

Abstract
Pseudomonas aeruginosa utilizes a type III secretion system (T3SS) to damage eukaryotic host cells and evade phagocytosis. Transcription of the T3SS regulon is controlled by ExsA, a member of the AraC/XylS family of transcriptional regulators. ExsA-dependent transcription is coupled to type III secretory activity through a cascade of three interacting proteins (ExsC, ExsD, and ExsE). Genetic data suggest that ExsD functions as an antiactivator by preventing ExsA-dependent transcription, ExsC functions as an anti-antiactivator by binding to and inhibiting ExsD, and ExsE binds to and inhibits ExsC. T3SS gene expression is activated in response to low-calcium growth conditions or contact with host cells, both of which trigger secretion of ExsE. In the present study we reconstitute the T3SS regulatory cascade in vitro using purified components and find that the ExsD·ExsA complex lacks DNA binding activity. As predicted by the genetic data, ExsC addition dissociates the ExsD·ExsA complex through formation of an ExsD·ExsC complex, thereby releasing ExsA to bind T3SS promoters and activate transcription. Addition of ExsE to the purified system results in formation of the ExsE·ExsC complex and prevents ExsC from dissociating the ExsD·ExsA complex. Although purified ExsA is monomeric in solution, bacterial two-hybrid analyses demonstrate that ExsA can self-associate and that ExsD inhibits self-association of ExsA. Based on these data we propose a model in which ExsD regulates ExsA-dependent transcription by inhibiting the DNA-binding and self-association properties of ExsA.
Many pathogenic and symbiotic Gram-negative bacteria express a highly conserved type III secretion system (T3SS) involved in the assembly of a macromolecular needle complex (injectisome) and translocation of toxins into eukaryotic host cells (23). Each species translocates a unique set of toxins that promote various aspects of their pathogenic/symbiotic lifestyles such as invasion, intracellular survival, or evasion of host defense mechanisms (7). Pseudomonas aeruginosa is an important opportunistic pathogen most commonly seen in patients with burn wounds, cystic fibrosis, or immunodeficiency. The T3SS of P. aeruginosa translocates the ExoS, ExoT, ExoU, and ExoY effector toxins into host cells, where they promote tissue destruction, evasion of phagocytosis, and dissemination from initial colonization sites (13).
Biosynthesis of the P. aeruginosa T3SS requires more than 30 gene products and is highly regulated at the transcriptional level (36). The primary regulator of T3SS gene expression is ExsA, a member of the AraC/XylS family of transcriptional activators (11). AraC/XylS proteins generally consist of two distinct functional regions, the amino-terminal (NTD) and carboxy-terminal (CTD) domains (10). The NTD of ExsA is required for self-association and binding to ExsD, a negative regulator of ExsA, and the CTD is responsible for DNA binding and transcriptional activation (3). A total of 10 ExsA-dependent promoters control T3SS gene expression. The best-studied promoter (PexoT) consists of two binding sites for monomeric ExsA: a high-affinity site (binding site 1) that overlaps the putative −35 region (2) and a lower-affinity site (binding site 2) centered at the −52 position with respect to the transcriptional start site. Binding of ExsA to site 2 is dependent upon prior occupation of binding site 1 by ExsA, suggestive of cooperative binding. Further evidence for cooperative binding is seen with the PexsC promoter, where protein-protein interactions mediated by the NTD of an ExsA monomer bound to site 1 are required for binding of a second ExsA molecule to binding site 2 (3). Once bound to the promoter at both sites 1 and 2, ExsA activates T3SS gene expression, primarily through recruitment of RNA polymerase to the promoter (31).
Transcriptional activation by ExsA is intimately coupled to type III secretory activity by three interacting proteins; ExsC, ExsD, and ExsE (recently reviewed in reference 4). Both ExsC and ExsD have two potential binding partners. ExsD is an antiactivator that forms a 1:1 complex with ExsA and inhibits the DNA binding activity of ExsA (21, 28). ExsC functions as an anti-antiactivator by binding to and inhibiting ExsD. The stoichiometry of the ExsD·ExsC complex is 2:2 with a dissociation constant (Kd) of 18 nM (9). ExsC also functions as a T3SS chaperone by forming a 2:1 complex (Kd of 1 nM) with ExsE. ExsE is a secreted regulator that prevents ExsC from associating with ExsD (27, 30). The current model proposes that the nonpermissive condition for T3SS gene expression (i.e., high-Ca2+ growth conditions) inhibits ExsA-dependent transcription because the binding equilibrium favors formation of the inhibitory ExsD·ExsA and ExsC·ExsE complexes (29, 30, 37). Inducing conditions (i.e., low calcium or contact of P. aeruginosa with host cells) activate type III secretory activity, resulting in secretion and/or translocation of ExsE. The resulting decrease in the intracellular concentration of ExsE favors formation of the ExsD·ExsC complex, thereby releasing ExsA to activate T3SS gene expression.
In the present study we show that ExsD inhibits both the DNA binding and self-association properties of ExsA. In addition, we report the reconstitution of the ExsADCE regulatory cascade in vitro using purified proteins. Our studies demonstrate that ExsC dissociates the ExsD·ExsA complex by forming a complex with ExsD, thereby releasing ExsA to bind to DNA. The combined data confirm the genetic model and demonstrate that most if not all of the essential components of the pathway have been identified.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Escherichia coli strains were maintained on Luria-Bertani (LB) agar plates containing antibiotics as necessary (kanamycin, 50 μg/ml; gentamicin, 15 μg/ml; ampicillin, 100 μg/ml; tetracycline, 12 μg/ml). P. aeruginosa strains were maintained on Vogel Bonner minimal medium (VBM) (33) with antibiotics as necessary (gentamicin, 100 μg/ml; carbenicillin, 300 μg/ml; tetracycline, 50 μg/ml). For T3SS gene expression studies, P. aeruginosa strains were cultured in tryptic soy broth supplemented with 100 mM monosodium glutamate and 1% glycerol (TSB++) and 2 mM EGTA as previously described (21).
Mutant construction.
To construct the ΔexsA mutant, a PCR-generated fragment containing the first 13 codons of exsA along with 1,600 bp of upstream flanking sequence was cloned into the EcoRI-BamHI sites of vector pEX18Gm (14), generating plasmid p2UY56UP. A second PCR-generated fragment containing the last 33 codons of exsA along with 2,500 bp of downstream flanking DNA was then cloned into the BamHI-HindIII sites of plasmid p2UY56UP, generating plasmid p2UY56ΔA. The ΔexsA allele on p2UY56ΔA was transferred to the chromosome of wild-type PA103 by homologous recombination as described previously (21). The ΔexsA exsD (ΔexsAD) double mutant was similarly constructed, except that the downstream flanking sequence (1,700 bp) was generated using chromosomal DNA from a PA103 ΔexsD mutant (21) as template in a PCR, and a primer incorporating a BclI site instead of BamHI was used since the ΔexsD allele contains the latter. The ΔexsAD double deletion on the resulting plasmid (p2UY59) was transferred to the chromosome of the ΔexsD strain by homologous recombination as above. The chromosomal deletions in both strains were confirmed by PCR using primers that anneal outside of the cloned sequences used in constructing plasmids p2UY56ΔA and p2UY59.
Protein expression and purification.
For the chromatin immunoprecipitation (ChIP) assays we used the ExsA expression vector p2UY95. This plasmid is a pJN105 derivative (25) in which the arabinose-inducible promoter and associated genes have been replaced with a mutant PlacUV5 promoter in which the −35 and −10 regions of PlacUV5 were changed from TTTACA and TATAAT to TTATCA and TACAGT, respectively. The mutant promoter drives expression of exsA (the cloned exsA fragment starts 310 bp upstream of the translational start site). Further details regarding the construction of p2UY95 are available upon request.
For the monohybrid studies exsD and the native ribosome-binding site were PCR amplified using primers incorporating EcoRI and SacI restriction sites. The PCR fragment was cloned into the corresponding sites of pJN105. To eliminate the potential for α-complementation in E. coli strain SU101, the α peptide from pJN105 was removed by digesting the plasmid with SacI and PvuI. The restriction ends were blunt ended with T4 DNA polymerase, and the plasmid was religated, resulting in pJNexsDΔα.
The pET16bexsA (2), pET25bexsC (9), pET24aexsE (30), pCOLADuet-1exsD (3), pCOLADuet-1exsAD (3), pCOLADuet-1exsC, and pCOLADuet-1exsCD (37) expression constructs were described previously. To purify His-tagged ExsC (ExsCHis), ExsEHis, and the ExsD·ExsAHis, and ExsD·ExsCHis cocomplexes, cultures of E. coli Tuner (DE3) transformed with the corresponding expression plasmids were grown in LB broth containing the appropriate antibiotics (200 μg/ml ampicillin or 50 μg/ml kanamycin) and 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h at 30°C. Cells were harvested by centrifugation and suspended in Ni-nitrilotriacetic acid (NTA) binding buffer (20 mM Tris [pH 8.0], 200 mM NaCl, and 20 mM imidazole) supplemented with a protease inhibitor cocktail (complete mini, EDTA-free protease inhibitor cocktail; Roche, Indianapolis, IN). Following cell disruption via a French pressure cell, cell lysates were cleared by centrifugation (20,000 × g for 15 min at 4°C) and subjected to Ni2+ affinity chromatography as previously described (21). Silver-stained gels were subjected to densitometry analyses, and each protein preparation was found to be >90% homogeneous. Expression and purification of ExsAHis were described previously (2).
Further purification of the ExsD·ExsAHis and ExsD·ExsCHis complexes was performed in 20 mM Tris (pH 8.0), 200 mM NaCl, and 1 mM dithiothreitol (DTT) on a Sephacryl S-200HR gel filtration column (2.5 by 50 cm; 246 ml; Bio-Rad) with a flow rate of 1.5 ml/min. Peak protein fractions from the Ni2+ affinity columns were concentrated to 10 to 20 mg/ml (Ultra-15 spin concentrator; Amicon), and 500 μl was loaded onto the gel filtration column. Column fractions were assayed for protein by the Bradford assay using bovine serum albumin (BSA) protein standards to generate a standard curve (Bio-Rad, Hercules, CA).
SDS-PAGE, immunoblotting, and β-galactosidase assays.
Whole-cell lysate samples were generated by growing cells in LB broth (E. coli) or TSB++ (P. aeruginosa) to an absorbance (A600) of 0.5 (E. coli) or 1.0 (P. aeruginosa). Samples were prepared by sedimenting 1.25 ml of culture, suspending the pellet in 0.25 ml of SDS-PAGE sample buffer, and sonicating for 10 s. Samples were analyzed by SDS-PAGE, followed by either silver staining, Coomassie staining, or immunoblotting using antibodies directed against ExsA, ExsC, ExsD, or LexA (Invitrogen).
The activity of the PexsD-lacZ reporter in P. aeruginosa was determined as follows. Strains grown overnight on VBM agar at 37°C were suspended in TSB++, diluted to an absorbance (A600) of 0.1 in 5 ml of TSB++, and incubated at 30°C until the A600 reached 1.0. β-Galactosidase activity in E. coli SU101 (bacterial monohybrid strain) was determined as previously described (8). Briefly, strains carrying the indicated expression plasmids (LexA, LexA-chloramphenicol acetyltransferase [CAT], LexA-ExsA, and pExsD or the corresponding vector control [pJN105]) were shaken overnight at 30°C in LB broth containing the appropriate antibiotics. IPTG (50 μM) and/or arabinose (0.5%) was added to induce expression of the LexA fusion partners and ExsD, respectively. The next day cultures were diluted 100-fold in the same medium and shaken at 30°C until the A600 reached 0.5. β-Galactosidase assays for both E. coli and P. aeruginosa were performed as previously described (21).
EMSAs.
Electrophoretic mobility shift assays (EMSAs) were performed as previously described (2). Briefly, DNA probes containing ExsA-dependent promoters (200 bp), as well as a nonspecific fragment from the coding region of pscF (160 bp), were generated by PCR and end labeled with 20 μCi of [γ32P]ATP (GE Healthcare) and 10 U of polynucleotide kinase (New England Biolabs) (2). EMSA reactions (19 μl) containing end-labeled specific and nonspecific probes (0.25 nM each), 25 ng/μl poly(2′-deoxyinosinic-2′-deoxycytidylic acid) (Sigma-Aldrich, St. Louis, MO), and ExsA DNA binding buffer (10 mM Tris [pH 7.9], 50 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, 5% glycerol, and 100 μg/ml bovine serum albumin) were incubated for 5 min at 25°C. ExsAHis, ExsCHis, ExsEHis, ExsD·ExsAHis, and/or ExsD·ExsCHis was added to the indicated concentrations in a final reaction volume of 20 μl and incubated at 25°C for 15 min. Samples were subjected to electrophoresis on 5% or 10% polyacrylamide glycine gels (10 mM Tris [pH 8.0], 380 mM glycine, 1 mM EDTA) at 4°C. Proteins were visualized by silver staining. Imaging of the labeled probes was performed using an FLA-7000 phosphorimager (Fujifilm) and Multigage, version 3.0, software (Fujifilm) for data analyses.
ChIP assay.
P. aeruginosa strains were grown with vigorous aeration in TSB++ to an optical density at 600 nm (OD600) of 0.6 at 30°C. Protein-DNA complexes were cross-linked by the addition of formaldehyde (final concentration of 1%), and cultures were shaken at 150 rpm for 20 min at 30°C. Reactions were quenched by the addition of 5 ml of 1.0 M glycine (pH 8.0). The cells were harvested by centrifugation (2,000 × g for 10 min at 4°C), washed two times with ice cold phosphate-buffered saline (PBS), and suspended in 500 μl of lysis buffer (10 mM Tris-HCl [pH 8.0], 50 mM NaCl, 20% sucrose, 10 mM EDTA) containing protease inhibitor cocktail (complete mini, EDTA-free protease inhibitor cocktail; Roche, Indianapolis, IN) and 2 mg/ml lysozyme. After 30 min on ice, 500 μl of 2× immunoprecipitation buffer (100 mM Tris-HCl [pH 7.0], 300 mM NaCl, 2% Triton X-100, 40 mM EDTA) was added to the reaction mixture and incubated at 37°C for 10 min, followed by 2 min on ice. Samples were sonicated, and unbroken cells were sedimented by centrifugation (16,000 × g for 5 min at 4°C). The supernatant was passed through a 0.22-μm-pore-size filter and subjected to immunoprecipitation. Anti-ExsA polyclonal antibody (2 μg) was incubated with the samples for 4 h at 4°C, followed by the addition of protein A magnetic beads (25 μl; New England Biolabs) for 45 min at 4°C. Beads were washed three times with 1× immunoprecipitation buffer and two times with TE buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA). Beads were suspended in 100 μl of TE buffer, and the formaldehyde cross-links were reversed by incubation for 8 h at 65°C. The eluate (5 μl) was used in PCRs with primers that amplify a ∼200 bp region of the PexsD, PexsC, and PfleQ promoters (5′-ATACGAATTCTTCCAGCCAGTCCTATTTCA and 5′-GACAGGTACCCCTGCTCCATTCTCTGCCTTG for PexsD, 5′-TGATGAATTCGCCTCCTAAAGCTCAG and 5′-ATACGAATTCTTCCAGCCAGTCCTATTTCAC for PexsC, and 5′-TTAGGTACCCACCAGATGTTCGGATAAGT and 5′-TTAGAATTCCGAATGGGTCTCGCTCGACC for PfleQ). The resulting PCR products were visualized on an agarose gel stained with ethidium bromide.
Ni-affinity copurification.
Purified proteins were incubated in 10 μl of ExsA DNA binding buffer for 2 h at 4°C. The volume was then adjusted to 50 μl with Ni-NTA binding buffer, and 30 μl of packed Ni-NTA agarose beads (Qiagen; Germantown, MD) was added. After 30 min the resin was sedimented (30 s at 2,000 × g) and washed twice with 300 μl of wash buffer (NTA binding buffer with 60 mM imidazole), and bound protein was eluted with 50 μl of elution buffer (NTA binding buffer with 500 mM imidazole). Unbound and bound protein samples were subjected to SDS-PAGE and silver staining.
RESULTS
Reconstitution of the ExsADCE regulatory cascade in vitro.
To test the model for regulation of ExsA activity by ExsD, ExsC, and ExsE, we developed an in vitro system using purified components. ExsAHis, ExsCHis, and ExsEHis were purified by Ni2+-affinity chromatography as previously described (2, 9, 30, 37). It should be noted that native ExsD is insoluble when expressed in E. coli and that neither amino- nor carboxy-terminal histidine-tagged forms of ExsD complement an exsD mutant (data not shown). For this reason ExsD was coexpressed with either ExsCHis or ExsAHis from the dual expression vector pCOLADuet-1 in E. coli and purified by Ni2+ affinity chromatography. Both the purified ExsD·ExsCHis and ExsD·ExsAHis complexes were soluble based on resistance to high-speed sedimentation (100,000 × g for 30 min), and both eluted from gel filtration columns as single included peaks with apparent molecular masses of 186 and 97 kDa, respectively (data not shown), which is consistent with previous studies (20, 28).
Genetic and biochemical data indicate that the purified ExsD·ExsAHis cocomplex is unable to bind to T3SS promoters in vitro (3, 21, 28). We also find that the ExsD·ExsAHis cocomplex lacks DNA binding activity in EMSAs compared to ExsAHis (Fig. 1, lanes 3 versus lanes 6). Binding of ExsAHis to the PexsC and PexoT promoter probes is characterized by the appearance of two distinct shift products that result from the binding of one and two monomers of ExsA to the promoter probes, shift products 1 and 2, respectively (Fig. 1, lanes 3). The difference between the migration rate of shift product 2 for the PexsC compared to shift product 2 for PexoT results from bending of the PexsC promoter DNA by ExsA (2). To further examine the difference in the DNA binding activities of ExsAHis and ExsD·ExsAHis, we attempted to determine the apparent equilibrium constant (Keq) for binding to the PexoT promoter probe using quantitative EMSA. The concentration of ExsAHis required to shift 50% of the promoter probe was 30 nM (Fig. 2). In contrast, the maximum amount of ExsD·ExsAHis that could be added to the reaction mixture (400 nM) resulted in shifting of only 25% of the probe. It is unlikely that the shifting observed at high concentrations of ExsD·ExsAHis is due to binding of the complex to the probe since the mobility of shift product 1 (Fig. 2A, lane 9) is identical to shift product 1 seen with ExsAHis alone (lane 2). The modest shifting observed at high concentrations of ExsD·ExsAHis likely reflects trace amounts of ExsAHis present as either a contaminant and/or dissociated from the ExsD·ExsAHis complex.
FIG. 1.
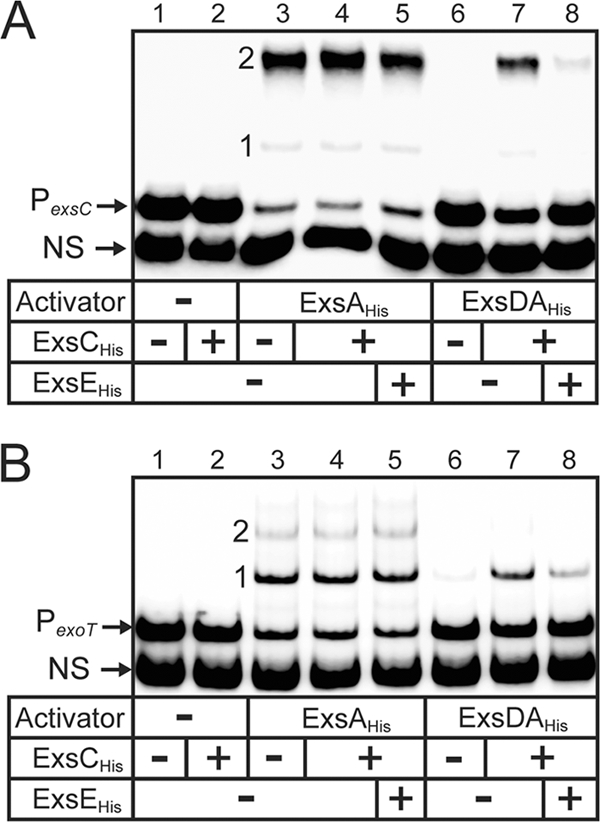
Reconstitution of the ExsADCE regulatory cascade in vitro. Electrophoretic mobility shift assays were performed by incubating ExsAHis (18 nM; lanes 3 to 5) or the ExsD·ExsAHis complex (18 nM; lanes 6 to 8) alone or with ExsCHis (180 nM; lanes 2, 4, 5, 7, and 8) and/or ExsEHis (720 nM; lanes 5 and 8) for 20 min at 4°C. DNA binding activity was then examined by adding radiolabeled nonspecific (NS) and specific probes derived from the PexsC (A) or PexoT (B) promoters for 15 min. Samples were subjected to electrophoresis and phosphorimaging. Shift products 1 and 2 for each of the promoter fragments are indicated.
FIG. 2.
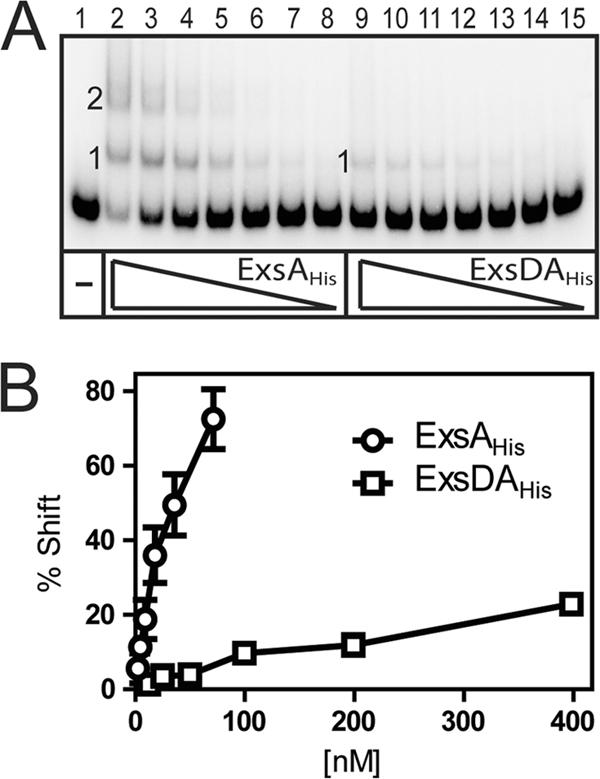
Binding affinity of ExsAHis and the ExsD·ExsAHis complex to the PexoT promoter. (A) The PexoT promoter fragment and a nonspecific probe (0.25 nM each) were incubated with increasing concentrations of ExsAHis (2 to 70 nM) or ExsD·ExsAHis complex (25 to 400 nM) for 15 min followed by electrophoresis and phosphorimaging. (B) Binding curve for ExsAHis and ExsD·ExsAHis to the PexoT promoter. The percentage of shifted probe (y axis) was plotted as a function of the protein concentration (x axis). The reported values are the averages of three independent experiments.
The observation that the ExsD·ExsAHis cocomplex lacks DNA binding activity is based on negative data. To address the possibility that the DNA binding activity of ExsA is lost during purification of the ExsD·ExsAHis complex, we tested the integrity of ExsAHis by dissociating the ExsD·ExsAHis complex with ExsCHis. Whereas incubation of the ExsD·ExsAHis complex with a 10-fold molar excess of ExsCHis restored DNA binding activity toward the PexsC (Fig. 1A, lane 6 versus lane 7) and PexoT (Fig. 1B, lane 6 versus lane 7) promoter probes, ExsCHis had no effect on the DNA binding activity of ExsAHis (Fig. 1A and B, lane 3 versus lane 4). Importantly, the relative mobilities of the shifted bands were identical using both ExsAHis and the ExsD·ExsAHis complex coincubated with ExsCHis. This finding suggests that ExsCHis functions solely by dissociating the ExsD·ExsAHis complex and does not form a complex with ExsA.
To prove that ExsC dissociates the ExsD·ExsA complex, ExsCHis was incubated with the ExsD·ExsAHis complex and then subjected to native polyacrylamide gel electrophoresis alongside the ExsD·ExsAHis and ExsD·ExsCHis complexes as standards. As expected, incubation of the ExsD·ExsAHis complex with ExsCHis resulted in the appearance of the ExsD·ExsCHis complex (Fig. 3A, lane 3 versus lane 4) and a reduction in the amount of ExsD·ExsAHis complex (Fig. 3A and B, lanes 2 versus lanes 4). Immunoblotting using antibodies specific for ExsA, ExsD, and ExsC demonstrates that the upper band in lane 4 contains only ExsCHis and ExsD (Fig. 3B). These data confirm that ExsCHis dissociates the ExsD·ExsAHis complex through formation of a complex with ExsD.
FIG. 3.
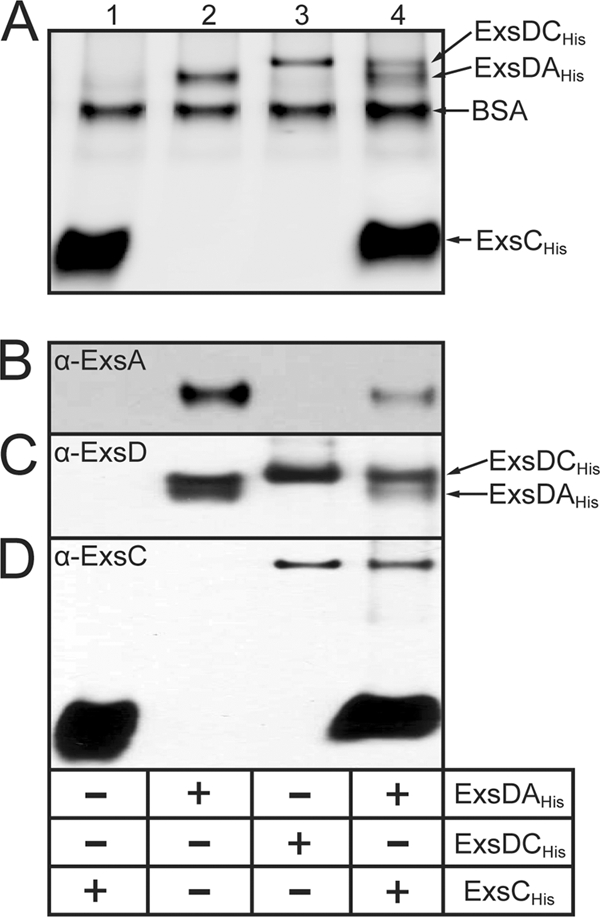
ExsCHis dissociates ExsD·ExsAHis by forming a complex with ExsD. ExsCHis (lanes 1), the ExsD·ExsAHis complex (lanes 2), the ExsD·ExsCHis complex (lanes 3), or the ExsD·ExsAHis complex and ExsCHis (lanes 4) were incubated for 20 min at 4°C under the conditions used for the EMSAs presented in Fig. 1. Reaction mixtures were electrophoresed through a nondenaturing polyacrylamide gel and subjected to silver staining (A) or Western blotting with antibodies directed against ExsA (B), ExsD (C), or ExsC (D).
The final protein in the regulatory cascade, ExsE, forms a complex with ExsC (27, 30) and can prevent formation of and/or dissociate the ExsD·ExsC complex. To test this prediction ExsCHis was first preincubated with an excess of ExsEHis, and then added to the ExsD·ExsAHis complex. This treatment prevented ExsCHis from dissociating the ExsD·ExsAHis complex, as evidenced by reduced DNA binding by ExsA (Fig. 1, lanes 8). These data are consistent with the current model for regulation of ExsA activity by ExsCDE and represent the first biochemical reconstitution of the ExsA regulatory cascade.
ExsD inhibits the DNA binding activity of ExsA in vivo.
To determine whether ExsD also inhibits the DNA binding activity of ExsA in vivo, we performed a chromatin immunoprecipitation (ChIP) assay. The ideal comparison would be between cells grown under high-Ca2+ conditions, where ExsD inhibits ExsA-dependent transcription, to T3SS inducing conditions (low Ca2+), where ExsC inhibits the negative regulatory activity of ExsD and where ExsA is bound to promoter DNA. The fact that ExsA levels increase 3- to 4-fold under inducing conditions (9), however, would make it difficult to determine whether an increase in DNA binding activity reflected increased ExsA expression or a lack of inhibition by ExsD. For this reason we designed a system in which ExsA expression remains constant and then assessed whether ExsD influences the DNA binding activity of ExsA. To this end ΔexsA and ΔexsA ΔexsD (ΔexsAD) mutants carrying a chromosomally integrated ExsA-dependent transcriptional reporter (PexsD-lacZ) were transformed with either a vector control (pJN105) or a plasmid constitutively expressing low levels of ExsA (pExsA). The resulting strains were grown under noninducing conditions for T3SS gene expression and assayed for β-galactosidase activity. As expected, the activity of the PexsD-lacZ reporter was low in the absence of exsA (Fig. 4A, pJN105). Introduction of pExsA into the ΔexsA mutant resulted in only a modest increase in PexsD-lacZ reporter activity (∼2-fold) due to the negative regulatory function of ExsD. In contrast, PexsD-lacZ reporter activity increased dramatically in the ΔexsAD mutant transformed with pExsA. Immunoblots of whole-cell lysates confirmed that the steady levels of ExsA expression were similar in both the ΔexsA and ΔexsAD strains transformed with pExsA (Fig. 4B). This finding suggests that the ability of ExsA to activate transcription differs in these two backgrounds.
FIG. 4.
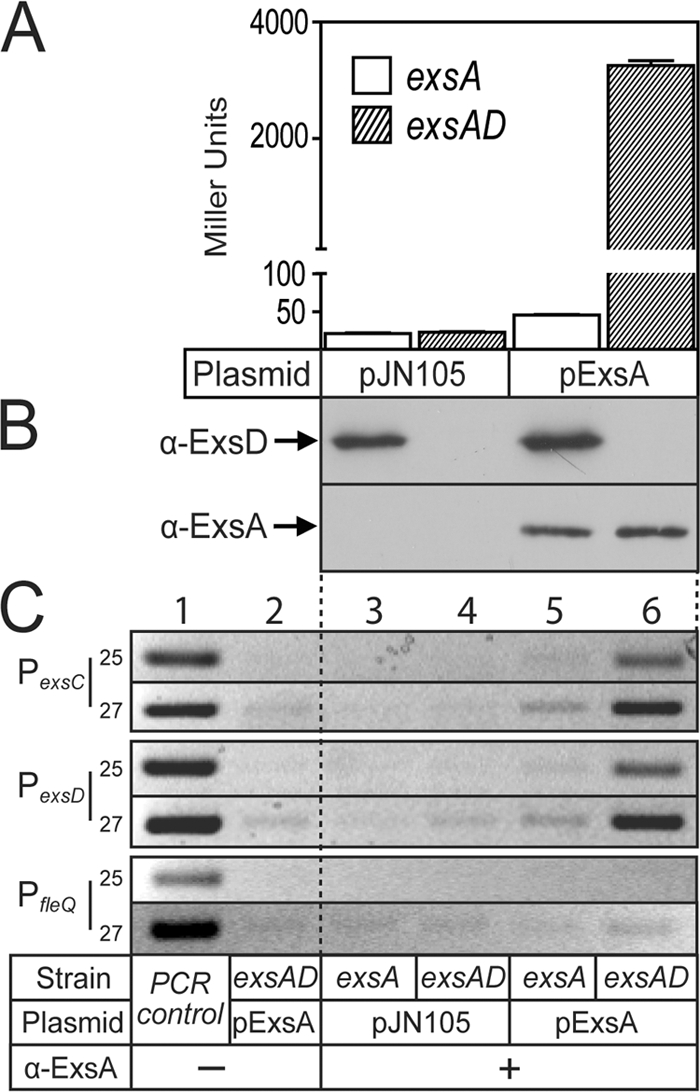
ExsD inhibits the DNA binding activity of ExsA in vivo. (A and B) An exsA mutant or an exsA exsD double mutant carrying the PexsD-lacZ reporter was transformed with a vector control (pJN105) or an expression plasmid (p2UY95, labeled pExsA in the figure) that constitutively expresses low levels of ExsA. The resulting strains were grown under noninducing conditions for T3SS gene expression and assayed for β-galactosidase activity (A) or protein expression levels (B) by performing immunoblotting of whole-cell lysates using the indicated antibodies. The reported values are the averages of three independent experiments, and error bars indicate the standard errors of the means. (C) ChIP assays performed in the presence or absence of ExsD. Cells were treated with formaldehyde to cross-link ExsA to the DNA and processed for ChIP assays using polyclonal anti-ExsA antibody. The immunoprecipitate was then used in a PCR with primers designed to amplify 200-bp regions of the PexsC, PexsD, and PfleQ promoters. The PCRs were programmed to run for 25 or 27 extension cycles, as indicated on the figure. The resulting PCR products were separated on an agarose gel and stained with ethidium bromide. P. aeruginosa chromosomal DNA was used as a positive control (lane 1) for the PCR, and reaction mixtures lacking antibody served as negative controls (lane 2) for chromosomal contamination.
To determine if the difference in expression of the PexsD-lacZ reporter correlated with changes in the DNA binding activity of ExsA, cells from log-phase cultures were treated with formaldehyde to cross-link ExsA to chromosomal DNA. The cellular DNA was then sonicated to generate 500- to 1,000-bp fragments and subjected to immunoprecipitation with polyclonal ExsA antibody. Following reversal of the formaldehyde cross-links by heat treatment, the cellular DNA coprecipitating with ExsA was used as template in a PCR with primers that amplify the ExsA-dependent PexsD or PexsC promoters and the ExsA-independent PfleQ promoter as a negative control. Strong PCR products representing the PexsD or PexsC promoters were seen in samples isolated from cells expressing ExsA in the absence of ExsD (Fig. 4C, lane 6), and the amounts of these products were significantly reduced in samples isolated from strains expressing ExsD (lane 5). PCR products were absent in reaction mixtures lacking ExsA (Fig. 4C, lanes 3 and 4) or ExsA antibody (lane 2), or when primers to PfleQ (lanes 3 to 6) were used. These combined data confirm that ExsD specifically prevents ExsA from binding to the PexsC and PexsD promoters both in vitro and in vivo.
ExsD dissociated from the ExsD·ExsCHis complex by ExsEHis is unable to inhibit the DNA binding activity of ExsA.
The regulatory model posits that secretion of ExsE triggers ExsC-dependent dissociation of the ExsD·ExsA complex and activation of T3SS gene expression. To determine whether ExsD released from the ExsD·ExsC complex inhibits ExsA activity, we performed EMSAs with the ExsD·ExsCHis complex. As described earlier, incubation of the PexsC or PexoT promoter probes with ExsAHis results in the appearance of shift products 1 and 2 (Fig. 5, lanes 2 and 7). Addition of the ExsD·ExsCHis complex or ExsEHis had no effect on ExsAHis binding activity (Fig. 5, lanes 3, 4, 8, and 9). Surprisingly, the DNA-binding activity of ExsA was also unaffected when it was incubated with both the ExsD·ExsCHis complex and ExsEHis (Fig. 5, lanes 5 and 10). One explanation for the latter finding is that ExsE is unable to disassociate the ExsD·ExsCHis complex under the conditions required for the EMSA. To examine this further, we employed metal affinity chromatography to separate histidine-tagged proteins (ExsAHis, ExsCHis, and ExsEHis) and cocomplexes (ExsD·ExsAHis and ExsD·ExsCHis) from untagged ExsD. Reaction mixtures identical to those used in the EMSA reactions were incubated with Ni-NTA affinity resin, and bound proteins were eluted with imidazole. For reaction mixtures containing ExsD·ExsCHis or ExsD·ExsCHis and ExsAHis, the majority of the ExsD was present in the bound fraction (Fig. 5B, lane 1 versus 2 and lane 5 versus 6). In contrast, the reaction mixture containing the ExsD·ExsCHis complex incubated with ExsEHis resulted in a complete loss of ExsD in the bound fraction (lane 3 versus lane 4). This finding demonstrates that ExsEHis dissociates the ExsD·ExsCHis complex under the conditions used for the EMSAs. Nevertheless, ExsD released from the ExsD·ExsCHis complex is unable to interact with ExsAHis, as evidenced by the absence of ExsD in the bound fraction following incubation of the ExsD·ExsCHis complex with ExsEHis and ExsAHis (lanes 7 and 8). These data suggest that ExsD released from the ExsD·ExsCHis complex in vitro is unable to form a complex with ExsAHis.
FIG. 5.
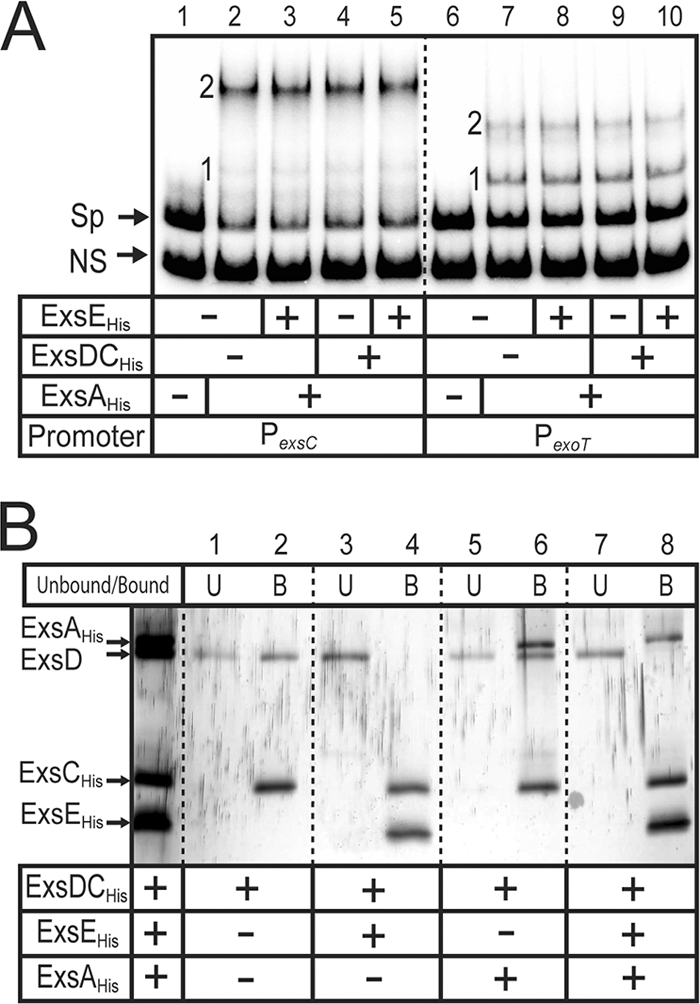
ExsD dissociated from the ExsD·ExsCHis complex does not bind to ExsAHis. (A) ExsAHis (18 nM) was incubated with the ExsD·ExsCHis complex (180 nM) (lanes 4, 5, 9, and 10) and/or ExsEHis (720 nM) (lane 3, 5, 8, and 10) for 20 min at 4°C. The protein mixes were incubated for 15 min with radiolabeled nonspecific (NS) and specific probes derived from the PexsC (lanes 1 to 5) or PexoT (lanes 6 to 10) promoters. Samples were subjected to electrophoresis and phosphorimaging. Shift products 1 and 2 for each of the promoter fragments are indicated. (B) The DNA binding reaction mixtures from panel A were incubated with Ni-NTA agarose and washed, and the unbound (U) and bound (B) fractions were separated on an SDS-PAGE gel and stained with silver. A standard consisting of all four proteins is included in the left lane.
ExsD prevents ExsA self-interaction.
Purified ExsAHis is monomeric in solution and binds to DNA as a monomer (2). When ExsA is bound to the PexsC promoter, however, protein-protein interactions mediated by the amino-terminal domain (NTD) of ExsA bound to site 1 are thought to facilitate recruitment of a second ExsA monomer to binding site 2 (3). To further examine the potential for ExsA self-association, we employed the LexA monohybrid system. LexA is a transcriptional repressor that must dimerize in order to bind DNA. In the monohybrid system each monomer of the LexA homodimer binds to a half-site within an operator to repress transcription of a β-galactosidase reporter. To test for ExsA self-association, the dimerization domain of LexA was replaced with ExsA or the amino-terminal domain of ExsA. Compared to LexA lacking the dimerization domain, expression of both the LexA-ExsA and LexA-NTD fusions resulted in strong repression (26- and 5-fold, respectively) of the β-galactosidase reporter (Fig. 6A). This level of inhibition was greater than that of a LexA-chloramphenicol acetyltransferase (CAT) fusion (3-fold), which is known to multimerize (Fig. 6A). These findings indicate that ExsA self-associates under some conditions and that self-association is mediated by the NTD.
FIG. 6.
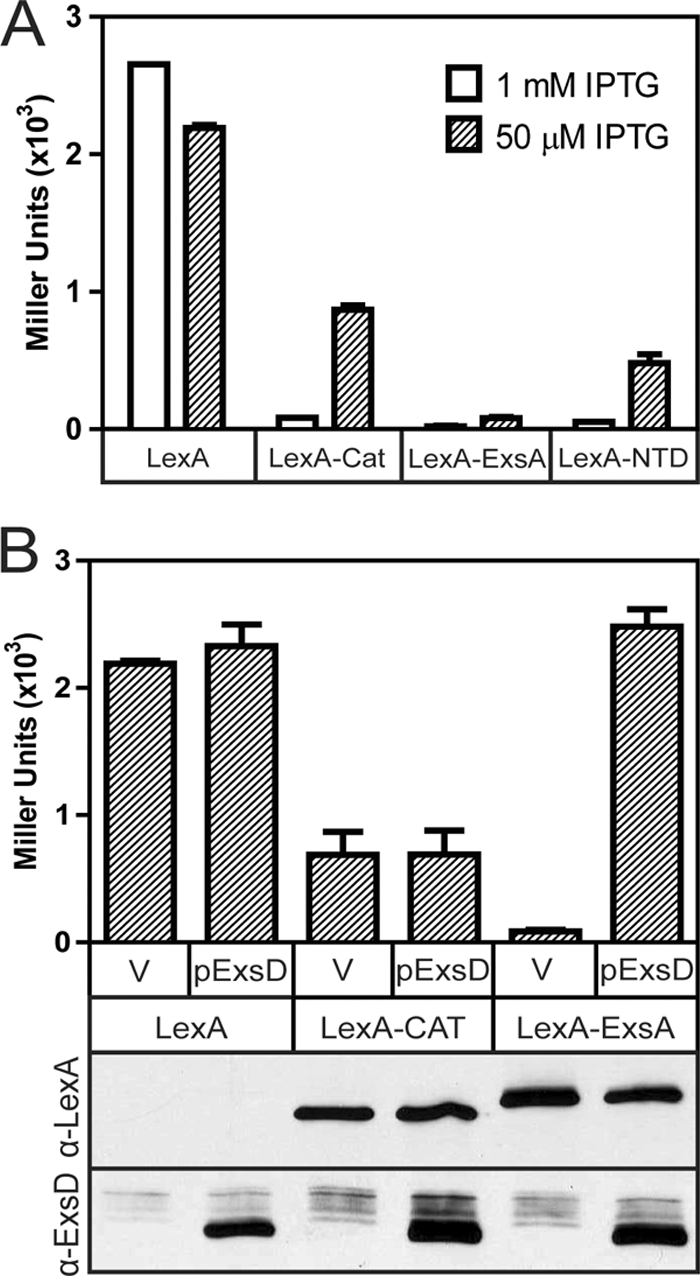
ExsD inhibits the self-association activity of ExsA. (A) Detection of ExsA self-association in the LexA monohybrid assay. E. coli SU101 (a reporter strain with a LexA-repressible lacZ reporter) was transformed with an IPTG-inducible plasmid expressing LexA1-87 lacking a dimerization domain (residues 1 to 87) or LexA1-87 fused to chloramphenicol acetyltransferase (LexA-CAT), ExsA (LexA-ExsA), or the amino-terminal domain of ExsA (LexA-NTD). The resulting strains were grown in the presence of 50 or 1,000 μM IPTG and assayed for β-galactosidase expression (reported in Miller units). (B) ExsD inhibits ExsA self-association. The strains from panel A were transformed with either a vector control (V) or an arabinose-inducible ExsD expression plasmid (pJNexsDΔα, labeled as pExsD in the figure), grown in the presence of 50 μM IPTG and 0.5% arabinose, and assayed for β-galactosidase activity. Whole-cell lysates from the same strains were analyzed by immunoblotting using anti-LexA or anti-ExsD antiserum. We presume that LexA1-87, which lacks a dimerization domain, is not stably expressed. The reported values for the data in both panels are the averages of three independent experiments, and error bars indicate the standard errors of the means.
Since ExsD was previously shown to interact with the NTD (3), we tested the possibility that ExsD functions by preventing ExsA from self-associating using the monohybrid system. In strains carrying the negative (LexA) or the positive (LexA-CAT) controls, coexpression of ExsD had no effect on the activity of the β-galactosidase reporter (Fig. 6B). In contrast, coexpression of ExsD completely disrupted the repressive activity of the LexA-ExsA fusion. To ensure that the LexA-fusions and ExsD were expressed at similar levels in each strain, cell lysates were subjected to immunoblotting using antibodies directed against either LexA or ExsD. The steady-state expression levels of the LexA-CAT and LexA-ExsA fusions were comparable and unaffected by coexpression of ExsD (Fig. 6B). These data suggest that ExsD inhibits ExsA self-association independently from any effect on DNA binding activity.
DISCUSSION
The ExsADCE regulatory cascade responds to known environmental signals (low Ca2+ and host cell contact) and directly controls the DNA binding activity of ExsA. In the current study we used purified components to demonstrate that ExsE, ExsC, ExsD, and ExsA are sufficient to reconstitute the regulatory cascade in vitro (Fig. 1). This finding is consistent with the fact that the binding affinity of ExsC is greater for ExsE than ExsD (37). Additionally, our data suggest that the affinity of ExsD is greater for ExsC than for ExsA. As such, changes in the concentration of ExsE (whether in a cell or a test tube) determine whether formation of the ExsD·ExsC or ExsD·ExsA complex is favored. The ExsADCE cascade is the major regulatory mechanism that controls T3SS gene expression but does not function in isolation. T3SS gene expression is complex and influenced by several global regulatory pathways that seem to function upstream of the ExsADCE cascade. These upstream pathways include the alternative sigma factors AlgU and RpoS (15, 35), the Vfr cyclic AMP (cAMP) receptor protein (34), cyclic diguanylate signaling (17), multiple two-component regulatory systems (12, 16, 32), and an RNA binding protein (RsmA) that regulates gene expression at the posttranscriptional level (12, 18). Our understanding of these pathways is limited in that the signals to which they respond and the mechanisms by which they influence T3SS gene expression are largely unknown. Although our data demonstrate that the ExsADCE cascade can function as an independent unit, we cannot exclude the possibility that these other regulatory factors fine-tune the cascade by changing the expression levels and/or binding affinities of ExsADCE.
ExsA-dependent promoters consist of two adjacent binding sites (termed sites 1 and 2) for monomeric ExsA (2, 3), consistent with the finding that purified ExsA is monomeric in solution. Although ExsA can bind DNA as a monomer, binding occurs in a cooperative fashion whereby binding of monomeric ExsA to binding site 1 is required for occupation of site 2 by a second ExsA monomer (3). The finding that the amino-terminal domain of ExsA is required for cooperative binding to the PexsC promoter was previously used as evidence that protein-protein interactions between two ExsA monomers facilitate cooperative binding (3). We now provide further evidence of ExsA self-association using the LexA monohybrid system and show that the amino-terminal domain is sufficient for self-association (Fig. 6A). It is not clear why purified ExsA is monomeric in solution and yet demonstrates a strong interaction in the monohybrid system. One possibility is that ExsA is actually dimeric under physiological conditions and that the experimental conditions used for ExsA purification and in vitro assays favor dissociation to the monomeric state. Alternatively, it is possible that the affinity of ExsA for itself is intrinsically low and that DNA binding, either in the context of the native protein or as a LexA fusion, increases the local concentration of ExsA, thereby promoting self-association.
ExsD is only one of two described antiactivators that target members of the AraC family of transcriptional activators, the other being OspD1 from Shigella flexneri (26). Antiactivators from other systems (non-AraC targets) function by one of three mechanisms: preventing self-association of the activator (5), occluding the DNA binding domain from interacting with DNA (19, 22, 24), or inducing conformational changes that alter the structure of the DNA binding domain (6). ExsD inhibits ExsA-dependent transcription by employing at least two of these mechanisms. The first is inhibition of the self-association properties of ExsA (Fig. 6B). This is not surprising as we have shown that ExsD binds to the amino-terminal domain of ExsA and that the amino-terminal domain is required for the self-association and cooperative binding properties of ExsA (3). Our data cannot distinguish whether ExsD prevents self-association from occurring in solution and/or dissociates promoter-bound ExsA that is already self-associated. In either case, our data are consistent with a model in which inhibition of self-association by ExsD would disrupt the cooperative binding properties of ExsA. The second mechanism of inhibition by ExsD comes from the observation that the ExsD·ExsA complex is unable to form shift product 1 in EMSAs (Fig. 1, lanes 6). Since generation of shift product 1 is not dependent upon cooperative binding (3), this finding demonstrates that ExsD also interferes with the DNA binding activity of ExsA. Whether inhibition by ExsD results from steric hindrance of the DNA binding motif or from an induced structural change in ExsA will be the subject of future studies. Finally, the fact that ExsD inhibits the DNA binding activity of ExsA raises the question as to whether inhibition of ExsA self-association by ExsD is physiologically significant since self-association seems to occur only when ExsA is bound to DNA.
Our studies thus far have focused on the mechanism of inducing T3SS gene expression by the ExsA-ExsD-ExsC-ExsE cascade. The model states that ExsE secretion triggers ExsC-dependent dissociation of the ExsD·ExsA complex and subsequent activation of T3SS gene expression and is consistent with the in vitro data presented in Fig. 1. In theory, the cascade should also work in reverse to inhibit T3SS gene expression following exposure to nonpermissive environmental conditions (i.e., high Ca2+). Under this scenario termination of secretion would result in an increase in the intracellular concentration of ExsE. There are two potential mechanisms by which ExsE could inhibit T3SS gene expression: sequestration of ExsC and/or dissociation of the ExsC·ExsD complex. In either case, the outcome would promote formation of the inhibitory ExsD·ExsA complex. A previous study demonstrated that addition of ExsE to the ExsD·ExsC complex results in the release of ExsD (37). We also found that incubation of ExsAHis and the ExsD·ExsCHis complex with ExsEHis resulted in the release of free ExsD. Nevertheless, the newly released ExsD was unable to bind to ExsAHis (Fig. 5). This was not entirely unexpected as the ExsD·ExsA complex forms only when both proteins are coexpressed in E. coli (28) but does not form when the proteins are expressed separately in E. coli and then mixed (28) (E. D. Brutinel and T. L. Yahr, unpublished data). The purified ExsD is not grossly misfolded because the ExsD·ExsC complex is formed when ExsD is mixed with purified ExsC (37). In principle, the ExsD required to form the ExsD·ExsA complex could be generated through liberation from the ExsD·ExsC complex and/or de novo protein synthesis. The fact that ExsD is unable to bind ExsA unless both proteins are simultaneously expressed would suggest that de novo synthesis of ExsD is required for inhibition of T3SS gene expression. ExsD appears to have three fates when synthesized: ExsA binding, ExsC binding, or self-association into a trimer (1, 37). One possibility to account for the data is that ExsD or ExsA preferentially binds to a folding intermediate of the cognate partner to form the 1:1 stoichiometric complex. Once folding proceeds past a critical point, however, ExsD or ExsA would no longer be capable of interacting with its partner. Another possibility is that ExsD dissociated from the 2:2 stoichiometric complex with ExsC is released in a dimeric conformation that is unable to interact with ExsA. A final explanation is that ExsD in the trimeric state is incompatible for ExsA binding. Regardless of the mechanism, the implication of these findings is that ExsE does not inhibit T3SS gene expression by dissociating the ExsD·ExsC complex because the ExsD that is released is inactive. Instead, our data indicate that the primary role of ExsE is to sequester ExsC from ExsD.
Acknowledgments
We thank Mark Urbanowski for constructing the ΔexsA and ΔexsA exsD mutants and for critical input.
This study was supported by the National Institutes of Health (grant RO1-AI055042-07).
Footnotes
Published ahead of print on 11 December 2009.
REFERENCES
- 1.Bernhards, R. C., X. Jing, N. J. Vogelaar, H. Robinson, and F. D. Schubot. 2009. Structural evidence suggests that antiactivator ExsD from Pseudomonas aeruginosa is a DNA binding protein. Protein Sci. 18:503-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brutinel, E. D., C. A. Vakulskas, K. M. Brady, and T. L. Yahr. 2008. Characterization of ExsA and of ExsA-dependent promoters required for expression of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 68:657-671. [DOI] [PubMed] [Google Scholar]
- 3.Brutinel, E. D., C. A. Vakulskas, and T. L. Yahr. 2009. Functional domains of ExsA, the transcriptional activator of the Pseudomonas aeruginosa type III secretion system. J. Bacteriol. 191:3811-3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brutinel, E. D., and T. L. Yahr. 2008. Control of gene expression by type III secretory activity. Curr. Opin. Microbiol. 11:128-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chai, Y., J. Zhu, and S. C. Winans. 2001. TrlR, a defective TraR-like protein of Agrobacterium tumefaciens, blocks TraR function in vitro by forming inactive TrlR:TraR dimers. Mol. Microbiol. 40:414-421. [DOI] [PubMed] [Google Scholar]
- 6.Chen, G., P. D. Jeffrey, C. Fuqua, Y. Shi, and L. Chen. 2007. Structural basis for antiactivation in bacterial quorum sensing. Proc. Natl. Acad. Sci. U. S. A. 104:16474-16479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coburn, B., I. Sekirov, and B. B. Finlay. 2007. Type III secretion systems and disease. Clin. Microbiol. Rev. 20:535-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daines, D. A., M. Granger-Schnarr, M. Dimitrova, and R. P. Silver. 2002. Use of LexA-based system to identify protein-protein interactions in vivo. Methods Enzymol. 358:153-161. [DOI] [PubMed] [Google Scholar]
- 9.Dasgupta, N., G. L. Lykken, M. C. Wolfgang, and T. L. Yahr. 2004. A novel anti-anti-activator mechanism regulates expression of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 53:297-308. [DOI] [PubMed] [Google Scholar]
- 10.Egan, S. M. 2002. Growing repertoire of AraC/XylS activators. J. Bacteriol. 184:5529-5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frank, D. W., and B. H. Iglewski. 1991. Cloning and sequence analysis of a trans-regulatory locus required for exoenzyme S synthesis in Pseudomonas aeruginosa. J. Bacteriol. 173:6460-6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodman, A. L., B. Kulasekara, A. Rietsch, D. Boyd, R. S. Smith, and S. Lory. 2004. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev. Cell 7:745-754. [DOI] [PubMed] [Google Scholar]
- 13.Hauser, A. R. 2009. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 7:654-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoang, T. T., R. R. Karkhoff-Schweizer, A. J. Kutchma, and H. P. Schweizer. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77-86. [DOI] [PubMed] [Google Scholar]
- 15.Hogardt, M., M. Roeder, A. M. Schreff, L. Eberl, and J. Heesemann. 2004. Expression of Pseudomonas aeruginosa exoS is controlled by quorum sensing and RpoS. Microbiology 150:843-851. [DOI] [PubMed] [Google Scholar]
- 16.Kuchma, S. L., J. P. Connolly, and G. A. O'Toole. 2005. A three-component regulatory system regulates biofilm maturation and type III secretion in Pseudomonas aeruginosa. J. Bacteriol. 187:1441-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kulasakara, H., V. Lee, A. Brencic, N. Liberati, J. Urbach, S. Miyata, D. G. Lee, A. N. Neely, M. Hyodo, Y. Hayakawa, F. M. Ausubel, and S. Lory. 2006. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. Proc. Natl. Acad. Sci. U. S. A. 103:2839-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lapouge, K., M. Schubert, F. H. Allain, and D. Haas. 2008. Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol. Microbiol. 67:241-253. [DOI] [PubMed] [Google Scholar]
- 19.Liu, D., R. Ishima, K. I. Tong, S. Bagby, T. Kokubo, D. R. Muhandiram, L. E. Kay, Y. Nakatani, and M. Ikura. 1998. Solution structure of a TBP-TAF(II)230 complex: protein mimicry of the minor groove surface of the TATA box unwound by TBP. Cell 94:573-583. [DOI] [PubMed] [Google Scholar]
- 20.Lykken, G. L., G. Chen, E. D. Brutinel, L. Chen, and T. L. Yahr. 2006. Characterization of ExsC and ExsD self-association and heterocomplex formation. J. Bacteriol. 188:6832-6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCaw, M. L., G. L. Lykken, P. K. Singh, and T. L. Yahr. 2002. ExsD is a negative regulator of the Pseudomonas aeruginosa type III secretion regulon. Mol. Microbiol. 46:1123-1133. [DOI] [PubMed] [Google Scholar]
- 22.Mol, C. D., A. S. Arvai, R. J. Sanderson, G. Slupphaug, B. Kavli, H. E. Krokan, D. W. Mosbaugh, and J. A. Tainer. 1995. Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell 82:701-708. [DOI] [PubMed] [Google Scholar]
- 23.Mueller, C. A., P. Broz, and G. R. Cornelis. 2008. The type III secretion system tip complex and translocon. Mol. Microbiol. 68:1085-1095. [DOI] [PubMed] [Google Scholar]
- 24.Navarro-Aviles, G., M. A. Jimenez, M. C. Perez-Marin, C. Gonzalez, M. Rico, F. J. Murillo, M. Elias-Arnanz, and S. Padmanabhan. 2007. Structural basis for operator and antirepressor recognition by Myxococcus xanthus CarA repressor. Mol. Microbiol. 63:980-994. [DOI] [PubMed] [Google Scholar]
- 25.Newman, J. R., and C. Fuqua. 1999. Broad-host-range expression vectors that carry the l-arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene 227:197-203. [DOI] [PubMed] [Google Scholar]
- 26.Parsot, C., E. Ageron, C. Penno, M. Mavris, K. Jamoussi, H. d'Hauteville, P. Sansonetti, and B. Demers. 2005. A secreted anti-activator, OspD1, and its chaperone, Spa15, are involved in the control of transcription by the type III secretion apparatus activity in Shigella flexneri. Mol. Microbiol. 56:1627-1635. [DOI] [PubMed] [Google Scholar]
- 27.Rietsch, A., I. Vallet-Gely, S. L. Dove, and J. J. Mekalanos. 2005. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 102:8006-8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thibault, J., E. Faudry, C. Ebel, I. Attree, and S. Elsen. 2009. Anti-activator ExsD forms a 1:1 complex with ExsA to inhibit transcription of type III secretion operons. J. Biol. Chem. 284:15762-15770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Urbanowski, M. L., E. D. Brutinel, and T. L. Yahr. 2007. Translocation of ExsE into Chinese hamster ovary cells is required for transcriptional induction of the Pseudomonas aeruginosa type III secretion system. Infect. Immun. 75:4432-4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Urbanowski, M. L., G. L. Lykken, and T. L. Yahr. 2005. A secreted regulatory protein couples transcription to the secretory activity of the Pseudomonas aeruginosa type III secretion system. Proc. Natl. Acad. Sci. U. S. A. 102:9930-9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vakulskas, C. A., K. M. Brady, and T. L. Yahr. 2009. Mechanism of transcriptional activation by Pseudomonas aeruginosa ExsA. J. Bacteriol. 191:6654-6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ventre, I., A. L. Goodman, I. Vallet-Gely, P. Vasseur, C. Soscia, S. Molin, S. Bleves, A. Lazdunski, S. Lory, and A. Filloux. 2006. Multiple sensors control reciprocal expression of Pseudomonas aeruginosa regulatory RNA and virulence genes. Proc. Natl. Acad. Sci. U. S. A. 103:171-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogel, H. J., and D. M. Bonner. 1956. Acetylornithinase of Escherichia coli: partial purification and some properties. J. Biol. Chem. 218:97-106. [PubMed] [Google Scholar]
- 34.Wolfgang, M. C., V. T. Lee, M. E. Gilmore, and S. Lory. 2003. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev. Cell 4:253-263. [DOI] [PubMed] [Google Scholar]
- 35.Wu, W., H. Badrane, S. Arora, H. V. Baker, and S. Jin. 2004. MucA-mediated coordination of type III secretion and alginate synthesis in Pseudomonas aeruginosa. J. Bacteriol. 186:7575-7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yahr, T. L., and M. C. Wolfgang. 2006. Transcriptional regulation of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 62:631-640. [DOI] [PubMed] [Google Scholar]
- 37.Zheng, Z., G. Chen, S. Joshi, E. D. Brutinel, T. L. Yahr, and L. Chen. 2007. Biochemical characterization of a regulatory cascade controlling transcription of the Pseudomonas aeruginosa type III secretion system. J. Biol. Chem. 282:6136-6142. [DOI] [PubMed] [Google Scholar]