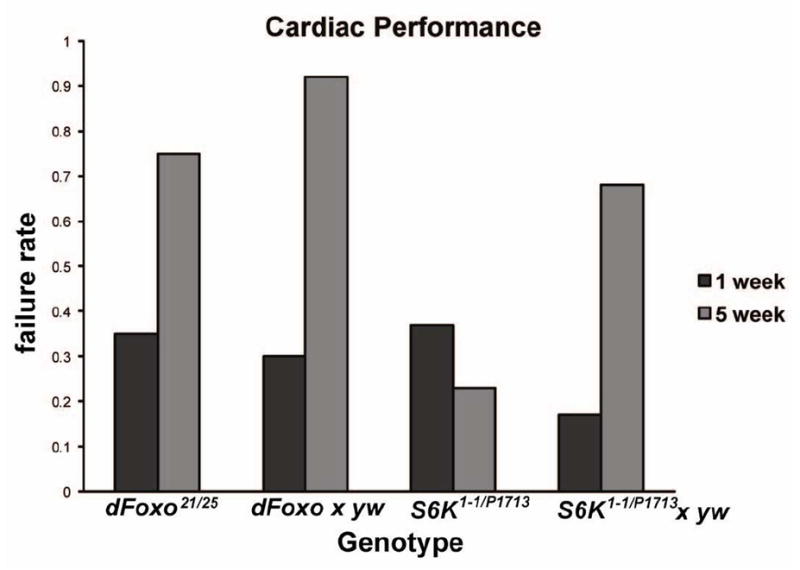
dFoxo21/25 flies show cardiac stress resistance similar to wild-type flies (yw data not shown) and heterozygotes (dFoxo × yw) at both 1 week and 5 week of age. dS6K mutants show no increase in stress-induced failure rates between one and five weeks of age (χ2=11, p<0.001). Heterozygotes (S6KP1713 × yw) show a normal decline in cardiac stress resistance between 1 and 5 weeks.
dS6K is a kinase that acts downstream of both insulin and TOR signaling to phosphorylate the ribosomal S6 protein and regulate translation (Ruvinsky and Meyuhas, 2006). Viable heteroallelic dS6K mutants (S6K1-1/S6KP1713; Montagne et al., 1999) show improved late-life cardiac performance, with stress-induced failure rates at five weeks of age that have not increased compared to those at 1 week, and are significantly less than in heterozygotes (genotype-by-age, chi-square χ2=11, p<0.001), which display normal age-related decline in cardiac stress resistance (Fig. 1).
dTOR modulates cardiac functional aging tissue-autonomously
Reduced dTOR function has been previously shown to extend lifespan in multiple species (Kapahi et. al., 2004; Powers et. al., 2006; Pan et. al., 2007) and protect youthful cardiac performance to advanced ages (Luong et al., 2006). In an effort to ascertain the relationship between insulin/Foxo signaling and TOR/S6K signaling in controlling cardiac age-related change, we have altered expression levels of these proteins, both singly and in combination, in myocardial tissue and assayed hearts for changes in stress response during the aging process. Using the myocardial cell-specific Gal4 driver GMH5 (Wessells et al., 2004), we expressed UAS-dTOR (Hennig and Neufeld, 2002) in the heart throughout adult life. Increased cardiac dTOR expression resulted in increased stress-induced failure rate already at young ages (Fig. 2A, genotype-by-age, χ2=17, p<0.0001). This high failure rate continued to increase during the aging process, remaining higher than controls at each time point. In flies, as in mammals, TOR activity is negatively regulated by the amino-acid responsive protein complex TSC1 and TSC2 (Gao et al., 2002). Co-overexpression of dTSC1&2 (Potter et al, 2001) in the heart throughout life greatly reduced the slope of age-related decline in cardiac stress response. dTSC-overexpressing hearts were identical in stress-induced failure rate to controls at young ages, but exhibited only a minimal increase in their stress induced failure rate with increased age (Fig. 2A, genotype-by-age, χ2=10, p<0.01; see also Suppl. Table 1 for control outcrosses). We conclude that TOR activity in myocardial tissue is important for regulating cardiac stress sensitivity, and that reduction in TOR activity promotes maintenance of youthful heart function during aging.
Figure 2. Cardiac TOR over-expression results in an increase in stress-induced heart failure.

A) Cardiac TOR over-expression (Hennig and Neufeld, 2002) resulted in an increase in cardiac stress-induced failure rate at young ages compared to the control group (blue) (week 1: χ2=17, p<0.0001). The failure rate at each time point remained higher than that of control flies (GMH5 × yw). Cardiac dTSC1-2 over-expression (yellow) generated similar cardiac failure rates to the control group at young ages, but significantly reduced age-related decline (genotype-by-age, χ2=10, p<0.01). B) Cardiac over-expression of UAS-dnS6K × GMH5 did not significantly affect the slope of age-related decline compared to controls. C) Adipose over-expression of UAS-dnTOR (brown), UAS-dnS6K × yw (blue), and UAS-dnS6K × Isp2-Gal4 did not significantly affect the slope of age-related decline compared to controls. Expressing UAS-dnS6K × dilp2-Gal4 (light blue) impeded age-related decline in cardiac stress failure rates (genotype-by-age, χ2=12, p<0.001). UAS-dnS6K × dilp2-Gal4 had a higher failure rate at week one compared to the control (dark blue) (χ2=29, p<0.0001) although this decreased over time while the control group increased (genotype-by-age, χ2=54, p<0.0001). Statistics: two-way ANOVA followed by a Bonferroni comparison.
dS6K can modulate cardiac functional aging tissue-non-autonomously
Since mutations in dS6K reduce age-related cardiac dysfunction, we examined whether tissue-specific reduction of dS6K in the heart could provide benefits to cardiac aging. We expressed a Gal4-inducible dominant-negative dS6K construct (Barcelo and Stewart, 2002) with the heart-specific GMH5-Gal4 driver. The progeny of UAS-dnS6K × GMH5 flies had an identical stress-induced failure rate at young ages to that of flies containing either GMH5 or UAS-dnS6K constructs alone (Fig. 2B). The GMH5 × yw and UAS-dnS6K × yw progeny displayed an increased failure rate with age similar to that of other controls (Suppl. Table 1). Interestingly, UAS-dnS6K × GMH5 progeny displayed an increase in their failure rate with age at a slightly shallower slope than their controls. However, the failure rate of the UAS-dnS6K × GMH5 progeny was not significantly different from that of UAS-dnS6K × yw flies. This suggests that this dnS6K is either less active in the heart than other tissues (see below), or dS6K activity in myocardial tissue does not account much for the effect of systemic reduction of dS6K in cardiac aging, and that that dS6K may (primarily) play an indirect role via other tissues.
In order to determine how S6K might act indirectly in other tissues to regulate cardiac aging, we first expressed dominant negative versions of each gene with an adipose-specific driver, lsp2-Gal4 (Cherbas et al., 2003; Henning and Neufeld, 2002). Neither dominant-negative dTOR (Hennig and Neufeld, 2002) nor dominant-negative dS6K had a significant effect on the slope of age-related decline when expressed in the fat body (Fig. 2C; Suppl. Table 1), as compared to outcrossed dominant-negative constructs without the presence of an inducing driver construct. Although a negative result with overexpression constructs is not conclusive by itself, manipulation of dTOR and dS6K in the fatbody does not alter cardiac aging under normal (dietary) conditions.
In vertebrates, S6K responds to glucose levels to control proliferation of pancreatic β͂-cells, thus regulating insulin production (Briaud et al., 2003). Since flies produce insulin-like peptides (DILPs) from specialized neuronal cells (Rulifson et al., 2002), we tested the possibility that dominant-negative S6K may affect heart function via the neuronal insulin-like peptide producing cells (IPCs). We utilized an IPC-specific driver (dilp2-Gal4; Ikeya et al., 2002) to express dnS6K and measured stress-induced heart failure rate. The progeny of dnS6K × dilp2-Gal4 flies showed a somewhat higher failure rate than controls at early ages. However, this failure rate did not increase with age and was even seen to be lower in five week-old flies compared to one-week old flies (Fig. 2C, genotype-by-age, χ2=12, p<0.001). This profile is strikingly similar to that seen in loss-of-function dS6K mutants (Fig. 1) and in flies with ablated IPCs (Wessells et al., 2004), suggesting that interference with S6K in the IPCs might affect cardiac function by virtue of lowering systemic DILP levels. To assess this possibility, we measured the mRNA levels of DILP2 in flies expressing dnS6K in the IPCs. We found that expression of dnS6K in the IPCs leads to a reduction in the mRNA levels for dilp2 (Fig. 3A, unpaired, two-tailed t-test, p<0.01, n=3). This reduction is similar to that seen in dS6K mutant flies (Fig. 3A). Consistent with this result, we measured ‘blood’ glucose levels and found a significant increase compared to controls (Fig. 3B, unpaired, two-tailed t-test, p<0.01, n=8). The reduction in dip2 levels in these flies is not a consequence of loss of IPCs, as flies expressing dnS6K driven by dipl2-Gal4 exhibit normal size IPC cell clusters (Fig. 3C, D). We also expressed dnS6K in the adipose tissue and did not see a change in dilp2 RNA levels (data not shown). Thus, reduction of dS6K activity in IPCs by multiple methods leads to lowered dilp2 RNA levels, which correlates with resistance to pacing-induced cardiac failure with age non-autonomously. We do not know at this point, whether it is the reduction of dilp2 expression or of another factor that is critical in modulating age-dependent cardiac performance via the IPCs in a S6K-dependent manner.
Figure 3. S6K modulates heart aging by regulating DILP2 levels.

A) Decreased dilp2 mRNA levels in the S6K mutant and by IPC expression of dnS6K. (p<0.05, unpaired, two-tailed t-test, n=3). Decreased dilp2 expression levels with dnS6K expression are shown fold change relative to control. B) Expression of dnS6K in the IPCs leads to increased glucose levels (p<0.001, unpaired, two-tailed t-test, n=8) compared to control. Glucose levels are shown fold change compared to control. These experiments have been performed 2 times independently. C, D) Expression of dnS6K in the IPCs does not lead to a change in the number of IPCs. Anterior is on the left and posterior on the right. The 2 sets of the IPC clusters are marked by circles and the IPCs by small arrows (7 per cluster). (C) Control genotype: (dilp2-Gal4) yw; dilp2-Gal4, UAS-GFP/+. (D) Experimental genotype: (dilp2-Gal4; UAS-dnS6K) yw; dilp2-Gal4, UAS-GFP/+; UAS-dnS6K/+. Images at 400X and Bar is 0.1 mm.
Taken together, these results are consistent with a model where both dTOR and dFoxo act directly in myocardial tissue to regulate cardiac aging. dS6K appears to be of lesser importance within the heart itself, but plays an indirect, permissive role in the IPCs controlling the amount of dilp2 or other factors the heart is exposed to. Action of dTOR and dS6K in adipose tissue appears to be dispensable for the regulation of cardiac functional aging under normal dietary conditions, but seems to mediate effects on the heart when the flies are fed a high fat diet (R. Birse; R. B. and S. O., unpublished).
d4eBP acts downstream of dFoxo and dTOR in a tissue-autonomous fashion to modulate cardiac functional aging
We wanted to know what downstream factors in cardiac tissue were necessary for dTOR and dFoxo to exert their effects on cardiac functional aging. Since dTOR activity promotes rapid functional aging and dFoxo activity slows functional aging, we looked for candidate downstream factors that are both negatively regulated by dTOR activity, and positively regulated by dFoxo activity. One such candidate is 4eBP, which, in flies as in mammals, acts to reduce levels of mRNA translation in the cell by binding to the translation initiation factor, Eif4e (Jastrzebski et al., 2007), which is known to modulate aging in worms (Syntichaki et. al., 2006). d4eBP is regulated transcriptionally by dFoxo (in flies; Junger et al., 2003; Puig et al., 2003), and its activity is controlled post-transcriptionally by TOR-mediated phosphorylation (Beretta et al., 1996; Brunn et al., 1997; Burnett et al., 1998; Pause et al., 1994). d4eBP has also been shown to interact genetically with dFoxo to control stress response and lifespan (Tettweiler et al., 2005).
d4eBP null mutant flies exhibit an early increase in stress-induced failure rate compared to controls, in which the P-element causing the mutation has been reverted (Fig. 4A, genotype-by-age, χ2=15, p<0.001) We then investigated whether d4eBP and its regulatory target, dEif4e, were sufficient to control cardiac functional aging in flies. Cardiac-specific expression of UAS-d4eBP (Miron et al., 2001) in adult flies dramatically reduces age-related decline in cardiac performance. Stress-induced failure rate of UAS-d4eBP × GMH5 progeny was as low at five weeks as at one week (Fig. 4B, genotype-by-age, χ2=1, p = 0.4). Expression of UAS-Eif4e in the myocardium, as well as cardiac-specific expression of three independently generated UAS-inducible insertions upstream of the dEif4e locus (eIF-4E, Krupp et al., 2005), also abrogated a gradual decline in cardiac stress response. However, in this case, flies throughout the five weeks of testing all showed a maximal failure rate in response to stress, with one-week-old flies showing high stress-induced failure rate normally associated with five-week old flies (Fig. 4B, χ2=22, p<0.0001).
Figure 4. Cardiac overexpression of d4eBP and deif4e flattens the slope of stress-induced failure rate during aging, while tissue-specific knockdowns of dFOXO and d4eBP increase stress-induced heart failure.

A) d4eBP null mutants exhibit a high failure rate at young ages and have a significantly different pattern of age-related change to revertant controls (genotype-by-age, χ2=15, p<0.001). B) Cardiac over-expression of d4eBP showed a significantly flattened slope of stress-induced failure. Indeed, the change in failure rate with age is no longer statistically significant (genotype-by-age, χ2=1, p =0.4). Expression of dEif4e also showed a flattened slope throughout the time period, but showed a higher failure rate compared to controls at each time point (at week one χ2=22, p<0.0001). Three independently generated upstream insertions of a UAS-inducible expression element immediately upstream of the 5’UTR of the dEif4e locus were crossed to GMH5-Gal4 and produced similar results. Results shown are from dEif4eGS2783 × GMH5-Gal4. UAS-dMyc showed no significant effect on the slope of age-related functional decline in cardiac tissue compared to the controls. C) Cardiac RNAi knockdowns of both Foxo and 4eBP showed significantly higher failure rates at week one compared to their respective controls (UAS-dfoxo-RNAi × GMH5: χ2=6, p<0.02; UAS-4eBP-RNAi × GMH5: χ2=14, p<0.001). No significant change in the high failure rate occurred in either genotype throughout five weeks. D) Cardiac co-expression of d4eBP and dTOR in flies had a similar profile of age-related failure as expression of d4eBP alone (genotype-by-age, χ2=1, p=0.3). Likewise, cardiac co-expression of deif4e and dTSC (yellow) was not significantly different from dEif4e over-expression alone (genotype-by-age, χ2=0.5, p=0.5). E) Co-expression of dFoxo and d4eBP was not significantly different than expressing d4eBP alone (genotype-by-age, χ2=3, p=1.0). UAS-dEif4e;UAS-dfoxo × GMH5 flies showed a higher failure rate than UAS-dEif4e;UAS-dfoxo × yw at 1 week (χ2=41, p<0.0001). No significant change in stress-induced cardiac failure of UAS-dEif4e; UAS-dfoxo × GMH5 flies occurred during five weeks of aging. F) A repetition of cardiac expression of dfoxo-RNAi produced similar results with a significantly higher failure rate at 1 week of age than is seen during cardiac expression of d4eBP or outcross controls (F-test, p<0.001). Co-overexpression of d4eBP with dFoxoRNAi completely eliminated the adverse effect of dFoxo RNA reduction. Statistics: two-way ANOVA followed by a Bonferroni comparison (except in F).
We next asked whether these phenotypes were specific to the 4eBP/Eif4e complex and its targets, or whether any protein capable of altering growth and cellular translation could generate the same effect. We expressed the Drosophila Myc gene in the myocardium and tested cardiac stress response over time. Myc is a highly conserved regulator of cellular growth and translation (de la Cova and Johnston, 2006). In Drosophila, dMyc has recently been demonstrated to be a direct target of dFoxo, and to act downstream of both dTOR and dFoxo to regulate ribosome biosynthesis in response to nutritional levels (Teleman et al., 2008). However, UAS-dMyc (Johnston et al., 1999) showed no effect on cardiac functional aging when overexpressed in the heart (Fig. 4B). We conclude that dEif4e activity is specific and sufficient to prematurely induce symptoms of cardiac functional aging, while its negative regulator, 4eBP, is sufficient to slow or even block such symptoms.
Since overexpression of either dFoxo or d4eBP decreases pacing-induced heart failure at old age, we asked if tissue-specific RNAi knockdown could prematurely increase pacing-induced heart failure. Cardiac expression of RNAi constructs for either dFoxo or d4eBP increased stress-induced cardiac failure rates significantly at young ages compared to controls. Furthermore, little age-related decline occurred in such flies, with one-week-old flies showing a similar stress response as five-week-old flies (Fig. 4C, UAS-dFoxo-RNAi × GMH5: χ2=6, p<0.02; UAS-4eBP-RNAi × GMH5: χ2=14, p<0.001). By contrast, outcrossed control strains carrying the same RNAi constructs without an inducible driver showed a normal pattern of age-related increase in stress-induced failure rate with age (Fig. 4C, see also Suppl. Table 1). We conclude that lowering either dFoxo or d4eBP function is sufficient to induce hearts to respond to stress as if they were already aged.
Since d4eBP expression has the same effect on cardiac stress response during aging as does inhibition of dTOR activity, we wanted to confirm whether d4eBP acts downstream of dTOR in this context or whether the two effects were independent. If the effects of d4eBP and dTOR are independent, then co-expression of both genes in the heart should result in an intermediate phenotype. Conversely, if the two act in a linear pathway, then the phenotype of flies co-expressing both genes should resemble the phenotype of the more downstream gene. Flies co-expressing d4eBP and dTOR had slightly elevated stress-induced failure rates as young flies. This failure rate did not increase with age, however, and even declined somewhat (Fig. 4D, genotype-by-age, χ2=1, p=0.3). This phenotype is similar to that of flies overexpressing d4eBP alone (Fig. 4B), consistent with the idea that d4eBP acts downstream of dTOR to regulate cardiac functional aging. A complementary experiment also produced results supportive of this model. Co-overexpression of dEif4e and the dTOR antagonist dTSC1-2 produced a phenotype identical to that of dEif4e overexpression alone (Fig. 4B, D, genotype-by-age, χ2=0.5, p=0.5 ).
Since d4eBP is a transcriptional target of dFoxo, we also tested whether d4eBP might account for the beneficial effects of dFoxo expression in aging hearts. To ask this question, we co-expressed the d4eBP binding partner dEif4e along with dFoxo. If the primary role of dFoxo in slowing cardiac functional aging is to up-regulate 4eBP, thus lowering dEif4e activity, then this co-expression should phenotypically mimic expression of dEif4e alone. If dFoxo acts instead through multiple independent targets, the prediction would be that heart performance would still benefit from dFoxo expression even in the presence of abundant dEif4e function. Flies co-overexpressing dEif4e and dFoxo exhibited elevated stress-induced failure rates already at one week of age and failure rates remained at a high level at later ages (Fig. 4E, one-week; χ2=41, p<0.0001). This profile matches exactly the phenotype of flies expressing dEif4e alone in heart tissue (Fig. 4B). Meanwhile, flies carrying both expression constructs without an inducible driver had a wild-type profile of age-related decline (Fig. 4E). These results are consistent with a model where up-regulation of dEif4e is sufficient to bypass the effects of dFoxo expression on cardiac functional aging.
We also tested whether dFoxo and d4eBP may have beneficial effects separately from their relationship with each other. We co-expressed the two and asked whether any additive benefit would result from combining the two, or whether, conversely, d4eBP expression would already provide the maximum possible benefit that dFoxo could provide to aging cardiac tissue. Flies co-expressing d4eBP and dFoxo in cardiac tissue exhibited a similar profile of slowed functional aging to flies expressing either gene alone (Fig. 4B, E and Wessells et. al., 2004; genotype-by-age, χ2=3, p=1.0). Furthermore, co-overexpression of d4eBP rescues the high failure rate phenotype caused by cardiac expression of dFoxoRNAi in one week old flies (Fig. 4F).
An inherent caveat of co-overexpression studies is that it is difficult to rule out the possibility that the effects seen can be attributed to unexpected differences in expression/activity levels between constructs. However, in this case, expression of several insulin/TOR pathway components in five different cardiac over-expression combinations (progeny of dEif4e/dFoxo and dFoxo/d4eBP -Fig. 4E; dTSC/dEif4e and d4eBP/dTOR -Fig. 4D; dFoxo-RNAi/d4eBP -Fig. 4F) all produce results that argue in the same direction, suggesting that coincidental differences in expression level are unlikely to be a deciding factor. We conclude that overexpression of d4eBP alone is capable of reproducing the entire effect of dFoxo expression on cardiac functional aging, confirming the previously established epistatic relationship between dFoxo and d4eBP (Junger et al., 2003).
Expression of dEif4e in the heart mimics the age-related increase in cardiac arrhythmias
The cardiac response to electrical pacing-induced stress has been a useful marker for measuring age-related decline of cardiac function in fly populations. Several other measurements of cardiac function have also been found to decline in aging flies (Ocorr et al., 2007a-c; Paternostro et al., 2001; Taghli-Lamallem et al., 2008; Wessells and Bodmer, 2007), and these correlate well with indices of vertebrate cardiac functional changes during aging (Jones, 2006; Judge and Leeuwenburgh, 2007). If d4eBP and dEif4e are critical factors in regulating the rate of cardiac aging, then the expectation would be that a change in d4eBP or dEif4e function should affect multiple indices of cardiac performance in aging flies.
We measured the effects of dEif4e expression in the myocardium on the age-dependence of both heart rate and incidence of arrhythmias. When compared to out-crossed controls, hearts overexpressing dEif4e exhibit a significant increase in heart period (corresponding to a lower heart rate) at young ages, which is similar to that observed at older ages (Fig. 5A, B). This increase in heart period is due to an increase in both the systolic as well as the diastolic interval during a heart beat (Fig. 5C, D).
Figure 5. Effects of heart-specific overexpression of dEif4E.

A, B) Representative M-mode records showing the movement of the heart tube walls (y axis) over time (x axis). Blue bars indicate the diastolic diameter of each heart and the red bars indicate the systolic diameter. Records for 1-week old flies (A) show predominately regular heart beat patterns as the GMH5 heterozygotes (as in Ocorr et al., 2007a), whereas most flies overexpressing UAS-dEif4e in the heart show arrhythmic heart beat patterns. Approximately 50% of the UAS-dEif4e heterozygotes exhibited slightly arrhythmic patterns, which may reflect a partial leakiness of the UAS transgene in the absence of the GMH5 driver. Records for 4 week old flies (B) all show an increased arrhythmicity, but flies with heart-specific overexpression of dEif4e exhibited a dramatic increase in the incidence of sustained systoles (fibrillation). C) Overexpression of dEif4e resulted in a significant increase in the systolic intervals (SI) in both young (p<0.01) and old flies (p<0.05) compared to controls (one-way ANOVA and Tukey’s multiple comparison posttest). There was no significant effect of age on SI (p>0.05, two-way ANOVA). D) Overexpression of dEif4e also significantly increased diastolic intervals (DI) in young flies (p≪0.01, one-way ANOVA and Tukey’s posttest). There was no age-dependent effect on DI for the group as a whole but there was a significant effect of age on the DI of the control groups (p<0.05, two-way ANOVA). E) The arrhythmias observed in the M-modes in (A) can be quantified as the arrhythmicity index (AI), which is the standard deviation of all heart periods in each record normalized to the median heart period for each fly (Ocorr et al., 2007a). The average AI for all the flies in each genotype is shown. This index shows a significant age-dependence (p<0.05, two-way ANOVA) and is also significantly higher in 4 week old flies with heart-specific overexpression of dEif4e compared to controls (p<0.05, two-way ANOVA and Bonferroni posttest). The AI is also significantly increased in hearts from 1 week old flies where dEif4e is overexpressed compared to GMH5 controls (p<0.05, Student’s t-test). The mean AI in outcrossed controls (UAS-dEif4e/+) is intermediate between the other groups consistent with what is seen in the M-modes. All error bars are +/− SEM, for each bar n>20 flies, and data are from a single experiment.
Next, we determined the incidence of arrhythmias, which normally increases steadily with age (Ocorr et al., 2007a). The level of cardiac arrhythmia is calculated as the standard deviation of all the heart periods recorded for a fly normalized to its median heart period, thereby generating an ‘arrhythmia index’ that quantitatively represents the level of cardiac beat-to-beat variation (Ocorr et al., 2007a). We find that increased cardiac dEif4e expression at young ages caused an elevated incidence of arrhythmias, similar to that normally observed in old flies (Fig. 5A, B, E). Remarkably, at older ages these arrhythmias increase even further with elevated dEif4e levels (Fig. 5A, B, E), suggesting an accelerated cardiac deterioration at advanced ages. The increased arrhythmia index in these flies was primarily due to an increased variability of the diastolic intervals (Suppl. Fig. 1). Additionally, bouts of fibrillation occurred with high frequency in dEif4e overexpressing hearts, especially in older flies (Fig. 5B), and such events did not occur in control flies until after three weeks of age. Taken together, these results indicate that the presence of high levels of dEif4e in cardiac tissue is sufficient to promote premature manifestation of several markers of age-related cardiac functional decline.
Discussion
Invertebrate model systems have become an informative arena in which to study regulation of “functional aging” or “healthspan”, in addition to the long-standing utility of flies and worms as models for research into regulation of lifespan. Indeed, interventions that extend lifespan do not necessarily extend functionality of critical tissues (Bhandari et al., 2007; Burger and Promislow, 2006). Conversely, tissue-specific interventions can protect organ functionality with minimal effect on lifespan (Wessells et al., 2004). In an effort to further understand the regulation of functional aging of cardiac tissue, we have used the fly system to test direct genetic interventions in cardiac tissue, as well as indirect interventions that affect cardiac function by virtue of their roles in other tissues. We find that d4eBP is a critical target downstream of both dFoxo and dTOR in cardiac tissue. Overexpression of d4eBP is sufficient to protect cardiac function against functional decline during aging, and can rescue the effects of TOR overexpression in the heart. Likewise, the binding target of d4eBP, dEif4e, is sufficient to hasten cardiac aging when expressed in the myocardium, and such expression can counteract the benefits of impeding either insulin or TOR signaling in heart tissue. Thus, these results highlight the promise of using Drosophila genetics to determine how the insulin and TOR pathways interact to regulate the function of tissues and organs, including the heart (model in Fig. 6).
Figure 6. Model of autonomous and non-autonomous effects of insulin/TOR signaling.

dFoxo and dTOR both act systemically in adipose tissue to promote growth and regulate lifespan. Both of these proteins also act autonomously in target tissues of insulin signaling, such as the heart, where dFoxo and dTOR are both necessary for normal cardiac senescence to occur. A critical target of both dTOR and dFoxo in this context is d4e-BP, which also acts autonomously in cardiac tissue to regulate cardiac senescence. By contrast, dS6K seems to play a prominent indirect role in the regulation of cardiac aging by virtue of its activity in the insulin-producing cells and the expression of insulin-like peptides, which in turn, signal to the heart.
Since our results are derived from overexpression and co-overexpression studies, we cannot formally conclude that d4eBP is fully epistatic to dTOR and dFoxo in this context. However, it is clear that overexpression of d4eBP has a potent protective effect on long-term maintenance of cardiac function during aging and represents a target of great potential for therapeutic interventions to protect the aging heart.
The Drosophila insulin-TOR pathways have numerous effects on cellular metabolism and maintenance, including fat storage (Bohni et al., 1999; Clancy et al., 2001; Luong et al., 2006; Tatar et al., 2001), glucose metabolism (Luong et al., 2006; Rulifson et al., 2002), oxidative stress sensitivity (Patel and Tamanoi, 2006; Wang et al., 2005), starvation stress sensitivity (Teleman et al., 2005; Tettweiler et al., 2005), autophagy (Scott et al., 2004), and cell growth (Colombani et al., 2003; Junger et al., 2003; Oldham et al., 2000; Puig et al., 2003; Zhang et al., 2000). Indeed, d4eBP is a critical mediator of the effects of dFoxo on cardiac functional aging. Additionally, 4eBP may be a generalized mediator of Foxo function as part of a feedback loop that controls its own translation and that of the insulin receptor (Marr et al., 2007). Each of these effects is likely to play a significant role in lifespan regulation. Which of these effects are critical for the regulation of cardiac functional aging? Neither resistance to starvation or oxidative stress are necessary processes for extension of cardiac functional aging, since hypomorphic dTOR mutations that do not enhance resistance to either starvation nor oxidative stress nonetheless extend cardiac function to advanced ages (Luong et al., 2006). Cardiac expression of another important target of dFoxo and dTOR, the cell growth regulator dMyc (Teleman et al., 2008) also had no effect on cardiac aging.
Interestingly, cardiac expression of a dominant-negative dS6K, another regulator of ribosomal activity, did not significantly slow cardiac performance decline with age, even though S6K can act systemically to influence cardiac aging, as it does for lifespan in worms by regulation of translation levels (Hansen et al., 2007). Thus, it does not seem that tissue-specific regulation of a cell growth program per se is the predominant factor controlling cardiac functional aging. Rather, downregulation of translation by d4eBP appears to have effects in cardiac tissue that are uniquely aging-related and distinguishable from other potent translational regulatory proteins.
An attractive candidate for the primary mechanism downstream of d4eBP in the aging heart may be regulation of fatty acid metabolism. Changes in fatty acid substrate utilization have been associated with age in rodent hearts (Sample et al., 2006). Expression of genes involved in fatty-acid metabolism are up-regulated both in dietary-restricted rodent hearts (Dhahbi et al., 2006; Linford et al., 2007) and in fasted Drosophila larvae (Bauer et al., 2004; Gershman et. al., 2007), while such genes are down-regulated during normal aging (Linford et al., 2007). Mutations in dTOR that slow cardiac functional aging also alter fatty acid metabolism (Luong et al., 2006). dFoxo also directly regulates lipid metabolism through its transcriptional target, dLip4 (Vihervaara and Puig, 2008). Furthermore, mutations in Drosophila fatty acid transporter genes dramatically alter late-life cardiac performance (S. M. and R. J. W., unpublished observation).
Even though cardiac interference with dS6K function did not affect cardiac aging, dS6K hypomorphic mutants exhibit dramatic improvement in late-life cardiac function. Tissue-specific expression of dnS6K in IPCs was able to replicate the cardiac benefits of dS6K partial loss-of-function mutants, suggesting that S6K activity can dramatically alter cardiac function with age by acting non-autonomously. Moreover, IPC expression of dnS6K leads to a reduction in the mRNA levels for dilp2. These findings are reminiscent of ablating the IPC neurons, which also leads to a block of the age-dependent increase in pacing-induced heart failure (Wessells et al., 2004). However, it is not clear at this point what the exact roles of the different DILPs are in modulating aging (Broughton et al., 2005; Min et al, 2008), and whether there is redundancy between them. As it may be, regulation of insulin signaling is clearly critical autonomously within the heart, and possibly also non-autonomously within the IPCs via S6K activity, in the control of cardiac aging.
In conclusion, we favor a model (Fig. 6) in which d4eBP/dEif4e act autonomously within the heart to modulate cardiac functional aging. There are also non-autonomous influences mediated by (adipose) tissues and secondary signaling hormones that may modulate overall lifespan and/or specifically the age-dependent decline of heart function. Importantly, insulin-like peptides themselves are some of these secondary signals, and insulin-TOR signaling has an important tissue-autonomous role in regulation of cardiac functional aging, which is likely via a d4eBP/dEif4e output, as our data suggest. dS6K, on the other hand, also plays a non-autonomous role in regulating expression of insulin-like peptide 2, and possibly other factors, in the insulin-producing cells. Whether it is the reduction in dilp2 expression or of another factor that is responsible in abrogating the functional decline of cardiac function with age remains to be determined.
Given the complex and flexible role of Foxo and TOR in regulating the response of metazoans to changing environmental conditions, discovery of limited, tissue-specific, effects of these proteins on the aging process offers exciting therapeutic possibilities. By increasing levels of 4eBP activity in post-mitotic adult cardiac tissue, and keeping them high throughout life, it may be possible to prevent or reduce the cardiac functional decline that comes about as a consequence of normal aging, while avoiding systemic alterations in metabolism.
Experimental Procedures
Stress-induced cardiac failure
Flies were aged in the same way as for lifespan. Once each week, a minimum of 50 males and 50 females were removed from the cohort and subjected to an electrical pacing protocol as previously described (Wessells and Bodmer, 2004). The percentage of flies that responded to pacing by entering either fibrillation or arrest was charted and expressed as “% failure rate”. These fibrillation events resemble stress-induced “sudden death” in vertebrates, and are not necessarily reflective of resting heart dysfunction. Rather, they are a marker for stress sensitivity. As gender did not significantly affect failure rate, combined male and female data was analyzed by multivariate regression for genotype by age effect.
Q-RT-PCR Analysis
Total RNA was extracted from six adult flies per genotype (done in triplicate biological samples) by using TRIzol (Invitrogen) and purified with the RNeasy kit (Qiagen, Valencia, CA) for adult flies. After treatment with DNaseI, first-strand cDNA was transcribed with SuperScript III (Invitrogen) by using oligo(dT) primer, followed by second-strand synthesis. Quantitative PCR was carried out by using the LightCycler FastStart DNA Master PLUS SYBR Green I kit (Roche), using primers that spanned the DILP2 and Actin 5C introns. DILP2 message levels were normalized to the actin control (primer sequences and cycle conditions available upon request).
Metabolic Assay
Glucose levels were detected using a glucose oxidase assay (Pointe) as described (Luong et al., 2006).
Image-based Analysis of Fly Heart Physiology
Image analysis of beating, semi-intact heart preparations from 1, 2, 3, 4, and 5 week-old adults was performed according to Ocorr et al. (2007a). M-modes were generated using a MatLab (The MathWorks, Natick, MA) -based image analysis program. Briefly, a 1 pixel-wide region is defined in a single frame of a high-speed digital movie that encompasses both edges of the heart tube; identical regions are then cut from all consecutive movie frames and aligned horizontally. This provides an edge trace that documents the movement of the heart tube walls in the y axis over time in the x axis.
Heart periods (HP) are defined as the time between the ends of two consecutive diastolic intervals. The “arrhythmia index” (AI) was calculated as the standard deviation of all recorded HPs for an individual fly, normalized to the median HP for that fly (Ocorr et al., 2007a). Large standard deviations in HP for a single fly are a reflection of non-rhythmic contraction/relaxation cycles.
Supplementary Material
References
- Barcelo H, Stewart MJ. Altering Drosophila S6 Kinase activity is consistent with a role for S6 Kinase in growth. Genesis. 2002;34:83–85. doi: 10.1002/gene.10132. [DOI] [PubMed] [Google Scholar]
- Bauer M, Hamm A, Pankratz MJ. Linking nutrition to genomics. J Biol Chem. 2004;385:593–596. doi: 10.1515/BC.2004.073. [DOI] [PubMed] [Google Scholar]
- Beretta L, Gingras AC, Svitkin YV, Hall MN, Sonenberg N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. Embo J. 1996;15:658–664. [PMC free article] [PubMed] [Google Scholar]
- Bhandari P, Jones MA, Martin I, Grotewiel MS. Dietary restriction alters demographic but not behavioral aging in Drosophila. Aging Cell. 2007;6:631–637. doi: 10.1111/j.1474-9726.2007.00320.x. [DOI] [PubMed] [Google Scholar]
- Bohni R, Riesgo-Escovar J, Oldham S, Brogliolo W, Stocker H, Andruss BF, Beckingham K, Hafen E. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1–4. Cell. 1999;97:865–875. doi: 10.1016/s0092-8674(00)80799-0. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Briaud I, Lingohr MK, Dickson LM, Wrede CE, Rhodes CJ. Differential activation mechanisms of Erk-1/2 and p70(S6K) by glucose in pancreatic beta-cells. Diabetes. 2003;52:974–983. doi: 10.2337/diabetes.52.4.974. [DOI] [PubMed] [Google Scholar]
- Broughton S, Alic N, Slack C, Bass T, Ikeya T, Vinti G, Tommasi AM, Driege Y, Hafen E, Partridge L. Reduction of DILP2 in Drosophila triages a metabolic phenotype from lifespan revealing redundancy and compensation among DILPs. PLoS ONE. 2008;3(11):e3721. doi: 10.1371/journal.pone.0003721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, Houghton PJ, Lawrence JC, Jr, Abraham RT. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science. 1997;277:99–101. doi: 10.1126/science.277.5322.99. [DOI] [PubMed] [Google Scholar]
- Burger JM, Promislow DE. Are functional and demographic senescence genetically independent? Exp Gerontol. 2006;41:1108–1116. doi: 10.1016/j.exger.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci U S A. 1998;95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherbas L, Hu X, Zhimulev I, Belyaeva E, Cherbas P. EcR isoforms in Drosophila: testing tissue-specific requirements by targeted blockade and rescue. Development. 2003;130:271–284. doi: 10.1242/dev.00205. [DOI] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Colombani J, Raisin S, Pantalacci S, Radimerski T, Montagne J, Leopold P. A nutrient sensor mechanism controls Drosophila growth. Cell. 2003;114:739–749. doi: 10.1016/s0092-8674(03)00713-x. [DOI] [PubMed] [Google Scholar]
- de la Cova C, Johnston LA. Myc in model organisms: a view from the flyroom. Seminars in cancer biology. 2006;16:303–312. doi: 10.1016/j.semcancer.2006.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhahbi JM, Tsuchiya T, Kim HJ, Mote PL, Spindler SR. Gene expression and physiologic responses of the heart to the initiation and withdrawal of caloric restriction. J Gerontol A Biol Sci Med Sci. 2006;61:218–231. doi: 10.1093/gerona/61.3.218. [DOI] [PubMed] [Google Scholar]
- Gao X, Zhang Y, Arrazola P, Hino O, Kobayashi T, Yeung RS, Ru B, Pan D. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol. 2002;4:699–704. doi: 10.1038/ncb847. [DOI] [PubMed] [Google Scholar]
- Gershman B, Puig O, Hang L, Peitzsch RM, Tatar M, Garofalo RS. High-resolution dynamics of the transcriptional response to nutrition in Drosophila: a key role for dFOXO. Physiol. Genomics. 2007;29:24–34. doi: 10.1152/physiolgenomics.00061.2006. [DOI] [PubMed] [Google Scholar]
- Giannakou ME, Goss M, Junger MA, Hafen E, Leevers SJ, Partridge L. Long-lived Drosophila with overexpressed dFOXO in adult fat body. Science. 2004;305:361. doi: 10.1126/science.1098219. [DOI] [PubMed] [Google Scholar]
- Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007;6:95–110. doi: 10.1111/j.1474-9726.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- Hars ES, Qi H, Ryazanov AG, Jin S, Cai L, Hu C, Liu LF. Autophagy regulates ageing in C. elegans. Autophagy. 2007;3:93–95. doi: 10.4161/auto.3636. [DOI] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Helfand SL, Rogina B. Molecular genetics of aging in the fly: is this the end of the beginning? Bioessays. 2003;25:134–141. doi: 10.1002/bies.10225. [DOI] [PubMed] [Google Scholar]
- Hennig KM, Neufeld TP. Inhibition of cellular growth and proliferation by dTOR overexpression in Drosophila. Genesis. 2002;34:107–110. doi: 10.1002/gene.10139. [DOI] [PubMed] [Google Scholar]
- Hwangbo DS, Gersham B, Tu MP, Palmer M, Tatar M. Regulation of aging and neuronal insulin transcription by dFOXO in fat body of adult Drosophila. Nature. 2004a;480:652–656. [Google Scholar]
- Hwangbo DS, Gershman B, Tu MP, Palmer M, Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature. 2004;429:562–566. doi: 10.1038/nature02549. [DOI] [PubMed] [Google Scholar]
- Ikeya T, Galic M, Belawat P, Nairz K, Hafen E. Nutrient-dependent expression of insulin-like peptides from neuroendocrine cells in the CNS contributes to growth regulation in Drosophila. Curr Biol. 2002;12:1293–1300. doi: 10.1016/s0960-9822(02)01043-6. [DOI] [PubMed] [Google Scholar]
- Jastrzebski K, Hannan KM, Tchoubrieva EB, Hannan RD, Pearson RB. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors. 2007;25:209–226. doi: 10.1080/08977190701779101. [DOI] [PubMed] [Google Scholar]
- Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–790. doi: 10.1016/s0092-8674(00)81512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SA. Ageing to arrhythmias: conundrums of connections in the ageing heart. The Journal of pharmacy and pharmacology. 2006;58:1571–1576. doi: 10.1211/jpp.58.12.0002. [DOI] [PubMed] [Google Scholar]
- Judge S, Leeuwenburgh C. Cardiac mitochondrial bioenergetics, oxidative stress, and aging. Am J Physiol Cell Physiol. 2007;292:C1983–1992. doi: 10.1152/ajpcell.00285.2006. [DOI] [PubMed] [Google Scholar]
- Junger MA, Rintelen F, Stocker H, Wasserman JD, Vegh M, Radimerski T, Greenberg ME, Hafen E. The Drosophila Forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J Biol. 2003;2:20. doi: 10.1186/1475-4924-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK. Common aging pathways in worms, flies, mice and humans. J Exp Biol. 2007;210:1607–1612. doi: 10.1242/jeb.004887. [DOI] [PubMed] [Google Scholar]
- Krupp JJ, Yaich LE, Wessells RJ, Bodmer R. Identification of genetic loci that interact with cut during Drosophila wing margin development. Genetics. 2005;170:1775–1795. doi: 10.1534/genetics.105.043125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, Kim S, Lee J, Park J, Lee G, Kim Y, Kim JM, Chung J. ATG1, an autophagy requlator, inhibits cell growth by negatively regulating S6 kinase. EMBO Rep. 2007;8:360–365. doi: 10.1038/sj.embor.7400917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003;115:489–502. doi: 10.1016/s0092-8674(03)00889-4. [DOI] [PubMed] [Google Scholar]
- Linford NJ, Beyer RP, Gollahon K, Krajcik RA, Malloy VL, Demas V, Burmer GC, Rabinovitch PS. Transcriptional response to aging and caloric restriction in heart and adipose tissue. Aging Cell. 2007;6:673–688. doi: 10.1111/j.1474-9726.2007.00319.x. [DOI] [PubMed] [Google Scholar]
- Luong N, Davies C, Wessells R, Graham S, King MT, Veech R, Bodmer R, Oldham SM. Activated FOXO-mediated insulin resistance is blocked by reduction of TOR activity. Cell Metabolism. 2006;4:133–142. doi: 10.1016/j.cmet.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Marr MT, 2nd, D’Alessio JA, Puig O, Tjian R. IRES-mediated functional coupling of transcription and translation amplifies insulin receptor feedback. Genes Dev. 2007;21:175–183. doi: 10.1101/gad.1506407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- Min KJ, Yamamoto R, Buch S, Pankratz M, Tatar M. Drosophila lifespan control by dietary restriction independent of insulin-like signaling. Aging Cell. 2008;7:199–206. doi: 10.1111/j.1474-9726.2008.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron M, Verdu J, Lachance PED, Birnbaum MJ, Lasko PF, Sonenberg N. The translational inhibitor 4E-BP is an effector of PI(3)K/Akt signalling and cell growth in Drosophila. Nature Cell Biol. 2001;3:596–601. doi: 10.1038/35078571. [DOI] [PubMed] [Google Scholar]
- Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6-kinase: A regulator of cell size. Science. 1999;285:2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- Neufeld TP. Contribution of Atg1-dependent autophagy to TOR-mediated cell growth and survival. Autophagy. 2007;3:477–479. doi: 10.4161/auto.4348. [DOI] [PubMed] [Google Scholar]
- Ocorr K, Reeves N, Wessells R, Fink M, Chen HS, Akasaka T, Yasuda S, Metzger JM, Giles W, Posakony J, Bodmer R. KNCQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. PNAS. 2007a;104:3943–3498. doi: 10.1073/pnas.0609278104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocorr K, Perrin L, Lim HY, Qian L, Wu X, Bodmer R. Genetic control of heart function and aging in Drosophila. Trends Cardiovasc Med. 2007b;17:177–182. doi: 10.1016/j.tcm.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocorr K, Akasaka T, Bodmer R. Age-related cardiac disease model of Drosophila. Mech Ageing Dev. 2007c;128:112–116. doi: 10.1016/j.mad.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham S, Hafen E. Insulin/IGF and target of rapamycin signaling: a TOR de force in growth control. Trends Cell Biol. 2003;13:79–85. doi: 10.1016/s0962-8924(02)00042-9. [DOI] [PubMed] [Google Scholar]
- Oldham S, Montagne J, Radimerski T, Thomas G, Hafen E. Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev. 2000;14:2689–2694. doi: 10.1101/gad.845700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell. 2007;6:111–119. doi: 10.1111/j.1474-9726.2006.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PH, Tamanoi F. Increased Rheb-TOR signaling enhances sensitivity of the whole organism to oxidative stress. J Cell Sci. 2006;119:4285–4292. doi: 10.1242/jcs.03199. [DOI] [PubMed] [Google Scholar]
- Paternostro G, Vignola C, Bartsch DU, Omens JH, McCulloch AD, Reed JC. Age-associated cardiac dysfunction in Drosophila melanogaster. Circ Res. 2001;88:1053–1058. doi: 10.1161/hh1001.090857. [DOI] [PubMed] [Google Scholar]
- Pause A, Belsham GJ, Gingras AC, Donze O, Lin TA, Lawrence JC, Jr, Sonenberg N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature. 1994;371:762–767. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- Potter CJ, Huang H, Xu T. Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell. 2001;105:357–368. doi: 10.1016/s0092-8674(01)00333-6. [DOI] [PubMed] [Google Scholar]
- Powers RW, III, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological lifespan in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–184. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O, Marr MT, Ruhf ML, Tjian R. Control of cell number by Drosophila FOXO: downstream and feedback regulation of the insulin receptor pathway. Genes Dev. 2003;17:2006–2020. doi: 10.1101/gad.1098703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulifson EJ, Kim SK, Nusse R. Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science. 2002;296:1118–1120. doi: 10.1126/science.1070058. [DOI] [PubMed] [Google Scholar]
- Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci. 2006;31:342–348. doi: 10.1016/j.tibs.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Sample J, Cleland JG, Seymour AM. Metabolic remodeling in the aging heart. J Mol Cell Cardiol. 2006;40:56–63. doi: 10.1016/j.yjmcc.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7:167–178. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Simonsen A, Cumming RC, Finley KD. Linking lysosomal trafficking defects with changes in aging and stress response in Drosophila. Autophagy. 2007;3:499–501. doi: 10.4161/auto.4604. [DOI] [PubMed] [Google Scholar]
- Syntichaki P, Troulinaki K, Tavernarakis N. eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature. 2007;445:922–926. doi: 10.1038/nature05603. [DOI] [PubMed] [Google Scholar]
- Taghli-Lamallem O, Akasaka T, Hogg G, Nudel U, Yaffe D, Chamberlain JS, Ocorr K, Bodmer R. Dystrophin-deficiency in Drosophila reduces lifespan and causes a dilated cardiomyopathy phenotype. Aging Cell. 2008;7:237–249. doi: 10.1111/j.1474-9726.2008.00367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science. 2003;299:1346–1351. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 2001;292:107–110. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- Tee AR, Blenis J. mTOR, translational control and human disease. Semin Cell Dev Biol. 2005;16:29–37. doi: 10.1016/j.semcdb.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Teleman AA, Chen YW, Cohen SM. Drosophila melted modulates FOXO and TOR activity. Dev Cell. 2005;9:271–281. doi: 10.1016/j.devcel.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Teleman AA, Hietakangas V, Sayadian AC, Cohen SM. Nutritional Control of Protein Biosynthetic Capacity by Insulin via Myc in Drosophila. Cell Metab. 2008;7:21–32. doi: 10.1016/j.cmet.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Teleman AA, Maitra S, Cohen SM. Drosophila lacking microRNA miR-278 are defective in energy homeostasis. Genes Dev. 2006;20:417–422. doi: 10.1101/gad.374406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettweiler G, Miron M, Jenkins M, Sonenberg N, Lasko PF. Starvation and oxidative stress resistance in Drosophila are mediated through the eIF4E-binding protein, d4E-BP. Genes Dev. 2005;19:1840–1843. doi: 10.1101/gad.1311805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihervaara T, Puig O. dFOXO regulates transcription of a Drosophila acid lipase. J Mol Biol. 2008;376:1215–1223. doi: 10.1016/j.jmb.2007.12.042. [DOI] [PubMed] [Google Scholar]
- Wang L, Want X, Proud CG. Activation of mRNA translation in rat cardiac myocytes by insulin involves mutiple rapamysin-sensetive steps. Am J Physiol Heart Circ Physiol. 2000;278:H1056–1068. doi: 10.1152/ajpheart.2000.278.4.H1056. [DOI] [PubMed] [Google Scholar]
- Wang MC, Bohmann D, Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121:115–125. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Wessells RJ, Bodmer R. Screening assays for heart function mutants in Drosophila. Biotechniques. 2004;37:58–60. doi: 10.2144/04371ST01. [DOI] [PubMed] [Google Scholar]
- Wessells RJ, Bodmer R. Cardiac Aging. Semin Cell Dev Biol. 2007;18:111–116. doi: 10.1016/j.semcdb.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Wessells RJ, Fitzgerald E, Cypser JR, Tatar M, Bodmer R. Insulin regulation of heart function in aging fruit flies. Nat Genet. 2004;36:1275–1281. doi: 10.1038/ng1476. [DOI] [PubMed] [Google Scholar]
- Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 2000;14:2712–2724. doi: 10.1101/gad.835000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.