

Abstract
RNA polymerase I (Pol I) transcription in the yeast Saccharomyces cerevisiae is greatly stimulated in vivo and in vitro by the multiprotein complex, upstream activation factor (UAF). UAF binds tightly to the upstream element of the rDNA promoter, such that once bound (in vitro), UAF does not readily exchange onto a competing template. Of the polypeptides previously identified in purified UAF, three are encoded by genes required for Pol I transcription in vivo: RRN5, RRN9, and RRN10. Two others, p30 and p18, have remained uncharacterized. We report here that the N-terminal amino acid sequence, its mobility in gel electrophoresis, and the immunoreactivity of p18 shows that it is histone H3. In addition, histone H4 was found in UAF, and myc-tagged histone H4 could be used to affinity-purify UAF. Histones H2A and H2B were not detectable in UAF. These results suggest that histones H3 and H4 probably account for the strong binding of UAF to DNA and may offer a means by which general nuclear regulatory signals could be transmitted to Pol I.
Of the three nuclear RNA polymerases in eukaryotes, RNA polymerase I (Pol I) is responsible for rRNA synthesis. In the yeast Saccharomyces cerevisiae, rRNA synthesis is the only essential function of Pol I, because strains that were mutated or deleted for one of the large subunits of Pol I, but which express rRNA from the GAL7 promoter by RNA polymerase II (Pol II), are viable (1). By using this GAL7-rDNA construction, other mutants could be isolated that were specifically defective in Pol I transcription (rrn mutants) and hence were dependent on galactose-induced Pol II transcription of rDNA for growth (2).
In addition to identifying genes for subunits of Pol I not shared with the other polymerases, studies on the rrn mutants led to discovery of Pol I-specific transcription factors. Extracts from such rrn mutant strains were defective for specific in vitro transcription of a rDNA template, but a protein fraction from a wild-type extract could restore activity, providing an assay for purification of Pol I transcription factors. The wild-type RRN genes, which were obtained by genetic complementation of the mutants, were tagged with the influenza virus hemagglutinin (HA1) epitope, enabling construction of strains expressing only the epitope-tagged RRN genes, and greatly aiding in purification of the factors from these strains (3–5).
The rDNA promoter of yeast, like that of higher eukaryotes, consists of two elements: an essential core element of about 50 bp including the start site of transcription, and an upstream element extending to about −150, which is not essential but is required for a higher level of transcription (refs. 4 and 6; see also refs. 7 and 8). The core element supports a low level of transcription, for which core factor (a multisubunit complex consisting of Rrn6p, Rrn7p, and Rrn11p; refs. 3, 9, and 10), Rrn3p (5), and Pol I are required. In addition to these factors, upstream activation factor (UAF) and TATA box-binding protein, as well as the upstream element, are required for a high level of transcription from the yeast rDNA promoter (4, 11). Purified UAF previously was shown to contain three genetically defined subunits, Rrn5p, Rrn9p, and Rrn10p, and two uncharacterized subunits, p30 and p18 (4).
DNA in the eukaryotic nucleus is organized as chromatin, consisting mostly of regularly repeating nucleosomes in which DNA is wrapped around an octameric structure of core histones (for reviews, see refs. 12 and 13). The core histones, H2A, H2B, H3, and H4, are small (15–18 kDa) and very basic proteins, and are present in two copies of each per nucleosome. Their N-terminal tails contain multiple lysine residues, which are acetylated or deacetylated in various combinations to regulate nucleosome assembly or interactions with other proteins, including transcription factors. Nucleosome structures generally prevent access of RNA polymerase and protein factors to promoters and are used to maintain genes in inactive states; some special mechanisms appear to be used to remodel or disrupt the nucleosome structures, allowing initiation of transcription (for reviews, see refs. 14–17). Thus, histones, the components of the nucleosome that bind DNA tightly, generally are considered to be repressive in gene expression. In this paper, we report our finding that histones H3 and H4 are components of UAF, a transcription activator used for yeast rDNA transcription by Pol I.
MATERIALS AND METHODS
Strains and Plasmids.
Strains and plasmids used are listed in Table 1. Strain NOY847, in which RRN5 is tagged with hexa-histidine [(His)6] and a triple HA1 [(HA1)3]-epitope at the C terminus [“RRN5-(HA1)3-(His)6”], and HHF2 (encoding histone H4) is myc-tagged, was constructed as follows: First, NOY844, carrying (His)6-tagged and (HA1)3-tagged RRN5 on a HIS3-marked CEN plasmid, was crossed with MX1–4C (a gift from M. M. Smith, University of Virginia; ref. 18), in which both chromosomal loci encoding histones H3 and H4 are deleted and are complemented by a URA3-marked CEN plasmid carrying one of the loci, HHT1-HHF1. The resulting diploid was His+ Ura+ and Leu+. The diploid was sporulated, and the tetrads were dissected. Haploid segregants that were His+ [RRN5-(HA1)3-(His)6], Ura+ (HHT1-HHF1) and Leu+ (chromosomal disruption of RRN5) were screened for sensitivity to 5-fluoroorotic acid (5-FOA), indicating that the URA3 plasmid carrying HHT1-HHF1 was essential for viability because both chromosomal deletions of the histone H3, H4 loci were present. The resulting strain, NOY846, was transformed with pNOY436, a TRP1-marked CEN plasmid carrying HHT2 and myc-tagged HHF2 (see Table 1 for construction details). NOY847 was a transformant selected for 5-FOA resistance, indicating loss of the URA3 (pMS329) plasmid carrying wild-type HHT1 and HHF1. Thus all of the histone H4 in NOY847 is myc-tagged.
Table 1.
Yeast strains and plasmids used
| Designation | Description |
|---|---|
| Strains | |
| NOY577 | MATα ade2 ade3 leu2 ura3 trp1 his can1 rrn5Δ::TRP1, pNOY103 (URA3, GAL7-35S rDNA) (4) |
| NOY798 | NOY577 carrying pNOY402 [LEU2, RRN5-(HA1)3-(His)6] instead of pNOY103 |
| NOY700 | MATa ade2-1 ura3-1 his3-11 trp1-1 leu2-3, 112 can1-100 rrn5Δ::LEU2, pNOY103; same as NOY699 (4) except for mating type |
| NOY844 | NOY700 carrying pNOY434 [HIS3, RRN5-(HA1)3-(His)6] instead of pNOY103 |
| MX1-4C | MATα ura3-52 leu2-3, 112 trp1 his3 Δ (hht1-hhf1) Δ(hht2-hhf2), pMS329 (CEN4 URA3 SUP11 HHT1-HHF1) (18) |
| NOY846 | MATα ura3 his3 trp1 leu2-3,112 rrn5Δ::LEU2Δ(hht1-hhf1) Δ(hht2-hhf2), pNOY434 [HIS3 RRN5-(HA1)3-(His)6], pMS329 (URA3 SUP11 HHT1-HHF1) |
| NOY847 | NOY846 carrying pNOY436 [TRP1 HHT2, myc-HHF2) instead of pMS329 |
| Plasmids | |
| pNOY103 | High-copy number plasmid carrying GAL7-35S rDNA, ADE3, URA3, 2μ, (2) |
| pNOY330 | LEU2, CEN6, RRN5-(HA1)3 (4) |
| pNOY402 | LEU2, CEN6, RRN5-(HA1)3-(His)6 derivative of pNOY330 in which an oligonucleotide encoding His tag (GSSHHHHHHSSG) is inserted immediately after the third HA1 repeat |
| pRS313 | Yeast-E. coli shuttle vector (19); HIS3, CEN6, ARS4 |
| pNOY434 | HIS3, CEN6, ARS4, RRN5-(HA1)3-(His)6, the 1.7-kb SalI/SacI fragment from pNOY402 carrying the His-tagged, HA1-tagged RRN5 gene cloned between the XhoI and SacI sites of pRS313 |
| pRS314 | Yeast-E. coli shuttle vector (19); TRP1, CEN6, ARS4 |
| pCC66 | Plasmid clone carrying the HHT2-HHF2 locus (a gift from F. Winston, Harvard; ref. 20) |
| pNOY435 | A 2.1-kb SacI/EcoRI fragment carrying HHT2 and HHF2 was amplified by PCR and cloned between the SacI and EcoRI sites of pRS314 |
| pNOY436 | TRP1, CEN6, ARS4, HHT2, myc-HHF2; oligonucleotide encoding a myc tag sequence (SEQKLISEEDL) inserted between the first and second codons of HHF2 in pNOY435 by site-directed mutagenesis |
Purification of UAF.
Purification of UAF usually was carried out by using strain NOY798 according to the scheme shown in Fig. 1A. For preparation of extracts, cells were grown in yeast extract/peptone/glucose medium (1) to OD600 1.0 to 1.2. Cells were collected by centrifugation, washed in 200 mM Tris⋅HCl, pH 8.0/0.3 M (NH4)2SO4/10 mM MgCl2/10% glycerol/0.1% Tween-20/1.0 mM phenylmethylsulfonyl fluoride, centrifuged again, and stored at −70°C. Frozen cells were thawed in 6 ml of breakage buffer per gram of cell paste [buffer as above but 0.4 M (NH4)2SO4/10 mM imidazole], disrupted in a French pressure cell at 20,000 psi, and centrifuged at 100,000 × g for 1 hr. The supernatant was added to NiSO4-charged chelating Sepharose (Pharmacia) and mixed by rotation at 4°C for 1 hr. After low-speed centrifugation (1,000 × g, 5 min) the nickel-resin was poured into a column and washed with 1 vol of 20 mM Tris⋅acetate, pH 8.0/400 mM KCl/10 mM Mg-acetate/20% glycerol/0.1% Tween-20. (His)6-tagged UAF was eluted with the above buffer containing 250 mM imidazole. For large-scale purification, the elution was done directly onto a heparin-Sepharose cartridge (5 ml, Pharmacia). The heparin cartridge was detached from the nickel-resin column and eluted with a 0.4 M to 1 M KCl gradient [in 20 mM Tris⋅HCl, pH 8.0/10 mM MgCl2/20% glycerol/0.1% Tween-20/0.2 mM EDTA/0.5 mM DTT]. UAF was eluted at about 0.7 M KCl. Peak fractions were identified by immunoreactivity with 12CA5 anti-HA1 mAb (3). For anti-HA1 affinity chromatography, the anti-HA1 antibody was precipitated from ascites fluid with 55% (NH4)2SO4 (wt/vol), purified by gradient elution from Q Sepharose and crosslinked to NHS-activated Sepharose (Pharmacia). To pooled UAF-containing peak fractions from the heparin Sepharose column, protease inhibitors (see ref. 4) were added followed by mixing with anti-HA1 beads. The mixture was rotated in the cold for 2 hr. Beads then were washed, and UAF was eluted with HA1 peptide onto a heparin cartridge as described previously (4). The heparin cartridge was eluted by a KCl gradient as above and gave only a single protein peak. The peak fractions contained essentially pure UAF as determined by SDS/PAGE followed by silver staining. Purified UAF was trichloroacetic acid (TCA)-precipitated, applied to SDS/PAGE, transferred to a (poly)vinylidene difluoride membrane, and stained by Coomassie blue. The p18 band was excised and sequenced by the Macromolecular Structure Facility at Michigan State University.
Figure 1.
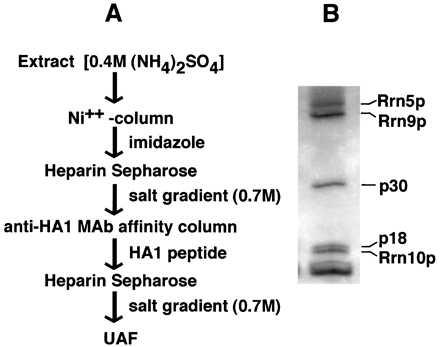
Purification of UAF. (A) Scheme for purification of UAF. (B) A silver-stained gel of a peak fraction from the final column. The polypeptide components of UAF previously identified (4) are indicated. Aprotinin added as carrier for TCA precipitation appears as a heavy band at the bottom of the gel. A band just above this heavy band may represent histone H4 (see Discussion). Separation was in a 10–15% SDS/pHast gel.
UAF also was prepared from a strain in which histone H4 was myc-tagged (NOY847) as described above through the nickel column step. UAF was applied to anti-HA1 beads and washed as described above, then batch eluted in 10 mM Tris⋅HCl, pH 8.0/0.4 M KCl/20% glycerol/0.05% Tween/0.5 mM DTT/protease inhibitors/0.2 mM phenylmethylsulfonyl fluoride/2 mg/ml HA1 peptide. Anti-myc affinity resin was prepared by first purifying 9E10 mAb from ascites fluid (Babco, Richmond, CA) and then crosslinking it to activated Sepharose as described above for anti-HA1 antibody. Control resin was prepared in parallel without the anti-myc antibody. The purified UAF was applied to anti-myc or control resin, washed in 10 mM Tris⋅HCl, pH 8.0/0.1 M KCl/20% glycerol/0.5% Tween/0.5 mM DTT, and batch-eluted twice in the same buffer as used for anti-HA1 elution above, with myc peptide instead of HA1 peptide, and the eluates were combined.
Pol I Transcription Assay.
UAF activity was assayed by using a reconstituted in vitro transcription system that will be described in detail elsewhere. Briefly, the reaction mixture contained linear template DNA that carried up to −210 bp relative to the rDNA transcription start site and produced a runoff transcript of about 550 bases, recombinant TATA box-binding protein, and Rrn3p purified from Escherichia coli, purified Pol I, and partially purified core factor that was free from UAF. Transcripts were labeled with α-[32P]GTP and quantified by PhosphorImager.
Analysis of Histones.
To analyze which core histones copurified with UAF, nickel affinity-purified UAF (as above) was chromatographed on a Superdex 200 sizing column in 20 mM Tris⋅HCl, pH 8.0/0.4 M KCl/20% glycerol/0.1% Tween-20/0.2 mM EDTA/0.5 mM DTT, and UAF-containing fractions were identified immunologically by using anti-HA1 antibody. The peak fractions were pooled, applied to heparin Sepharose, and step-eluted batch-wise in the same buffer but containing 1 M KCl. After TCA precipitation, the UAF-containing material was subjected to 12% SDS/PAGE and Western blotted. Blots were incubated overnight with rabbit polyclonal antibodies made against purified yeast core histones H2A and H2B (a gift of A. Carmen and M. Grunstein, University of California, Los Angeles), diluted 1:2,500 and 1:1,000, respectively, or with rabbit serum directed against the unacetylated and acetylated amino terminal tails of histone H3 (a gift of S. Y. Roth, University of Texas, M.D. Anderson Cancer Center, Houston), each diluted 1:500. Secondary goat antibody against whole rabbit IgG conjugated to alkaline phosphatase (Sigma No. A8025) was diluted 1:4,000.
Mouse mAb against an epitope common to all core histones (Boehringer), but which detected only H3 and H4 in our hands, was diluted to 1 μg/ml for overnight incubation. Secondary goat anti-mouse (against whole IgG) antibody (Sigma No. A3688) was diluted 1:4,000. Yeast histones for use as standards were prepared as described (21).
RESULTS
Identification of Histones H3 and H4 as Subunits of UAF.
We wanted to purify enough UAF to obtain amino acid sequences from the p30 and p18 subunits, which had not been identified genetically. We first constructed a yeast strain (NOY798) in which RRN5 is replaced with RRN5 tagged with hexa-histidine as well as with a triple HA1 epitope. Extracts were made from this strain, and UAF was purified according to the scheme described in Fig. 1A, which consists of a nickel column affinity step and an immunoaffinity purification that used anti-HA1 antibodies combined with two heparin Sepharose chromatographic steps. Fig. 1B shows one of the final heparin agarose fractions after analytical SDS/PAGE and silver staining. The subunit composition appeared to be the same as previously described, including the three genetically defined subunits Rrn9p, Rrn5p, and Rrn10p, and also p30 and p18. By using preparative SDS/PAGE, which enabled better separation of p18 from Rrn10p than that shown in Fig. 1B, p18 was isolated, and its N-terminal amino acid sequence was determined by a conventional Edman degradation. The 20-aa residues identified were the same as those of yeast histone H3. (The p30 band also was analyzed, but we have not succeeded in obtaining sequence information for this protein.)
The identification of p18 as histone H3 raised an obvious question, namely, if other histones, especially histone H4, are also present in UAF, because histone H3 usually is complexed with H4 in vivo. To examine this question, we first analyzed the purified UAF preparation shown in Fig. 1B in a higher percentage gel for improved separation in the low molecular weight range, and carried out Western immunoblot analysis by using a mAb directed against all four mammalian core histones. In our hands the antibody reacted most strongly with histones H3 and H4 in the preparations of yeast core histones used for standards (Fig. 2A). As expected, the p18 (histone H3) band was detectable in UAF. In addition, a band corresponding to histone H4 was evident in the purified UAF (Fig. 2A). (The amount of histone H4 relative to H3 in the purified UAF preparation analyzed in this way appears to be significantly less than that found in the standards. The question of stoichiometry of H4 relative to H3 needs further study.) Silver staining of the purified UAF preparation in a higher-percentage gel also visualized a band corresponding to histone H4 (Fig. 2B). A separate immunoblot analysis confirmed that Rrn10p migrated between histones H3 and H4 with a mobility similar to that of histone H2A (data not shown).
Figure 2.
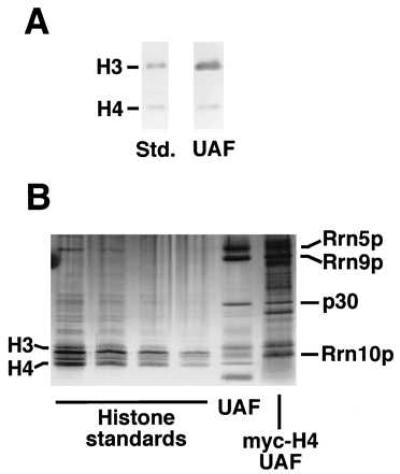
The presence of histone H4 in purified UAF. (A) Western immunoblot analysis of yeast core histone standards and the purified UAF preparation shown in Fig. 1. Separation was in 12% SDS/PAGE. The blot was probed with mAb against all four mammalian core histones, but which reacts mostly with yeast histones H3 and H4. (B) Silver-stained gel of yeast core histone standards, the UAF shown in Fig. 1, and UAF purified from a strain (NOY847) in which histone H4 is myc-tagged. The latter preparation was obtained without heparin Sepharose steps and shows several contaminating protein bands. The lanes of standards are 2-fold serial dilutions. The core histones in order of decreasing molecular weight are H3, H2B (the darkest band), H2A (faint), H3* (a fragment of H3, see ref. 21), and H4. Separation was in a 8–25% SDS/pHast gel. The heavy lowest band in the UAF lane is aprotinin (carrier), and Rrn10p appears as a diffuse band between H3 and H4.
To verify that histone H4 was part of the UAF complex, we constructed a strain (NOY847) in which histone H4 was myc-tagged in addition to the tagging of RRN5 with (His)6 and (HA1)3. From this strain, UAF was purified by nickel and anti-HA1 affinity chromatography but without the heparin steps used in the method described in Fig. 1A. When this UAF preparation was analyzed by SDS/PAGE and silver staining, the Rrn5p, Rrn9p, p30, and histone H3 bands were present. The histone H4 band was not present, but instead there was a band of increased molecular weight corresponding to myc-H4 (Fig. 2B, lane myc-H4 UAF). Immunoblotting that used anti-myc antibody confirmed that this band corresponded to myc-tagged histone H4 (data not shown). The Rrn10p band in myc-H4 UAF is obscured by the myc-tagged H4 band.
The above-described UAF preparation that contained myc-tagged histone H4 was applied to affinity resin with anti-myc antibody crosslinked to it, or to control resin. After washing, proteins bound via the myc-epitope were eluted with myc-peptide, and their UAF activities were assayed by in vitro transcription. This in vitro system has been reconstituted from purified components. Without UAF, a low basal level of activity was observed, whereas addition of UAF stimulated transcription (Fig. 3). The activities of the myc-peptide eluates indicate that the eluate from the anti-myc resin contained a significant amount of UAF relative to the eluate from the control resin (Fig. 3). Thus myc-tagged histone H4 is a stably bound component of UAF.
Figure 3.
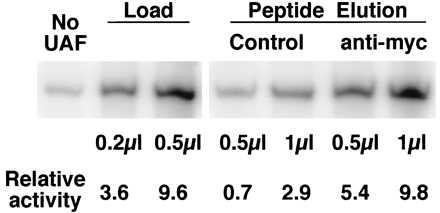
Transcription stimulatory activity of UAF preparations obtained from the myc-tagged histone H4 strain (NOY847) with and without a myc-affinity purification step. The UAF preparation (Load) containing myc-tagged histone H4 (shown in Fig. 2B) was incubated with control beads or with beads carrying anti-myc antibody crosslinked to them. After repeated washing, proteins were eluted with myc peptide. Samples of the indicated amounts were added to 20-μl reactions of a yeast Pol I in vitro transcription system that lacked UAF. Portions of a gel displaying the resulting transcripts are shown. The relative activities were corrected for activity in the absence of added UAF.
The association of histone H4 with UAF also was demonstrated by carrying out sizing column chromatography. The extract prepared from the strain carrying the myc-tagged H4 (NOY847) was subjected to nickel chelate chromatography and then applied to a Superdex 200 sizing column. Fractions were analyzed for UAF activity in rDNA transcription, and for the presence of Rrn5p (by using anti-HA1 antibodies), histone H3 (by using a mixture of antibodies against an unacetylated H3 peptide and an acetylated H3 peptide), and histone H4 (by using anti-myc antibodies) by SDS/PAGE followed by Western blot analysis. The peak position of histone H4 coincided with the peak positions of UAF activity, Rrn5p, and histone H3. No significant trailing of H4 relative to H3 or Rrn5p was observed, indicating that the association of H4 with other UAF components is strong (data not shown). From the position of size markers, the size of the UAF complex in this experiment was estimated to be roughly 250 kDa, which is the same, within error, as that previously published for a highly purified UAF preparation (4). From all of these experiments, we conclude that both histones H3 and H4 are tightly associated with other UAF components and UAF activity.
The Absence of Histones H2A and H2B in UAF.
Because histones H3 and H4 had been shown to be components of UAF, it was important to determine if the other core histones present in nucleosomes, H2A and H2B, also might be associated with UAF. For this experiment, UAF was purified from strain NOY798 expressing untagged, wild-type histones by using nickel chelate chromatography followed by Superdex 200 sizing column chromatography in the same way as described above. The UAF peak fractions from the sizing column were pooled and then concentrated by binding to heparin Sepharose followed by step elution with high salt. The purified UAF was analyzed by SDS/PAGE followed by immunoblot. Serial 2-fold dilutions of yeast core histones, in which H3, H2B, H2A, and H4 were of about equal intensity after Coomassie staining, served as standards. The blot was probed first with a mixture of two antibodies directed against yeast histones H2A and H2B. Histone H2A was detected with the greatest sensitivity among the standards; a band was visible in the 0.06-μl lane (Fig. 4). By contrast neither histones H2A nor H2B were detected in the UAF sample (Fig. 4). Next, the same blot was probed with antibodies directed against histone H3. The H3 band was evident in the UAF samples and in the lanes with the largest amounts of standards (Fig. 4). From the results, we estimated that histone H2A, if present, was below 0.4% of the level of H3 in UAF (see the legend to Fig. 4). Histone H2B in the standard preparation used in Fig. 2B was stained most strongly by silver. In contrast, no corresponding band was observed in the purified UAF preparation (Fig. 2B, lane UAF). By comparing UAF with the standard as was done for H2A, we estimated that we would have been able to detect histone H2B in an amount that was 10% of H3 in UAF. We conclude that the other core histones are not components of UAF.
Figure 4.
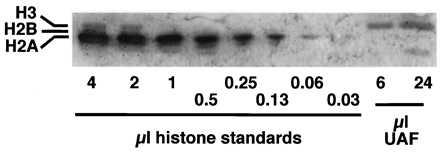
Western immunoblot analysis of histone H2A. The indicated amounts of yeast core histone standards or the purified UAF preparation shown in Fig. 1 were separated by 12% SDS/PAGE, blotted, and probed first for histones H2A (the strongest signal in the standards) and H2B, and subsequently for histone H3. (In the experiment shown here, UAF was concentrated by binding to heparin Sepharose followed by elution and TCA precipitation, whereas histone standards were directly analyzed. In separate experiments, yeast standards were subjected to the same treatments, that is, binding to heparin Sepharose followed by elution and TCA precipitation, and then analyzed. No significant change in relative amounts of histones was observed.) It can be seen that the immunoreactivity of the histone H3 band in 6 μl of UAF is greater than or equal to that of 4 μl of standard, whereas the histone H2A band in 0.06 μl of standard (64 times less than 4 μl) is greater than that in the 24-μl sample of UAF (4 times greater than 6 μl). Thus the ratio of histone H3 to H2A in the 24-μl sample is at least 256:1 (4 × 64), that is, histone H2A can be estimated to be present at less than 0.4% of the level of H3 in UAF.
DISCUSSION
We have presented evidence that the p18 component of the yeast Pol I activator complex, UAF, is histone H3. The N-terminal amino acid sequence, its mobility in SDS/PAGE, and the immunoreactivity of p18 are the same as histone H3. We then looked for and found histone H4 in UAF. The band in UAF that comigrated with histone H4 showed similar immunoreactivity as well. When histone H4 was tagged with the myc epitope, the band shifted as expected from the increase in molecular weight, and UAF activity could be bound to and then eluted from anti-myc affinity resin. [We attribute our failure to previously note the presence of histone H4 in pure UAF to diffuse bands in the low molecular weight region of our SDS/PAGE gels and to interference by artifacts visualized by silver staining or by a peptide used as carrier. Indeed, a band corresponding to histone H4 may be visible in figure 6a of the previous paper (4), and a likely band is evident just above the aprotinin band in Fig. 1 of this paper.] The other core histones, H2A and H2B, were not found in UAF; our detection limit was, for H2A, less than 1% of the abundance of H3, and for H2B less than 10%. Thus, it is unlikely that the presence of histones H3 and H4 in our UAF preparations is simply because of contamination by nucleosomes.
Histones H3 and H4 are tightly bound components of UAF. They were retained with UAF through a variety of purification steps, including mAb affinity chromatography directed against the triple HA1 tag on the Rrn5p subunit, nickel chelate chromatography directed against the hexa-histidine tag on Rrn5p, gradient elution from heparin Sepharose and from the cation exchanger MonoS, and molecular sizing chromatography. Thus, the interactions of histones H3 and H4 with the other UAF components appear to be distinguished from the recently described interactions between transcriptional repressors, such as Tup1 (21) or silencing proteins Sir3 and Sir4 (22), and the H3-H4 components of nucleosomes through the N-terminal tails of these histones. Instead, UAF containing histones H3 and H4 is reminiscent of chromatin assembly complex (CAC). In CAC, a histone H3-H4 tetramer was stably bound to chromatin assembly factor (CAF-1) so that it was retained with CAF-1 through an antibody affinity purification step and a glycerol gradient (23). However, CAC functions in initiating assembly of nucleosomes by depositing the H3-H4 tetramer onto newly replicated DNA and is not known to have any function related to transcription. By contrast, UAF is a transcriptional activator and is not known to have any function in nucleosome assembly or rearrangement.
UAF previously was shown to bind and commit a rRNA template to transcription in vitro. That is, UAF binding to the template was necessary for subsequent binding of TATA box-binding protein and core factor into a stable preinitiation complex. Once bound, UAF is not appreciably exchanged onto a second competing template, in the presence or absence of other factors (4). Thus UAF appears to bind strongly to DNA. The genetically identified subunits of UAF, Rrn5p, Rrn9p, and Rrn10p, all have isolectric points of less than 7. Therefore, histones H3 and H4 probably contribute substantially to UAF’s apparent high affinity for its binding site, and perhaps one (or more) of the other subunits is responsible for specificity of binding. In view of the observed tight in vitro binding of UAF to the upstream element of the promoter, there is a good possibility that a UAF-rDNA promoter complex might be formed at the time of DNA replication in vivo, and that the UAF-rDNA promoter complex, with or without other transcription factors, might remain present regardless of whether or not rDNA transcription is actively taking place. Thus, a rDNA repeat with bound UAF might correspond to a rDNA repeat proposed by Sogo and coworkers (24, 25) that is “activatable” but is distinct from a totally inactive repeat with a typical nucleosome beads structure. If this is, in fact, the case, regulation of rDNA transcription might take place without altering the number of such activatable repeats, for example, by causing modification of components of UAF bound to the promoter, histones H3 and H4, in particular (or components involved in subsequent steps that would lead to transcription initiation).
It is known that histones H3 and H4 are posttranscriptionally modified by acetylation of lysine residues in their amino-terminal tails (for recent reviews, see refs. 26 and 27). Although we have not yet determined their acetylation state in UAF, histones H3 and H4 would seem to offer a simple route by which general regulatory signals could be transmitted via UAF to control Pol I transcription, regardless of the question of reversibility of UAF binding to DNA.
It is known that the histone H3-H4 tetramer interacts with DNA and forms a structure similar to a nucleosome (28, 29), presumably by using positively charged residues on the H3-H4 tetramer surface lining the path of the DNA as analyzed for the nucleosome (30, 31). If histones H3 and H4 are present in UAF as a tetramer, and if UAF might wrap DNA like a nucleosome, as has been proposed for TFIID (32), are questions that remain to be addressed in future studies.
Acknowledgments
We thank Drs. M. M. Smith (University of Virginia), M. Grunstein (University of California-Los Angeles), F. Winston (Harvard University), C. D. Allis (University of Rochester), and S. Y. Roth (University of Texas, M. D. Anderson Cancer Center) for kindly providing us with yeast strains, plasmids, and antibodies, as described in this paper. Plasmid pNOY402 and strain NOY798 were constructed by D. A. Keys in this laboratory. We also thank Drs. M. Waterman and S. Arfin for critical reading of the manuscript, and D. Semanko for assistance in its preparation. This work was supported by U.S. Public Health Grant R37GM35949 from the National Institutes of Health.
ABBREVIATIONS
- UAF
upstream activation factor
- Pol I
RNA polymerase I
- HA1
influenza virus hemagglutinin
- TCA
trichloroacetic acid
References
- 1.Nogi Y, Yano R, Nomura M. Proc Natl Acad Sci USA. 1991;88:3962–3966. doi: 10.1073/pnas.88.9.3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nogi Y, Vu L, Nomura M. Proc Natl Acad Sci USA. 1991;88:7026–7030. doi: 10.1073/pnas.88.16.7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keys D A, Vu L, Steffan J S, Dodd J A, Yamamoto R T, Nogi Y, Nomura M. Genes Dev. 1994;8:2349–2362. doi: 10.1101/gad.8.19.2349. [DOI] [PubMed] [Google Scholar]
- 4.Keys D A, Lee B-S, Dodd J A, Nguyen T T, Vu L, Fantino E, Burson L M, Nogi Y, Nomura M. Genes Dev. 1996;10:887–903. doi: 10.1101/gad.10.7.887. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto R T, Nogi Y, Dodd J A, Nomura M. EMBO J. 1996;15:533–561. [PMC free article] [PubMed] [Google Scholar]
- 6.Choe S Y, Schultz M C, Reeder R H. Nucleic Acids Res. 1992;20:279–285. doi: 10.1093/nar/20.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musters W, Knol J, Maas P, Dekker A F, van Heerikhuizen H, Planta R J. Nucleic Acids Res. 1989;17:9661–9678. doi: 10.1093/nar/17.23.9661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kulkens T, Riggs D L, Heck J D, Planta R J, Nomura M. Nucleic Acids Res. 1991;19:5363–5370. doi: 10.1093/nar/19.19.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lalo D, Steffan J S, Dodd J A, Nomura M. J Biol Chem. 1996;271:21062–21067. doi: 10.1074/jbc.271.35.21062. [DOI] [PubMed] [Google Scholar]
- 10.Lin C W, Moorefield B, Payne J, Aprikian P, Mitomo K, Reeder R H. Mol Cell Biol. 1996;16:6436–6443. doi: 10.1128/mcb.16.11.6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steffan J S, Keys D A, Dodd J A, Nomura M. Genes Dev. 1996;10:2551–2563. doi: 10.1101/gad.10.20.2551. [DOI] [PubMed] [Google Scholar]
- 12.van Holde K E. Chromatin. New York: Springer; 1988. [Google Scholar]
- 13.Wolffe A. Chromatin: Structure and Function. London: Academic; 1992. [Google Scholar]
- 14.Paranjape S M, Kamakaka R T, Kadonaga J T. Annu Rev Biochem. 1994;63:265–297. doi: 10.1146/annurev.bi.63.070194.001405. [DOI] [PubMed] [Google Scholar]
- 15.Felsenfeld G. Nature (London) 1992;355:219–224. doi: 10.1038/355219a0. [DOI] [PubMed] [Google Scholar]
- 16.Felsenfeld G. Cell. 1996;86:13–19. doi: 10.1016/s0092-8674(00)80073-2. [DOI] [PubMed] [Google Scholar]
- 17.Pazin M J, Kadonaga J T. Cell. 1997;88:737–740. doi: 10.1016/s0092-8674(00)81918-2. [DOI] [PubMed] [Google Scholar]
- 18.Morgan B A, Mittman B A, Smith M M. Mol Cell Biol. 1991;11:4111–4120. doi: 10.1128/mcb.11.8.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sikorski R S, Heiter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark-Adams C D, Norris D, Osley M A, Frassler J S, Winston F. Genes Dev. 1988;2:150–159. doi: 10.1101/gad.2.2.150. [DOI] [PubMed] [Google Scholar]
- 21.Edmondson D G, Smith M M, Roth S Y. Genes Dev. 1996;10:1247–1259. doi: 10.1101/gad.10.10.1247. [DOI] [PubMed] [Google Scholar]
- 22.Hecht A, Laroche T, Strahl-Bolsinger S, Gasser S M, Grunstein M. Cell. 1995;80:583–592. doi: 10.1016/0092-8674(95)90512-x. [DOI] [PubMed] [Google Scholar]
- 23.Verreault A, Kaufman P D, Kobayashi R, Stillman B. Cell. 1996;87:95–104. doi: 10.1016/s0092-8674(00)81326-4. [DOI] [PubMed] [Google Scholar]
- 24.Conconi A, Widmer R M, Keller T, Sogo J M. Cell. 1989;57:753–761. doi: 10.1016/0092-8674(89)90790-3. [DOI] [PubMed] [Google Scholar]
- 25.Dammann R, Lucchini R, Koller T, Sogo J M. Nucleic Acids Res. 1993;21:2331–2338. doi: 10.1093/nar/21.10.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pazin M J, Kadonaga J T. Cell. 1997;89:325–328. doi: 10.1016/s0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- 27.Roth S Y, Allis C D. Cell. 1996;87:5–8. doi: 10.1016/s0092-8674(00)81316-1. [DOI] [PubMed] [Google Scholar]
- 28.Camerini-Otero R D, Sollner-Webb B, Felsenfeld G. Cell. 1976;8:333–347. doi: 10.1016/0092-8674(76)90145-8. [DOI] [PubMed] [Google Scholar]
- 29.Hayes J J, Clark D J, Wolffe A P. Proc Natl Acad Sci USA. 1991;88:6829–6833. doi: 10.1073/pnas.88.15.6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arents G, Moudrianakis E N. Proc Natl Acad Sci USA. 1993;90:10489–10493. doi: 10.1073/pnas.90.22.10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luger K, Mader A W, Richmond R K, Sargent D F, Richmond T J. Nature (London) 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 32.Hoffmann A, Chiang C-M, Oelgeschlager T, Xie X, Burley S K, Nakatani Y, Roeder R G. Nature (London) 1996;380:356–359. doi: 10.1038/380356a0. [DOI] [PubMed] [Google Scholar]